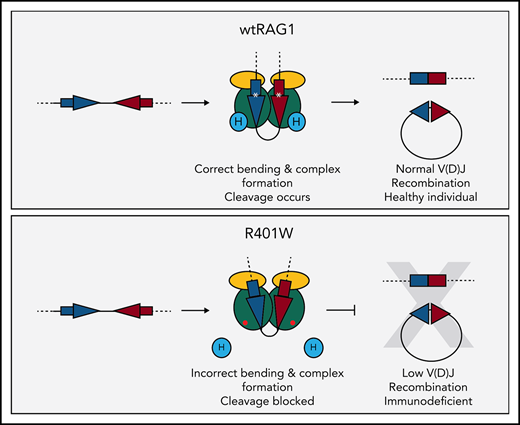
Key Points
A novel RAG1 mutation significantly reduces binding of the accessory protein, HMGB1, to diminish V(D)J recombination in vitro and in vivo.
Complementary knockdown studies of HMGB1 provide the first compelling evidence that HMGB1 is essential for V(D)J recombination in vivo.

Abstract
The Recombination Activating Genes, RAG1 and RAG2, are essential for V(D)J recombination and adaptive immunity. Mutations in these genes often cause immunodeficiency, the severity of which reflects the importance of the altered residue or residues during recombination. Here, we describe a novel RAG1 mutation that causes immunodeficiency in an unexpected way: The mutated protein severely disrupts binding of the accessory protein, HMGB1. Although HMGB1 enhances RAG cutting in vitro, its role in vivo was controversial. We show here that reduced HMGB1 binding by the mutant protein dramatically reduces RAG cutting in vitro and almost completely eliminates recombination in vivo. The RAG1 mutation, R401W, places a bulky tryptophan opposite the binding site for HMG Box A at both 12- and 23-spacer recombination signal sequences, disrupting stable binding of HMGB1. Replacement of R401W with leucine and then lysine progressively restores HMGB1 binding, correlating with increased RAG cutting and recombination in vivo. We show further that knockdown of HMGB1 significantly reduces recombination by wild-type RAG1, whereas its re-addition restores recombination with wild-type, but not the mutant, RAG1 protein. Together, these data provide compelling evidence that HMGB1 plays a critical role during V(D)J recombination in vivo.
Introduction
The ability of vertebrates to combat a vast range of potential pathogens critically relies on V(D)J recombination. This stochastically mixes and matches individual V, D, and J gene segments in the immunoglobulin and T-cell receptor loci to generate a huge array of variable exons, which collectively encode more than 107 different antigen binding sites.1 The importance of V(D)J recombination is demonstrated by mutations in the proteins involved in the cutting or joining steps of the reaction: the outcome is invariably combined immunodeficiency (CID).2
Only 2 proteins are essential for initiation of V(D)J recombination: RAG1 and RAG2. These lymphoid-specific proteins bind to recombination signal sequences (RSSs) that lie adjacent to V, D, and J gene segments and consist of conserved heptamer and nonamer sequences, separated by a relatively nonconserved 12 (±1)-bp or 23 (±1)-bp spacer.3 Recombination proceeds by RAG proteins bringing 2 complementary RSSs (ie, 12-RSS + 23-RSS) into a synaptic complex.4 Coupled cleavage of the RSSs is then achieved by RAG1 binding to the nonamer of 1 RSS, whereas RAG2 directs the RAG1 catalytic triad of acidic residues (D600, D708, E962) known as the DDE motif to nick the partner RSS precisely at the heptamer/coding junction5 ; this ultimately generates blunt double-strand breaks at the 2 RSSs and hairpin structures at the 2 coding ends. Subsequent processing and joining of the broken DNA ends is normally carried out by the classical nonhomologous end joining machinery.6
Complete loss of function of either RAG protein leads to T−, B− severe CID (SCID).2 Similarly, inactivating mutations in the nonhomologous end joining proteins also result in SCID, but in this case, the immunodeficiency is accompanied by increased cellular radio-sensitivity resulting from defects in DNA repair. Hypomorphic RAG mutations that retain some residual recombination activity result in Omenn syndrome, which, similar to SCID, generally manifests soon after birth. More recently, milder forms of CID have been reported in older children with less-inactivating RAG mutations. By studying the effects of individual RAG mutations, a strong correlation has been noted between the severity of the immunodeficiency and the importance of the mutated amino acids to RAG function.2
Although RAG1 and RAG2 are sufficient for cleavage of an RSS substrate in vitro,7 the high-mobility group box (HMGB) proteins, HMGB1 or HMGB2, were found to increase cutting by 7- to 100-fold8 and to decrease the Kd of the RAG complex for both 12- and 23-RSSs.9 HMGB proteins are an abundant group of nonspecific DNA-binding proteins that bind in the minor groove to facilitate DNA bending.10 Band shift assays showed that HMGB1/2 predominantly increases RAG binding to 23-RSSs, where it was proposed to bend the 23-bp spacer to bring the nonamer and heptamer sequences closer together,8 a prediction that was confirmed by recent single-molecule fluorescence experiments11 and structural studies.12,13 HMGB1 also stabilizes the bend at the 12-RSS to help to position the RAG complex for correct cleavage of its target site.13,14 HMGB1 has 2 DNA binding regions, HMG boxes A and B, and high-resolution structures of the RAG/RSS complex showed that a single HMG box (likely Box A) binds to the 12-RSS spacer, whereas 2 HMG boxes bind to the 23-RSS spacer, where the additional binding stabilizes a 150° DNA bend.12,13
Although the role of HMGB1/2 in V(D)J recombination has been thoroughly investigated in vitro, there is less evidence that HMGB1/2 is required in vivo. HMGB1 stimulates RAG cutting of nucleosome templates in vitro, implying that HMGB1/2 potentially increases the accessibility of RSSs to RAGs in vivo.15 Subsequently, recombination of an extrachromosomal substrate was found to increase by more than 3-fold when additional HMGB1/2 was provided by transfection.16 In contrast, HMGB1 knockout mice displayed normal levels of serum immunoglobulins and a complete T-cell receptor repertoire after birth, raising doubts about the requirement of HMGB1 for V(D)J recombination in vivo.17 It is possible, however, that HMGB2, which has 79% amino acid identity and is expressed in lymphoid tissues, substitutes for HMGB1 to promote recombination.
Here, we analyze the effects of compound heterozygous RAG1 mutations from an adult patient with primary immunodeficiency. One of these mutations permits residual RSS binding and recombination, accounting for the patient’s relatively mild phenotype. The other mutation, however, has profound effects on HMGB1 binding in vitro and on V(D)J recombination in vivo, providing strong evidence that HMGB1/2 is essential for efficient V(D)J recombination in vivo.
Methods
RAG constructs
To express RAG proteins in 293T cells, maltose binding protein-tagged core RAG1 and core RAG2 cDNAs were cloned into pEF-XC.18 Full-length RAG1 cDNA was cloned into pCS2MT (Clontech, Mountain View, CA) and used in transfection-based recombination assays. RAG1 mutations were generated using the NEB Q5 Site-Directed Mutagenesis Kit, using primers 8-15 (supplemental Table 1, available on the Blood Web site).
Extrachromosomal V(D)J recombination assay
NIH3T3 cells were seeded at 2 × 105 cells per well in a 6-well plate and transfected with 1 μg of the recombination substrate pJH299,19 160 ng wild-type (WT) or mutant pCS2MT-RAG1, and 320 ng pEFXC-RAG2, using polyethylenimine. Transfected cells were cultured for 48 hours; plasmid DNA was recovered by Hirt extraction.
Recombination levels were determined by nested quantitative polymerase chain reaction (qPCR). First-round amplification used Taq DNA Polymerase with primers DR55 and 1233 (1+2; supplemental Table 1). Thermocycling comprised 19 cycles of 15 seconds at 95°C, 15 seconds at 55°C, and 30 seconds at 68°C. qPCR reactions (10 μL) contained Luna Universal Probe qPCR Master Mix (NEB, Ipswich, MA), 1 μL first-round PCR product, 4 pmol SJ-F and SJ-R primers (3+4; supplemental Table 1), and 10 pmol JH299 SJ probe (7; supplemental Table 1), and were performed in a RotorGene 6000 cycler. Recombination was normalized to the total amount of recovered recombination substrate, using primers CAT-F and CAT-R (5+6; supplemental Table 1). qPCR reactions contained SYBR green master mix (Sensifast No ROX BIO-98005; Bioline, London, United Kingdom), 1 μL of 1:100 dilution of first-round PCR product, and 4 pmol of each primer. Analysis was with RotorGene-Q software, v 2.3.1.
Western blotting
NIH3T3 cells (1 × 106) were transfected with 0.8 μg WT or mutant pCS2MT-RAG1, 1.6 μg pEFXC-RAG2, and 500 ng pTkβ-galactosidase (Clontech). After 48 hours, extracts were made and western blotting performed as described.20 Incubations were as follows: 1:2000 dilution of anti-c-Myc antibody (9E10; Abcam, Cambridge, United Kingdom), 1:10 000 dilution of anti-goat antibody (5210-0170; Sera Care, MA), and 1:20 000 dilution of HRP-conjugated anti-rabbit antibody (A120-101P, Bethyl, TX). Blots were stripped and incubated with 1:2000 dilution of anti-β-galactosidase antibody (Z3781; Promega, Madison, WI) and 1:20 000 dilution of HRP conjugated anti-mouse antibody (715-035-151; Jackson ImmunoResearch, Cambridgeshire, United Kingdom). Signal detection was via SuperSignal WestPico Chemiluminescent Substrate (34080; Thermo Fisher, Waltham MA) and a Sysgene G:Box ChemiXRQ Imaging System.
Purification of RAG and HMGB1 proteins
Maltose binding protein-tagged WT or mutant core RAG1 and core RAG2 were purified from HEK293T cells, as described.21
The full-length rat HMGB1 expression plasmid, pETM11-HMGB1, has an N-terminal 6xHis tag, followed by a Tobacco Etch Virus protease cleavage site. The 6xHis-HMGB1 fusion protein was expressed in Escherichia coli and purified by affinity chromatography, using Ni-NTA, followed by removal of the His tag (except Figure 4A in order to retain the 6xHis tag for the supershift experiment) by Tobacco Etch Virus cleavage and purification via ion exchange chromatography, using a HiTrapQ HP column.
In vitro RAG cleavage and binding assays
RAG cleavage assays were performed using fluorescently labeled RSS probes (supplemental Methods), whereas binding assays were performed using radioactively labeled oligonucleotides to improve sensitivity. Oligonucleotide sequences are given in supplemental Table 1.
Generation of HMGB1 knockdown cells
CRISPRi was used to knockdown HMGB1 expression, using a catalytically dead Cas9 nuclease (dCas9) fused to a KRAB transcriptional repressor domain.22 Detailed descriptions of the plasmids and production of lentiviral particles are available in supplemental Methods.
NIH3T3 cells were seeded at 1 × 105 cells per well of a 6-well plate. After 24 hours, the media was replaced with 1 mL of each lentiviral supernatant containing 6 μg/mL polybrene (TR-1003-G, Merck Kenilworth, NJ). Plates were centrifuged at 800g for 30 minutes at 32°C. At 24 hours posttransduction, the media was replaced with media containing 10 μg/mL blasticidin S HCl (sc-495389, Santa Cruz Biotechnology, Heidelberg, Germany) and 400 μg/mL zeocin (J67140.XF, Alfa Aesar, Haverhill, MA). HMGB1 knockdown was confirmed by western blotting, using a mouse monoclonal anti-HMGB1 antibody (sc-56698, Santa Cruz Biotechnology) and a mouse monoclonal anti-β-tubulin antibody as a loading control, both at a 1:1000 (TA347064; Origene, Rockville, MD). Antibiotics were withdrawn 24 hours before transfection for the recombination assay to minimize their effects.
Expression of HMGB1/2
HMGB1 and HMGB2 were reintroduced, using lentiviruses encoding full-length HMGB2 and the truncated proteins, HMGB1ΔC and HMGB2ΔC, fused to puromycin N-acetyltransferase separated by a self-cleaving P2A peptide sequence. Both HMGB1ΔC and HMGB2ΔC (aa 1-172) lack the C-terminal acidic tail and express at much higher levels than their full-length counterparts, and HMGB1ΔC has higher DNA and RAG binding activity.23 Detailed descriptions of plasmids and lentiviral transductions are in supplemental Methods.
Results
A detailed clinical description of the patient is provided in a recent publication (patient 14).24 Briefly, the patient presented at age 34 years with established bronchiectasis, recurrent chest infections, normal immunoglobulin levels, and mild T-cell lymphopenia. After genetic testing using a gene panel-based approach, he was found to have biallelic mutations in RAG1: c.1210C>T p.Arg404Trp and c.1520G>A p.Arg507Gln.
To dissect how these RAG1 mutations (Figure 1A) affect V(D)J recombination, recombination levels were assayed by transfecting a reporter plasmid and RAG expression vectors into nonlymphoid cells. In this reporter plasmid, the 12- and 23-RSSs are initially in the same orientation; recombination causes the intervening sequence to be inverted, allowing quantification of its levels using PCR primers that become convergent only after recombination (Figure 1B).
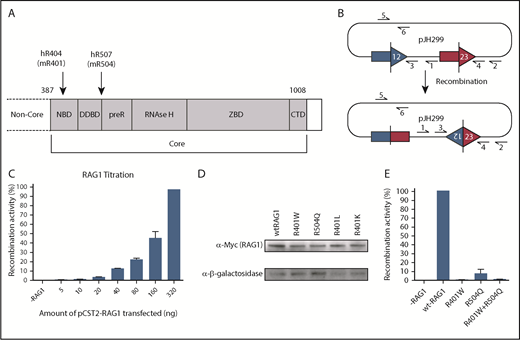
RAG1 mutations affect V(D)J recombination in vivo. (A) Schematic of RAG1 showing the main core RAG1 subdomains. The positions of the mutations in the patient are shown with the equivalent mouse amino acids in brackets beneath. (B) Plasmid pJH299 used to quantify V(D)J recombination in vivo. Here, 12- and 23-RSSs are indicated as triangles. After inversional recombination, primers 1+2 and 3+4 are used in a qPCR nested assay, with a hydrolysis probe across the RSS junction. Total plasmid amounts are measured using primers 5 and 6. (C) Increasing amounts of WT RAG1 expression plasmid were transfected into NIH3T3 cells, and the levels of recombination were determined. A direct correlation with the amount of RAG1 activity is observed. n = 3; error bars show standard error of the mean (SEM). (D) Western blot showing expression levels of the various RAG1 proteins in transfected NIH3T3 cells; a β-galactosidase expression vector was co-transfected as a loading control. (E) Recombination levels using WT and mutated RAG1 proteins, relative to WT RAG1. Recombination was normalized according to the total amount of recombination plasmid recovered; similar levels of RAG1 protein expression were verified by western blotting (D). n = 3; error bars show SEM. CTD, carboxy-terminal domain; DDBD, dimerization and DNA binding domain; NBD, nonamer binding domain; preR, pre-RNaseH domain; ZBD, zinc binding domain.
RAG1 mutations affect V(D)J recombination in vivo. (A) Schematic of RAG1 showing the main core RAG1 subdomains. The positions of the mutations in the patient are shown with the equivalent mouse amino acids in brackets beneath. (B) Plasmid pJH299 used to quantify V(D)J recombination in vivo. Here, 12- and 23-RSSs are indicated as triangles. After inversional recombination, primers 1+2 and 3+4 are used in a qPCR nested assay, with a hydrolysis probe across the RSS junction. Total plasmid amounts are measured using primers 5 and 6. (C) Increasing amounts of WT RAG1 expression plasmid were transfected into NIH3T3 cells, and the levels of recombination were determined. A direct correlation with the amount of RAG1 activity is observed. n = 3; error bars show standard error of the mean (SEM). (D) Western blot showing expression levels of the various RAG1 proteins in transfected NIH3T3 cells; a β-galactosidase expression vector was co-transfected as a loading control. (E) Recombination levels using WT and mutated RAG1 proteins, relative to WT RAG1. Recombination was normalized according to the total amount of recombination plasmid recovered; similar levels of RAG1 protein expression were verified by western blotting (D). n = 3; error bars show SEM. CTD, carboxy-terminal domain; DDBD, dimerization and DNA binding domain; NBD, nonamer binding domain; preR, pre-RNaseH domain; ZBD, zinc binding domain.
Having confirmed that recombination increases quantitatively according to RAG1 activity (Figure 1C), we introduced individual mutations into mouse RAG1 cDNA to alter the equivalent amino acids (R401W and R504Q) to those in the patient. Expression vectors for these RAG1 proteins were then transfected into NIH3T3 cells, together with the recombination reporter plasmid and a RAG2 expression vector. Although mutation R504Q gave detectable levels of recombination, equivalent to ∼2% to 5% of WT (Figure 1D-E), recombination with R401W was negligible.
Effects of mutation R504Q on RAG activity in vitro
To investigate the molecular basis of the reduced recombination, RAG binding and cutting assays were carried out in vitro. Core RAG proteins (amino acids 384-1008 of RAG1 and 1-382 of RAG2) are fully active in binding and cutting, but are more amenable to purification than full-length RAG proteins.21 Therefore, to enable our data to be compared with available high-resolution structures, the relevant mutations were introduced into mouse core RAG1 (which is 96.3% identical and 97.6% similar to human core RAG1), and were expressed together with core RAG2 in 293T cells. These were then purified and used in RAG cutting assays in which an end-labeled oligonucleotide carrying a 12- or 23-RSS was incubated with RAG proteins and full-length HMGB1, with or without the relevant 12- or 23- partner RSS. With R504Q, cutting is reduced by ∼15-fold compared with WT RAG1 at both 12- and 23-RSSs (Figure 2A), which mirrors the reduction in RSS binding (Figure 2B) and the observed decrease in recombination in vivo (Figure 1E).
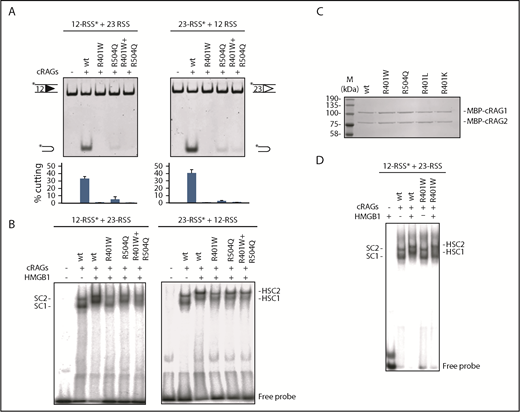
RAG1 mutations affect cutting and binding in vitro. (A) Cutting at a 12-RSS (left) or 23-RSS (right) is shown. An oligonucleotide carrying a consensus 12- or 23-RSS, as indicated, was incubated with equivalent amounts of core RAG2 (cRAG2) plus the core RAG1 (cRAG1) proteins shown (C) in the presence of HMGB1, with a 10-fold excess of unlabeled 23- or 12- partner RSS. For cutting reactions on the far right of each gel, equal amounts of R401W and R504Q were mixed to give the same total amount of RAG1 as the other lanes. The percentage cutting is indicated beneath the gels; an asterisk indicates the labeled oligonucleotide. (B) Binding to a labeled 12-RSS (left) and 23-RSS (right). Binding reactions were performed with cRAG2 and the cRAG1 proteins indicated. Complexes SC1 and SC2, as well as HSC1 and HSC2, are shown. (C) The levels of purified RAG proteins are equivalent. Purified, maltose binding protein-tagged cRAG1 and cRAG2 proteins were separated by electrophoresis and the gel stained with Coomassie blue. The various RAG1 proteins are indicated above each lane. (D) Comparison of R401W and WT RAG1 binding to a 12-RSS. RAG binding reactions were performed in the presence or absence of HMGB1, as indicated. Whereas R401W forms equivalent complexes to WT with the 12-RSS, in the absence of HMGB1, R401W does not form HSC1.
RAG1 mutations affect cutting and binding in vitro. (A) Cutting at a 12-RSS (left) or 23-RSS (right) is shown. An oligonucleotide carrying a consensus 12- or 23-RSS, as indicated, was incubated with equivalent amounts of core RAG2 (cRAG2) plus the core RAG1 (cRAG1) proteins shown (C) in the presence of HMGB1, with a 10-fold excess of unlabeled 23- or 12- partner RSS. For cutting reactions on the far right of each gel, equal amounts of R401W and R504Q were mixed to give the same total amount of RAG1 as the other lanes. The percentage cutting is indicated beneath the gels; an asterisk indicates the labeled oligonucleotide. (B) Binding to a labeled 12-RSS (left) and 23-RSS (right). Binding reactions were performed with cRAG2 and the cRAG1 proteins indicated. Complexes SC1 and SC2, as well as HSC1 and HSC2, are shown. (C) The levels of purified RAG proteins are equivalent. Purified, maltose binding protein-tagged cRAG1 and cRAG2 proteins were separated by electrophoresis and the gel stained with Coomassie blue. The various RAG1 proteins are indicated above each lane. (D) Comparison of R401W and WT RAG1 binding to a 12-RSS. RAG binding reactions were performed in the presence or absence of HMGB1, as indicated. Whereas R401W forms equivalent complexes to WT with the 12-RSS, in the absence of HMGB1, R401W does not form HSC1.
Mutation R401W significantly alters RAG activity
With R401W, RAG cutting is almost completely eliminated at both types of RSS (Figure 2A). Moreover, DNA binding was significantly altered. RAGs form 2 complexes with DNA: a faster migrating complex, SC1, which consists of 2 molecules of RAG1, and 1 of RAG2 and SC2, which contains 2 molecules of each protein. In the presence of HMGB1, these complexes are shifted to slower mobility complexes, HSC1 and HSC2,25 where HSC2 is thought to be active in RAG cleavage. At a 12-RSS, although RSS binding by R401W appears normal, forming SC1/SC2 complexes like WT RAG1 (Figure 2D) in the presence of HMGB1, HSC1 is substantially reduced and HSC2 is reduced by more than 50% (Figure 2B). Indeed, HSC1 is almost completely replaced by a faster-migrating complex that has the same mobility as SC1. This implies that HMGB1 does not bind efficiently to the R401W/12-RSS complex. With a 23-RSS, R401W forms similar complexes to WT RAG1, although HSC1 and HSC2 have slightly faster mobilities (which are more fully resolved in Figure 4D), suggesting altered conformations.
To better understand the molecular basis of this altered binding, the recent cryo-electron microscopy and X-ray crystal structures of the mouse and zebrafish RAG/RSS complexes12,13 were examined. Two areas of HMGB1 binding, representing Box A and Box B, were mapped to the 23-RSS spacer and correspond to 60° and 90° DNA bends.13 Although unresolved, a single region of HMGB1 density, likely Box A, was observed in the 12-RSS spacer, also at a 60° bend. Notably, the location of Box A at both RSSs is directly opposite the region occupied by R401 (Figure 3). It is likely that the nonamer binding domain of RAG1 (where R401 is located) establishes the 60° bend,8,26 as HMGB1 is recruited after RAG27 and Box A recognizes prebent DNA.28 Indeed, this bend may be a requirement for Box A binding, and it is possible that the bulky tryptophan in R401W disrupts this bending to reduce HMGB1 association.
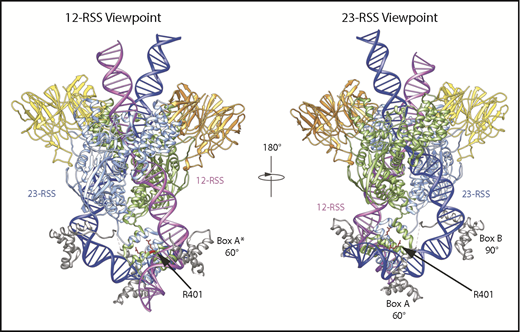
HMGB1 binding to the RAG/DNA complex. HMGB1 binding was determined from the published cryo-electron microscopy and X-ray crystal structures of mouse RAG/RSS complexes.12,13 Blue and green cartoons represent individual RAG1 monomers. (Left) HMGB1 Box A from the 23-RSS structure was superimposed over the density that likely corresponds to HMGB1 Box A at the 12-RSS, denoted by the asterisk.13 This falls directly opposite R401 of mouse RAG1, indicated in red. R401 contacts DNA in the 12-RSS spacer, close to the RSS nonamer. (Right) HMGB1 Box B binding is observed in the middle of the 23-RSS spacer, whereas an additional area of HMGB1 binding (Box A) lies opposite R401 (in red) at the 23-RSS. Data from PDB 6CG0.13
HMGB1 binding to the RAG/DNA complex. HMGB1 binding was determined from the published cryo-electron microscopy and X-ray crystal structures of mouse RAG/RSS complexes.12,13 Blue and green cartoons represent individual RAG1 monomers. (Left) HMGB1 Box A from the 23-RSS structure was superimposed over the density that likely corresponds to HMGB1 Box A at the 12-RSS, denoted by the asterisk.13 This falls directly opposite R401 of mouse RAG1, indicated in red. R401 contacts DNA in the 12-RSS spacer, close to the RSS nonamer. (Right) HMGB1 Box B binding is observed in the middle of the 23-RSS spacer, whereas an additional area of HMGB1 binding (Box A) lies opposite R401 (in red) at the 23-RSS. Data from PDB 6CG0.13
At the 23-RSS, Box B lies in the middle of the 23-RSS spacer, where it stabilizes a 90° bend. With this arrangement, it is possible Box B stabilizes HMGB1 binding to the RAG complex, whereas the R401W mutation again lies in the region that is bound by Box A (Figure 3, right), and where the bulky tryptophan could again destabilize Box A binding. Consistent with this, HSC2 and HSC1 formed with R401W at the 23-RSS have slightly faster mobilities (Figure 2B). Moreover, as bending of the 12- and 23-RSSs by 60° and 150°, respectively, is required to position the RSSs for efficient cleavage, the altered binding can explain R401W’s drastically reduced activity (Figure 2A).
R401W decreases HMGB1 binding
To test whether the altered RAG/RSS complexes indeed result from changes in HMGB1 binding, His-tagged HMGB1 was employed in RAG binding assays. This forms very similar complexes to those with untagged HMGB1 (Figure 4A); importantly, addition of an anti-His antibody supershifts the complexes formed with HMGB1, whereas the faster migrating complex formed with R401W is unchanged (Figure 4A). This therefore supports the idea that HMGB1 binds significantly less well to RAG/DNA complexes formed with R401W at 12-RSSs. These data further suggest that the addition of a second molecule of RAG2 to form HSC2 may stabilize HMGB1 binding, but as recombination is negligible, this HSC2-like complex appears to be nonfunctional.
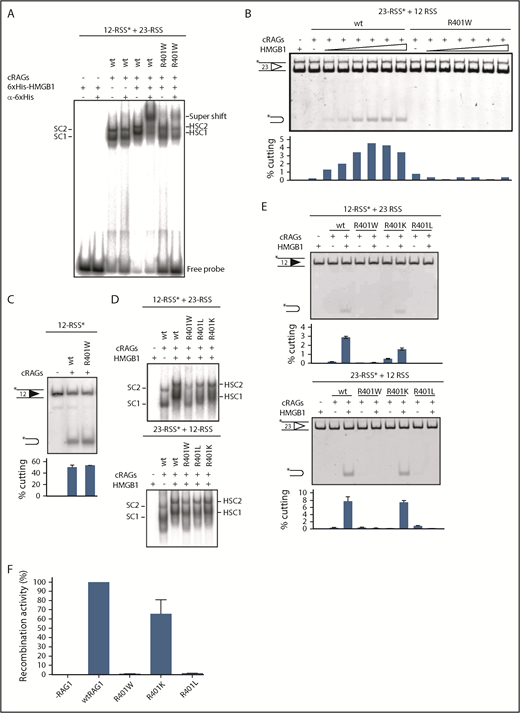
Amino acid substitutions progressively restore HMGB1 binding and RAG1 activity. (A) HMGB1 binds less well to R401W/12-RSS complexes. WT or R401W mutant RAG1 complexes were formed with a labeled 12-RSS in the presence or absence of His-tagged HMGB1. A 23-RSS oligonucleotide partner was present in all cases. Supershifted complexes formed on addition of an anti-His tag antibody are indicated. The mobilities of SC1, SC2, HSC1, and HSC2 are shown. (B) Increasing amounts of HMGB1 do not restore cutting with R401W. RAG cutting reactions were performed with WT RAG1 or R401W in the presence of increasing amounts of HMGB1 (0, 25, 50, 100, 200, 400, 800 nmoles). A 23-RSS substrate was used, as HMGB1 has the greatest effect at this type of RSS. It is notable that in the absence of HMGB1, cutting by WT RAG1 is not detectable (lane 2). (C) R401W has catalytic activity. RAG cutting reactions were performed using a single labeled 12-RSS in the presence of manganese. (D) HSC1 and HSC2 complexes become increasingly like WT RAG1 complexes with R401L and R401K. RAG binding to a labeled 12- or 23-RSS in the presence of an unlabeled partner RSS and HMGB1, as indicated. The gel was run for longer than Figure 2B to better separate the complexes; a cropped image is shown to highlight the mobility differences. (E) RAG cutting with R401W, R401L, and R401K. RAG cutting was performed with a labeled 12-RSS or 23-RSS, indicated by the asterisk, in the presence of unlabeled partner and the RAG1 proteins indicated, with or without HMGB1. The percentage cutting is given beneath the gel. (F) R401K recombination activity is close to WT RAG1 levels in vivo. Recombination was monitored using pJH299 and equivalent levels of RAG1 proteins (Figure 1B,D). Recombination levels are shown relative to WT RAG1. n = 3; error bars show SEM.
Amino acid substitutions progressively restore HMGB1 binding and RAG1 activity. (A) HMGB1 binds less well to R401W/12-RSS complexes. WT or R401W mutant RAG1 complexes were formed with a labeled 12-RSS in the presence or absence of His-tagged HMGB1. A 23-RSS oligonucleotide partner was present in all cases. Supershifted complexes formed on addition of an anti-His tag antibody are indicated. The mobilities of SC1, SC2, HSC1, and HSC2 are shown. (B) Increasing amounts of HMGB1 do not restore cutting with R401W. RAG cutting reactions were performed with WT RAG1 or R401W in the presence of increasing amounts of HMGB1 (0, 25, 50, 100, 200, 400, 800 nmoles). A 23-RSS substrate was used, as HMGB1 has the greatest effect at this type of RSS. It is notable that in the absence of HMGB1, cutting by WT RAG1 is not detectable (lane 2). (C) R401W has catalytic activity. RAG cutting reactions were performed using a single labeled 12-RSS in the presence of manganese. (D) HSC1 and HSC2 complexes become increasingly like WT RAG1 complexes with R401L and R401K. RAG binding to a labeled 12- or 23-RSS in the presence of an unlabeled partner RSS and HMGB1, as indicated. The gel was run for longer than Figure 2B to better separate the complexes; a cropped image is shown to highlight the mobility differences. (E) RAG cutting with R401W, R401L, and R401K. RAG cutting was performed with a labeled 12-RSS or 23-RSS, indicated by the asterisk, in the presence of unlabeled partner and the RAG1 proteins indicated, with or without HMGB1. The percentage cutting is given beneath the gel. (F) R401K recombination activity is close to WT RAG1 levels in vivo. Recombination was monitored using pJH299 and equivalent levels of RAG1 proteins (Figure 1B,D). Recombination levels are shown relative to WT RAG1. n = 3; error bars show SEM.
We next performed RAG cutting assays in the presence of increasing amounts of HMGB1 to test whether the weak HMGB1 binding is overcome at higher amounts of HMGB1, and whether this can restore activity of R401W. Although cutting by WT RAG1 gradually increases as more HMGB1 is added, the activity of R401W was negligible, irrespective of the amount of HMGB1 (Figure 4B).
HMGB1 is required for RAG activity under our reaction conditions (Figure 4B), but it is also possible that the inactivity of R401W is independent of reduced HMGB1 binding. To test this idea, RAG cutting assays were performed in the presence of manganese, which enables cutting of single RSSs.29 This contrasts to standard RAG cutting conditions in which the divalent cation is magnesium and cutting requires synaptic complex formation between complementary 12- and 23-RSSs. Using these less-stringent manganese conditions, cutting by R401W is readily observed at similar levels to WT protein (Figure 4C). Thus, the R401W mutant protein has catalytic activity, but this is not manifested in the absence of stable HMGB1 binding. This is also consistent with recent structural studies, outlined here, that show that HMGB1 is required to position RSSs correctly in the synaptic complex for RAG cutting.13
We next investigated how R401W reduces HMGB1 binding and reasoned that this could be a result of 2 nonmutually exclusive effects: First, the bulky tryptophan of R401W could sterically hinder HMGB1 binding and/or DNA bending within the RAG/DNA complex. Alternatively, the loss of arginine’s positive charge could disrupt RAG1 interactions with phosphate groups in the DNA spacer to reduce HMGB1 binding to the RAG/DNA complex.
To differentiate these possibilities, R401 was mutated to leucine, which has a similar length to arginine, but which lacks a positive charge. This new mutation, R401L, partially restores binding to generate HSC1 with lower mobility and increased amounts of HSC2 (Figure 4D); however, no cutting is observed under standard conditions (Figure 4E). Moreover, (H)SC1 is more smeared at a 12-RSS, and HSC2 levels are below those of WT RAG1.
To test whether a positive charge is required, R401 was substituted with lysine. This resulted in almost WT levels of cutting at both 12- and 23-RSSs (Figure 4E). Moreover, R401K formed very similar amounts of HSC1 and HSC2 to WT RAG1 at a 23-RSS. At a 12-RSS, however, HSC1 formed with R401K is more smeared than with WT RAG1 (Figure 4D), suggesting that the longer arginine side chain and/or its guanidinium group, which can form contacts in 3 directions compared with just 1 for lysine,30 might be required for RSS bending and stable HMGB1 binding.
To assess the effects of these new mutations on V(D)J recombination in vivo, we employed the transfection-based recombination assay. Whereas R401L catalyses only negligible levels of recombination, consistent with lower/less stable formation of HSC1 and HSC2 complexes in vitro, R401K restores recombination to almost WT levels, again correlating with in vitro cutting and formation of HSC1 and HSC2 (Figure 4F). Together, these data strongly support the idea that R401W disrupts association of HMGB1 with the RAG/RSS complex and highlight the critical role of HMGB1 binding for RAG cutting and recombination in vivo.
Knockdown of HMGB1 significantly decreases recombination
If HMGB1 is indeed essential for V(D)J recombination in vivo, we reasoned that in the absence of HMGB1, recombination will be negligible. We further predicted that if HMGB1 is restored, V(D)J recombination will be re-established in the presence of WT RAG1, but not R401W. To test these ideas, we employed a fusion protein between catalytically dead Cas9 (dCas9) and the KRAB repressor (dCas9-KRAB).22 This was targeted to the start site of the HMGB1 promoter, using a CRISPR guide RNA (Figure 5A), resulting in knockdown of HMGB1 by up to 90% (Figure 5B). V(D)J recombination is also reduced by an average of 80% with WT RAG1, whereas recombination with R401W remains negligible (Figure 5C), exactly as predicted by our model. On transduction of these cells with a lentivirus that expresses HMGB1 under the control of the EF1α promoter, HMGB1 levels are restored to 250% to 400% of WT cells. Also as predicted, V(D)J recombination activity is rescued with WT RAG1 and R401K, but remains negligible with R401W (Figure 5C). These studies therefore strongly suggest that HMGB1 is required for V(D)J recombination in vivo.
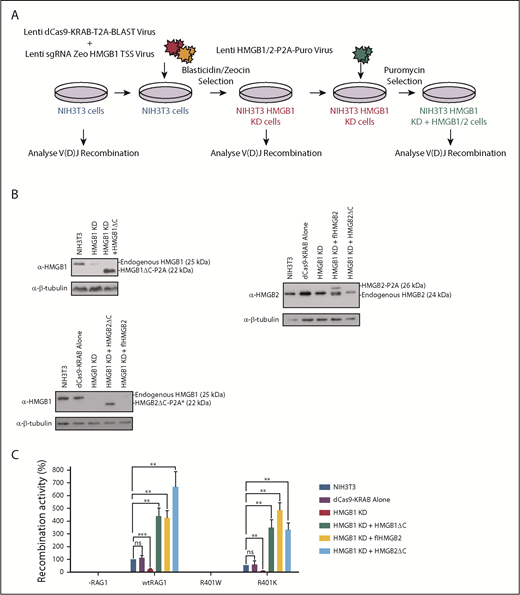
Knockdown of HMGB1 significantly reduces V(D)J recombination in vivo. (A) Schematic of the steps used in CRISPR/Cas9-mediated knockdown of HMGB1. Transduction with lentiviruses and antibiotic selection steps are shown. (B) Representative western blots showing the levels of HMGB1 and full-length (fl) and C-terminal deletion (ΔC) HMGB2 relative to a β-tubulin control. Antibodies that specifically recognize HMGB2ΔC are unavailable, as the epitope lies at the C terminus. However, the core regions of HMGB1 and HMGB2 are 85.5% identical, and the anti-HMGB1 antibody cross-reacts with HMGB2 (indicated by the asterisk). (C) V(D)J recombination levels with the RAG1 proteins shown in WT NIH3T3 cells (blue bars), when the KRAB repressor as introduced in the absence of guide RNAs (purple bars), when HMGB1 has been knocked down (red bars), or when HMGB1 (dark green bars) or HMGB2 full-length (light green bars) or HMGB2ΔC (turquoise bars) have been reintroduced. n = 3; error bars show SEM. The changes in recombination activity are statistically significant. Student t test; **P = .01; ***P = .001. The endogenous HMGB2 in NIH3T3 cells could account for residual recombination activity after HMGB1 knockdown. ns, nonspecific.
Knockdown of HMGB1 significantly reduces V(D)J recombination in vivo. (A) Schematic of the steps used in CRISPR/Cas9-mediated knockdown of HMGB1. Transduction with lentiviruses and antibiotic selection steps are shown. (B) Representative western blots showing the levels of HMGB1 and full-length (fl) and C-terminal deletion (ΔC) HMGB2 relative to a β-tubulin control. Antibodies that specifically recognize HMGB2ΔC are unavailable, as the epitope lies at the C terminus. However, the core regions of HMGB1 and HMGB2 are 85.5% identical, and the anti-HMGB1 antibody cross-reacts with HMGB2 (indicated by the asterisk). (C) V(D)J recombination levels with the RAG1 proteins shown in WT NIH3T3 cells (blue bars), when the KRAB repressor as introduced in the absence of guide RNAs (purple bars), when HMGB1 has been knocked down (red bars), or when HMGB1 (dark green bars) or HMGB2 full-length (light green bars) or HMGB2ΔC (turquoise bars) have been reintroduced. n = 3; error bars show SEM. The changes in recombination activity are statistically significant. Student t test; **P = .01; ***P = .001. The endogenous HMGB2 in NIH3T3 cells could account for residual recombination activity after HMGB1 knockdown. ns, nonspecific.
Paradoxically, HMGB1 knockout mice develop a normal immune system.17 Because HMGB2, which is 79% identical, substitutes for HMGB1 in RAG cutting assays in vitro,8 it seemed plausible that HMGB2 substitutes for HMGB1 in vivo. To test whether this is the case, we expressed HMGB2 in our HMBG1 knockdown cells and find indeed that HMGB2 restores recombination activity with WT RAG1, but not R401W (Figure 5C). Together, these studies provide compelling evidence that at least 1 of the highly related bending proteins, HMGB1/2, is required for V(D)J recombination in vivo.
Discussion
Previous studies of RAG mutations highlighted the amino acids that are important for RAG function per se; for example, that mediate RAG/DNA or RAG/RAG contacts.2,31 Here, a RAG1 mutation instead demonstrates the critical role of an accessory protein during V(D)J recombination. Indeed, a long-standing question is whether HMGB1/2 is required for V(D)J recombination in vivo. Although there is very good evidence that HMGB1 stimulates RAG cutting in vitro,8 and recent structural studies explained the mechanistic basis, relatively little evidence existed that HMGB1/2 plays this role in vivo, primarily because of inconclusive mouse knockout studies.17 Here, we show that the RAG1 mutation, R401W, which places a bulky tryptophan group exactly at the position of a critical 60° bend at both 12- and 23-RSSs, dramatically alters subsequent HMGB1 binding in vitro and almost eliminates V(D)J recombination in vivo. Moreover, when this tryptophan is replaced with lysine, HMGB1 binding and RAG cutting are restored to close to WT levels in vitro, correlating with an equivalent increase in recombination in vivo. These data, together with the significant decrease in recombination upon knockdown of HMGB1, provide strong evidence that HMGB1 is required for V(D)J recombination in vivo. Moreover, by showing that HMGB2 can substitute for HMGB1 in vivo, our data solve a long-standing discrepancy by demonstrating that either of the DNA bending proteins, HMGB1 or HMGB2, is required for V(D)J recombination.
By analyzing point mutations in each RAG1 allele individually from a patient with primary immunodeficiency, the relative contribution of each mutation to V(D)J recombination could be dissected. R401W has negligible RAG cutting and recombination activity. Although it binds DNA, and forms the expected complexes with RAG2, it does not form the expected complexes with HMGB1. This is particularly apparent at a 12-RSS, where HSC1 is substantially reduced and HSC2 is reduced by more than 50%; at a 23-RSS, HSC1 and HSC2 are formed, albeit with increased mobility. Importantly, changing R401 to leucine and then lysine causes HMGB1 binding to become progressively more like the WT pattern. These changes strongly correlate with changes in RAG activity both in vitro and in vivo. Previous studies showed that HMGB1 preferentially binds to a preformed RAG1/RSS complex in which RAG1 causes partial RSS bending.32 The observed alterations in HMGB1 binding to the RAG1/RSS complex could be explained because R401 binds a phosphate group in the DNA backbone directly opposite the region that is bound in the minor groove by the HMG boxes.13 R401 thus stabilizes a bend at the nonamer/RSS spacer junction at both RSSs, and consequently, when R401 is mutated to tryptophan, the bulky side chain likely disrupts this R401-phosphate interaction to destabilize HMGB1 Box A binding at both RSSs (Figure 6). Although a second region of HMGB1 binding (Box B) maps to the middle of the 23-RSS spacer to maintain HMGB1 association, binding to just this single site would not induce bending,13 correlating with reduced RAG cutting.
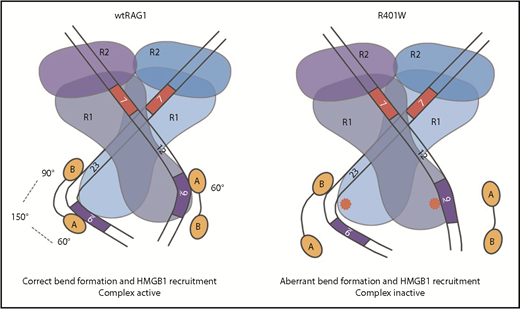
Schematic showing altered HMG binding to WT RAG1 and R401W. The DNA strands are shown with the heptamer, spacer, and nonamer indicated. A and B represent the likely positions of HMG Box A and B, respectively, and R1 and R2 represent RAG1 and RAG2. The bend angles within each RSS are shown.
Schematic showing altered HMG binding to WT RAG1 and R401W. The DNA strands are shown with the heptamer, spacer, and nonamer indicated. A and B represent the likely positions of HMG Box A and B, respectively, and R1 and R2 represent RAG1 and RAG2. The bend angles within each RSS are shown.
From Figures 2B and 4D, it is apparent that R401W binds to RSSs less efficiently than WT RAG1 in the presence of HMGB1. Recent studies of human RAG1 mutations suggested that this may be a result of this arginine (R404 in human RAG1) stacking with R443 in the other RAG1 monomer to alter the conformation of the nonamer binding domain, and consequently RAG1/RSS binding.33 Such interactions, however, do not necessarily exclude the effects described here on HMGB1 binding when this arginine is replaced by tryptophan.
The second RAG1 mutation, R504Q, retains residual recombination activity, although RSS binding and RAG cutting are reduced (Figure 2A-B). This is consistent with structural studies that show that R504 contacts the first 2 bases within the spacer after the heptamer,12 and it is possible that the loss of positive charge in R504Q destabilizes RAG binding. Notably, when we examined the effect of the RAG1 mutations on the patient’s TCRβ repertoire, Vβ14 and Vβ12 are overrepresented and Vβ2 and Vβ16 are underrepresented (data not shown). It is possible that weaker RAG binding to less conserved RSSs is exaggerated with R504Q.
Although the R504Q mutation reduces recombination activity, the patient nonetheless has a partially functional immune system, consistent with their late presentation. Notably, when RAG1 proteins with the R401W and R504Q mutations are mixed, intermediate levels of RAG cutting and recombination are observed (Figures 1E and 2A). This is possibly because R401W inhibits the formation of (partially) active RAG complexes by R504Q, but complexes with 2 R504Q proteins can still form, giving residual levels of recombination and the mild phenotype of the patient.
Reduced binding of HMGB1 to RAG/RSS complexes formed with the RAG1 R401W mutation strongly correlates with negligible activity in RAG cutting assays in vitro and undetectable recombination in vivo. This, coupled with the fact that knockdown of HMGB1 greatly reduces V(D)J recombination by WT RAG1, strongly implies that HMGB1 plays an essential role in V(D)J recombination in vivo.
For original data, please contact j.m.boyes@leeds.ac.uk. One table is shared in the supplemental Data, available with the online version of this article.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Marco Bianchi (San Raffaele University, Milan, Italy) for his kind gift of the HMGB1 expression plasmid, Richard Bayliss for helpful comments on the manuscript, and Alastair Smith for help with the statistics.
This work was supported by a University of Leeds Research Scholarship (D.T.T.) and Bloodwise grant 15042 (J.M.B.). S.S. was supported by the National Institute for Health Research Leeds Biomedical Research Centre.
The views expressed are those of the authors and not necessarily those of the National Health Service, the National Institute for Health Research, or the Department of Health.
Authorship
Contribution: D.T.T., S.S., and J.M.B. conceived the project, designed the experiments, and wrote the paper; and D.T.T., C.C., and D.L. performed the experiments.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Joan M. Boyes, School of Molecular and Cellular Biology, Faculty of Biological Sciences, University of Leeds, Leeds LS2 9JT, United Kingdom; e-mail: j.m.boyes@leeds.ac.uk