

Key Points
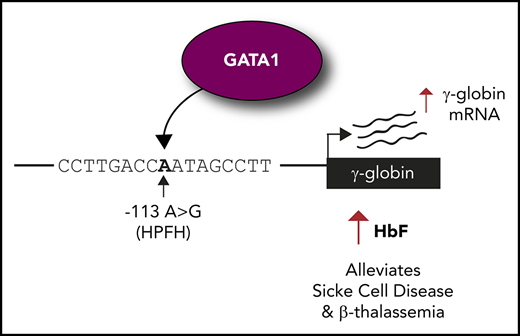
Introducing the −113A>G HPFH mutation into erythroid cells elevates fetal globin levels.
The −113A>G mutation creates a de novo site for the activator GATA1 but does not disrupt binding of fetal globin repressor BCL11A.

Abstract
β-hemoglobinopathies, such as sickle cell disease and β-thalassemia, result from mutations in the adult β-globin gene. Reactivating the developmentally silenced fetal γ-globin gene elevates fetal hemoglobin levels and ameliorates symptoms of β-hemoglobinopathies. The continued expression of fetal γ-globin into adulthood occurs naturally in a genetic condition termed hereditary persistence of fetal hemoglobin (HPFH). Point mutations in the fetal γ-globin proximal promoter can cause HPFH. The −113A>G HPFH mutation falls within the −115 cluster of HPFH mutations, a binding site for the fetal globin repressor BCL11A. We demonstrate that the −113A>G HPFH mutation, unlike other mutations in the cluster, does not disrupt BCL11A binding but rather creates a de novo binding site for the transcriptional activator GATA1. Introduction of the −113A>G HPFH mutation into erythroid cells using the clustered regularly interspaced short palindromic repeats (CRISPR)–CRISPR-associated protein 9 (Cas9) system increases GATA1 binding and elevates fetal globin levels. These results reveal the mechanism by which the −113A>G HPFH mutation elevates fetal globin and demonstrate the sensitivity of the fetal globin promoter to point mutations that often disrupt repressor binding sites but here create a de novo site for an erythroid activator.
Introduction
The fetal γ-globin gene is silenced around the time of birth and normal individuals express fetal hemoglobin (HbF) at levels of ∼1%. Hereditary persistence of fetal hemoglobin (HPFH) is a benign condition in which the duplicated fetal γ-globin genes (Aγ/Gγ) continue to be expressed and produce HbF throughout adulthood. Large β-globin locus deletions or single point mutations or small deletions within the γ-globin proximal promoter can cause HPFH. Coinheriting HPFH with a β-hemoglobinopathy alleviates disease symptoms.1 The majority of HPFH mutations cluster ∼115 and ∼200 bp upstream of the γ-globin transcription start site. Recently, it was shown that the 2 major HbF repressors, BCL11A2,3 and ZBTB7A,4 directly bind to and repress the γ-globin gene through the −115 and −200 sites, respectively, with HPFH mutations at these sites disrupting binding.5,6
Here, we focus on the −115 site of the γ-globin promoter and on a previously reported HPFH mutation: an A to G substitution 113 bp upstream of the γ-globin transcription start site7 (Figure 1A). The −113A>G HPFH mutation was identified as a heterozygous mutation in the Aγ-globin promoter of a 36-year-old Italian subject, boosting HbF levels to 6.5%.7 Although it has recently been shown that BCL11A binding is disrupted by the −117G>A, −114C>A, −114C>T, −114C>G, and 13-bp deletion (Δ13bp) HPFH mutations,5,6 the mechanism by which the −113A>G HPFH mutation operates is unknown. We demonstrate that this mutation does not disrupt the binding of the HbF repressor BCL11A, but rather creates a de novo binding site for the master erythroid regulator GATA18-10 to drive γ-globin expression and HbF production.
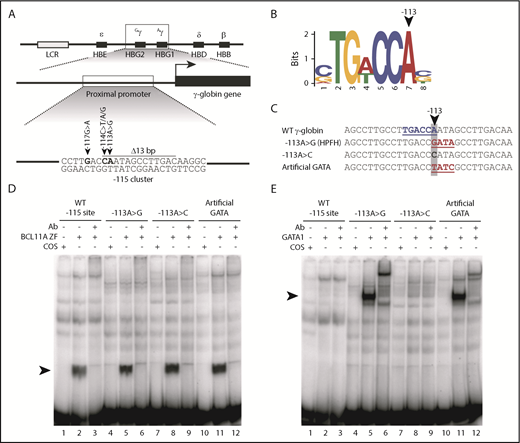
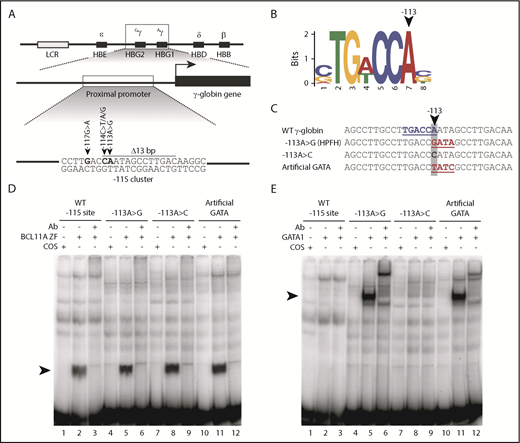
The −113A>G HPFH mutation does not disrupt BCL11A binding but rather creates a de novo binding site for the transcriptional activator GATA1 in vitro. (A) Five mutations and a 13-bp deletion (Δ13bp) at the −115 site of the γ-globin promoter have been reported to induce HPFH. (B) The −113A>G HPFH mutation alters a critical residue within the BCL11A in vivo binding motif TG(A/T)CCA.5,6 (C) The −113A>G HPFH mutation creates a “GATA” motif at the −115 site of the γ-globin promoter. The γ-globin promoter sequences (−128 to −100 bp) of other introduced mutations, −113A>C and −113A>T/−110A>C, which creates an “artificial GATA” site (TATC as the reverse complement), are also shown. The BCL11A TGACCA motif is highlighted in blue and GATA motifs are highlighted in red. (D) EMSA showing that BCL11A zinc fingers (ZF) bind to the wild-type (WT) −115 site of the γ-globin promoter (−128 bp to −100 bp) and that the −113A>G HPFH mutation, −113A>C mutation, and “artificial GATA” site do not disrupt BCL11A binding in vitro. Nuclear extracts were prepared from COS-7 cells transfected with a pcDNA3 empty vector as a control or from COS-7 cells transiently overexpressing BCL11A ZF (amino acids 740-835). A supershift of the BCL11A ZF:probe complex was performed with an antibody (Ab) against endogenous BCL11A. (E) GATA1 does not bind to the WT −115 site of the γ-globin promoter (−128 bp to −100 bp) or the −113A>C mutation, however, the −113A>G HPFH mutation and an “artificial GATA” site create a de novo binding site for GATA1 in vitro. Nuclear extracts were prepared from COS-7 cells transfected with a pcDNA3 empty vector as a control or from COS-7 cells transiently overexpressing full-length GATA1. A supershift of the GATA1:probe complex was performed with an Ab against endogenous GATA1.
The −113A>G HPFH mutation does not disrupt BCL11A binding but rather creates a de novo binding site for the transcriptional activator GATA1 in vitro. (A) Five mutations and a 13-bp deletion (Δ13bp) at the −115 site of the γ-globin promoter have been reported to induce HPFH. (B) The −113A>G HPFH mutation alters a critical residue within the BCL11A in vivo binding motif TG(A/T)CCA.5,6 (C) The −113A>G HPFH mutation creates a “GATA” motif at the −115 site of the γ-globin promoter. The γ-globin promoter sequences (−128 to −100 bp) of other introduced mutations, −113A>C and −113A>T/−110A>C, which creates an “artificial GATA” site (TATC as the reverse complement), are also shown. The BCL11A TGACCA motif is highlighted in blue and GATA motifs are highlighted in red. (D) EMSA showing that BCL11A zinc fingers (ZF) bind to the wild-type (WT) −115 site of the γ-globin promoter (−128 bp to −100 bp) and that the −113A>G HPFH mutation, −113A>C mutation, and “artificial GATA” site do not disrupt BCL11A binding in vitro. Nuclear extracts were prepared from COS-7 cells transfected with a pcDNA3 empty vector as a control or from COS-7 cells transiently overexpressing BCL11A ZF (amino acids 740-835). A supershift of the BCL11A ZF:probe complex was performed with an antibody (Ab) against endogenous BCL11A. (E) GATA1 does not bind to the WT −115 site of the γ-globin promoter (−128 bp to −100 bp) or the −113A>C mutation, however, the −113A>G HPFH mutation and an “artificial GATA” site create a de novo binding site for GATA1 in vitro. Nuclear extracts were prepared from COS-7 cells transfected with a pcDNA3 empty vector as a control or from COS-7 cells transiently overexpressing full-length GATA1. A supershift of the GATA1:probe complex was performed with an Ab against endogenous GATA1.
Study design
Electrophoretic mobility shift assays (EMSAs) were performed as previously described.11
The previously described HUDEP-2(ΔGγ) cell line5,12 was used to study the effects of the −113A>G, −113A>C, and “artificial GATA” site mutations. Cells were nucleofected with clustered regularly interspaced short palindromic repeats (CRISPR)–CRISPR-associated protein 9 (Cas9) plasmid pX458,13 encoding a single-guide RNA (sgRNA) for the −115 γ-globin site and a single-stranded donor oligonucleotide containing the desired mutation. Transfected cells were selected by fluorescent-activated cell sorting, and clonal populations with the desired modification were selected.
Total RNA was extracted and complementary DNA synthesized. Globin expression levels were determined by quantitative polymerase chain reaction (qPCR) as previously described.5 Expression was normalized to 18S ribosomal RNA.
High-performance liquid chromatography (HPLC) was performed as previously described.5
Chromatin immunoprecipitation (ChIP) was performed using antibodies against GATA1 (Cell Signaling Technology, Danvers, MA), BCL11A (Novus Biologicals, Centennial, CO), and control rabbit immunoglobulin G (Cell Signaling Technology).
See supplemental Methods (available on the Blood Web site) for more details.
Results and discussion
The −113A>G HPFH mutation not only occurs at the end of the core binding motif for BCL11A5,6 (Figure 1B) but it also creates a consensus binding site for the erythroid transcriptional regulator GATA18,14 (Figure 1C). We first carried out in vitro experiments to investigate the effect of the −113A>G mutation on BCL11A and GATA1 binding to a probe containing the −115 γ-globin promoter site. We designed 2 additional non-HPFH mutation control probes: a −113A>C mutation that alters the sequence at −113 but does not create a GATA motif, and a probe generating an “artificial GATA” site on the reverse strand (TATC on the upper strand with −113A>T and −110A>C substitutions) (Figure 1C). This last control not only affects the core BCL11A motif but also creates a consensus GATA1-binding site and thus, effectively tests whether introducing a de novo GATA1 site at this location is important or whether some other unrecognized changes drive the phenotype.
We found that BCL11A binds to the wild-type (WT) −115 γ-globin site in EMSAs and that neither the −113A>G HPFH mutation, the −113A>C nor the “artificial GATA” mutations impair BCL11A binding (Figure 1D). And, as expected, both the naturally occurring −113A>G HPFH mutation and the “artificial GATA” probes allow GATA1 binding, whereas the WT and −113A>C mutant probes do not (Figure 1E). These in vitro results were confirmed by cold competition EMSAs (supplemental Figure 1), and there was no evidence of simultaneous binding of BCL11A and GATA1 to the −115 γ-globin site via EMSA (supplemental Figure 2). These results suggest that −113A>G induces HPFH by creating a de novo site for the activator GATA1, rather than by disrupting BCL11A binding.
To investigate the in vivo mechanism, we used the CRISPR-Cas9 system to homozygously introduce these mutations into a previously described HUDEP-2(ΔGγ) cell line,12 which contains only 1 copy of the γ-globin gene per allele and expresses γ-globin at levels similar to unmodified HUDEP-2 cells.15 We targeted sgRNAs to the −115 γ-globin site and introduced −113A>G, −113A>C, and “artificial GATA” mutations to establish homozygous clonal populations (Figure 2A; supplemental Figure 3).
![Figure 2. The −113A>G HPFH mutation elevates γ-globin expression and creates a de novo binding site for GATA1 in vivo. (A) The CRISPR-Cas9 sgRNA target sites used to introduce the −113A>G (HPFH) and −113A>C mutations and the “artificial GATA” site into HUDEP-2(ΔGγ) WT cells. The protospacer adjacent motif (PAM) site is indicated along with the various modifications introduced. (B) γ-globin mRNA expression levels, expressed as a percentage (γ/γ+β) in HUDEP-2(ΔGγ) WT, −113A>G, −113A>C, and “artificial GATA” clonal populations. Eleven technical replicates are shown for the HUDEP-2(ΔGγ) WT parental clone, 10 clonal populations for −113A>G HPFH mutation, and 11 clonal populations for the −113A>C and “artificial GATA” motif. Shown is the mean plus or minus standard error of the mean (SEM). A nonparametric Mann-Whitney statistical test was used to determine statistical significance: P < .0001 (****), P = .0001 to .001 (***), P = .001 to .01 (**). (C) The percentage of HbF protein levels [(HbF/(HbF + HbA)] in HUDEP-2(ΔGγ) WT (n = 6), −113A>G (n = 10), −113A>C (n = 11) and “artificial GATA” (n = 11) clonal populations as determined by HPLC. The mean plus or minus SEM is represented. A nonparametric Mann-Whitney statistical test was used to determine statistical significance: P = .0001 to .001 (***). (D) GATA1 ChIP-qPCR in HUDEP-2(ΔGγ) WT (n = 7), −113A>G (n = 7), −113A>C (n = 5), and “artificial GATA” site (n = 5) clonal populations. The relative fraction of input, normalized to the positive control (+ctrl) locus ZFPM1 (mean plus or minus SEM), is represented. A nonparametric Mann-Whitney statistical test was used to determine statistical significance: P = .01 to .05 (*). Target loci include the −115 site of the γ-globin promoter, the KLF1 promoter as a +ctrl, and the −4.5 kb upstream region of the IFNB gene as negative control (-ctrl). n.s., nonsignificant. HbA, adult hemoglobin.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/133/8/10.1182_blood-2018-07-863951/3/m_blood863951f2.png?Expires=1769171590&Signature=GKTWtgmtox-9s-XWWE4nrakXmOoI~GkLPsJiPS~~uSpCJUGpSe~exA1dCsVz-~~KfesrzbdYTm5ot5J6z9OJL7HCrE7aLXk33wrt2t9h-NJUhgIbs9WZ-2BEEl9l67~2e5Wr61eXWwDou1CrMoLxXoPm~Wt4swCoPdDkRFTIDBl2w-Zx3sCMe6CyzZV9sClegmDEXNMEyaSTD6QglTjPIIOqgEdpgiLzmk47DyJtQFQy4DLyBrgI3yFjErOUXmtz6DAidJtE1tTT0RvxF7S53x1iKpsnpHXzGmL710~sSSYfqUF8xLgxSVpFSX~Pv3CTL~Wf1MmYJfgnsBr5kRxR8A__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
The −113A>G HPFH mutation elevates γ-globin expression and creates a de novo binding site for GATA1 in vivo. (A) The CRISPR-Cas9 sgRNA target sites used to introduce the −113A>G (HPFH) and −113A>C mutations and the “artificial GATA” site into HUDEP-2(ΔGγ) WT cells. The protospacer adjacent motif (PAM) site is indicated along with the various modifications introduced. (B) γ-globin mRNA expression levels, expressed as a percentage (γ/γ+β) in HUDEP-2(ΔGγ) WT, −113A>G, −113A>C, and “artificial GATA” clonal populations. Eleven technical replicates are shown for the HUDEP-2(ΔGγ) WT parental clone, 10 clonal populations for −113A>G HPFH mutation, and 11 clonal populations for the −113A>C and “artificial GATA” motif. Shown is the mean plus or minus standard error of the mean (SEM). A nonparametric Mann-Whitney statistical test was used to determine statistical significance: P < .0001 (****), P = .0001 to .001 (***), P = .001 to .01 (**). (C) The percentage of HbF protein levels [(HbF/(HbF + HbA)] in HUDEP-2(ΔGγ) WT (n = 6), −113A>G (n = 10), −113A>C (n = 11) and “artificial GATA” (n = 11) clonal populations as determined by HPLC. The mean plus or minus SEM is represented. A nonparametric Mann-Whitney statistical test was used to determine statistical significance: P = .0001 to .001 (***). (D) GATA1 ChIP-qPCR in HUDEP-2(ΔGγ) WT (n = 7), −113A>G (n = 7), −113A>C (n = 5), and “artificial GATA” site (n = 5) clonal populations. The relative fraction of input, normalized to the positive control (+ctrl) locus ZFPM1 (mean plus or minus SEM), is represented. A nonparametric Mann-Whitney statistical test was used to determine statistical significance: P = .01 to .05 (*). Target loci include the −115 site of the γ-globin promoter, the KLF1 promoter as a +ctrl, and the −4.5 kb upstream region of the IFNB gene as negative control (-ctrl). n.s., nonsignificant. HbA, adult hemoglobin.
The −113A>G HPFH mutation elevates γ-globin expression and creates a de novo binding site for GATA1 in vivo. (A) The CRISPR-Cas9 sgRNA target sites used to introduce the −113A>G (HPFH) and −113A>C mutations and the “artificial GATA” site into HUDEP-2(ΔGγ) WT cells. The protospacer adjacent motif (PAM) site is indicated along with the various modifications introduced. (B) γ-globin mRNA expression levels, expressed as a percentage (γ/γ+β) in HUDEP-2(ΔGγ) WT, −113A>G, −113A>C, and “artificial GATA” clonal populations. Eleven technical replicates are shown for the HUDEP-2(ΔGγ) WT parental clone, 10 clonal populations for −113A>G HPFH mutation, and 11 clonal populations for the −113A>C and “artificial GATA” motif. Shown is the mean plus or minus standard error of the mean (SEM). A nonparametric Mann-Whitney statistical test was used to determine statistical significance: P < .0001 (****), P = .0001 to .001 (***), P = .001 to .01 (**). (C) The percentage of HbF protein levels [(HbF/(HbF + HbA)] in HUDEP-2(ΔGγ) WT (n = 6), −113A>G (n = 10), −113A>C (n = 11) and “artificial GATA” (n = 11) clonal populations as determined by HPLC. The mean plus or minus SEM is represented. A nonparametric Mann-Whitney statistical test was used to determine statistical significance: P = .0001 to .001 (***). (D) GATA1 ChIP-qPCR in HUDEP-2(ΔGγ) WT (n = 7), −113A>G (n = 7), −113A>C (n = 5), and “artificial GATA” site (n = 5) clonal populations. The relative fraction of input, normalized to the positive control (+ctrl) locus ZFPM1 (mean plus or minus SEM), is represented. A nonparametric Mann-Whitney statistical test was used to determine statistical significance: P = .01 to .05 (*). Target loci include the −115 site of the γ-globin promoter, the KLF1 promoter as a +ctrl, and the −4.5 kb upstream region of the IFNB gene as negative control (-ctrl). n.s., nonsignificant. HbA, adult hemoglobin.
We analyzed γ-globin messenger RNA (mRNA) and HbF protein levels for each of the mutant HUDEP-2(ΔGγ) clones (Figure 2B-C). The −113A>G HPFH mutation and “artificial GATA” site significantly increased γ-globin mRNA and HbF levels, compared with WT HUDEP-2(ΔGγ) cells, demonstrating that creating a de novo GATA motif in the γ-globin promoter, either by virtue of a natural HPFH mutation or artificially, boosts HbF levels. The −113A>C mutation did not increase γ-globin mRNA expression or HbF production, compared with the WT, consistent with the fact that in vitro it neither disrupts BCL11A binding nor creates a de novo GATA1 site.
Next, we performed ChIP followed by qPCR for GATA1 in the HUDEP-2(ΔGγ) WT, −113A>G, −113A>C, and “artificial GATA” site clonal populations (Figure 2D). There was a significant increase in GATA1 binding to the −115 site of the γ-globin promoter in the −113A>G HPFH and “artificial GATA” clonal populations, compared with WT. There was no significant difference in GATA1 binding between the WT and −113A>C cell lines. We also tested the effect of the −113A>G mutation on in vivo binding of BCL11A by introducing the mutation into another previously described HUDEP-2(ΔGγ) WT clonal population where γ-globin is expressed,5 as we have previously found that BCL11A binding to the γ-globin promoter is only detectable when the γ-globin locus is open and accessible.5 We hypothesize that this is because BCL11A is in a transition phase where the necessary cofactors have not yet been recruited to silence the fetal globin gene. However, consistent with our in vitro data, we did not observe a reduction in binding of BCL11A in the presence of the −113A>G HPFH mutation (supplemental Figure 4).
Several different approaches to therapeutically reactivate fetal γ-globin are being explored that include targeting the erythroid-specific enhancer of BCL11A16-19 and forced chromatin looping through artificial transcription factors.20 An alternative approach is to boost HbF levels by introducing natural HPFH mutations by genome editing or base editing,21,22 mimicking the HPFH phenotype.5,12,23-25 We show that introducing −113A>G HPFH or an “artificial GATA” site into the γ-globin promoter could also be explored as a strategy as it increases GATA1 binding, reverses the γ- to β-globin switch, and elevates HbF to levels that could be therapeutically relevant. Mechanistically, we propose 2 possibilities regarding how the −113A>G HPFH mutation elevates HbF: GATA1 could directly activate fetal globin expression or alternatively GATA1 could indirectly activate fetal globin by displacing BCL11A binding to the −115 γ-globin site. Furthermore, the −113A>G HPFH mutation also disrupts binding of NF-Y, a transcriptional activator that can bind the distal CCAAT box at the −115 γ-globin site (supplemental Figure 5). Further studies should focus on understanding the interplay of NF-Y, BCL11A, and GATA1.
In summary, our results reveal that the −113A>G HPFH mutation does not disrupt binding of the repressor BCL11A, but rather creates a de novo binding site for the activator GATA1, driving HbF production. We have previously reported how HPFH mutations at positions −175 and −198 elevate HbF by creating de novo binding sites for the strong erythroid activators TAL123 and KLF1,12 respectively. GATA1, TAL1, and KLF1 are often considered “erythroid master regulators” and it is remarkable that mutations that create sites for each of these activators drive HPFH, in addition to sites that disrupt the major HbF repressors BCL11A and ZBTB7A.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The px458 plasmid was a gift from F. Zhang (Massachusetts Institute of Technology and Harvard University) (Addgene 48138). The authors acknowledge H. Lebhar and the University of New South Wales (UNSW) Recombinant Products Facility (UNSW Sydney) for HPLC assistance, and C. Brownlee and E. Johansson Beves from The Biological Resources Imaging Laboratory (BRIL; UNSW Sydney) for assistance with flow cytometry.
This work was supported by funding from the Australian National Health and Medical Research Council (APP1098391; M.C.). G.E.M was supported by an Australian Postgraduate Award. B.W. was supported by a University International Postgraduate Award. K.G.R.Q was supported by a UNSW Sydney Scientia Fellowship.
Authorship
Contribution: M.C., K.G.R.Q., G.E.M., and B.W. designed the study and experiments; G.E.M. performed the experiments and analyzed the data; R.K. and Y.N. provided HUDEP-2 cells; G.E.M., B.W., K.G.R.Q., and M.C. wrote the manuscript; K.G.R.Q. and M.C. supervised the study; and all authors read and approved the contents of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Merlin Crossley, UNSW Sydney, Level 3, E26, Bioscience South, Sydney, NSW, 2052, Australia; e-mail: m.crossley@unsw.edu.au.

![Figure 2. The −113A>G HPFH mutation elevates γ-globin expression and creates a de novo binding site for GATA1 in vivo. (A) The CRISPR-Cas9 sgRNA target sites used to introduce the −113A>G (HPFH) and −113A>C mutations and the “artificial GATA” site into HUDEP-2(ΔGγ) WT cells. The protospacer adjacent motif (PAM) site is indicated along with the various modifications introduced. (B) γ-globin mRNA expression levels, expressed as a percentage (γ/γ+β) in HUDEP-2(ΔGγ) WT, −113A>G, −113A>C, and “artificial GATA” clonal populations. Eleven technical replicates are shown for the HUDEP-2(ΔGγ) WT parental clone, 10 clonal populations for −113A>G HPFH mutation, and 11 clonal populations for the −113A>C and “artificial GATA” motif. Shown is the mean plus or minus standard error of the mean (SEM). A nonparametric Mann-Whitney statistical test was used to determine statistical significance: P < .0001 (****), P = .0001 to .001 (***), P = .001 to .01 (**). (C) The percentage of HbF protein levels [(HbF/(HbF + HbA)] in HUDEP-2(ΔGγ) WT (n = 6), −113A>G (n = 10), −113A>C (n = 11) and “artificial GATA” (n = 11) clonal populations as determined by HPLC. The mean plus or minus SEM is represented. A nonparametric Mann-Whitney statistical test was used to determine statistical significance: P = .0001 to .001 (***). (D) GATA1 ChIP-qPCR in HUDEP-2(ΔGγ) WT (n = 7), −113A>G (n = 7), −113A>C (n = 5), and “artificial GATA” site (n = 5) clonal populations. The relative fraction of input, normalized to the positive control (+ctrl) locus ZFPM1 (mean plus or minus SEM), is represented. A nonparametric Mann-Whitney statistical test was used to determine statistical significance: P = .01 to .05 (*). Target loci include the −115 site of the γ-globin promoter, the KLF1 promoter as a +ctrl, and the −4.5 kb upstream region of the IFNB gene as negative control (-ctrl). n.s., nonsignificant. HbA, adult hemoglobin.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/133/8/10.1182_blood-2018-07-863951/3/m_blood863951f2.png?Expires=1769171591&Signature=NEi7LWUlngO1hAYznl34EbaH73FQCWp9O3INuEky3wuzVBgnKadbn7CYQlsVzxZISevNmyskyBo6WaLuYI8umI8McsVvg-p9uYU4CpiwIoE7NzRnPDfGKUPMdXjNZXvh0ZrMMSjyC0K2MZDJT3Hyi2UmpRiMWhxIrc0IKXjVwBQ3GQvzm8NIWziR~DaDQVr-726rPm1MshkL5WImd-TAtW-7kO6KLc1wKNhRwIqGBtfQr8ADHgkGKLzhAhwn6-uqw5AzOAFElgKBXmLCWqgPb32kg6-nRSfomCKJWBcSNcslASAztQRVrIMMKW4lfvTi2I2z-X-YPneTjYf7Xfq4~w__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
