Key Points
bcas2 is a novel factor required for HSPC maintenance in zebrafish embryos.
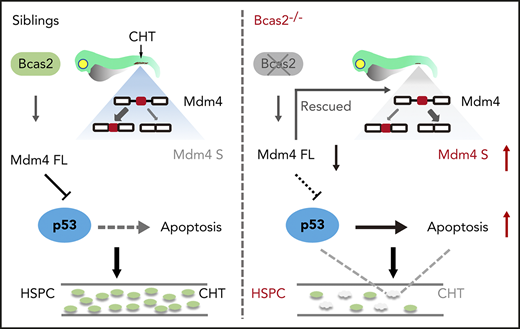
Deletion of bcas2 triggers alternative splicing of Mdm4 and upregulation of p53 activation during HSPC development.

Abstract
Hematopoietic stem and progenitor cells (HSPCs) originate from the hemogenic endothelium via the endothelial-to-hematopoietic transition, are self-renewing, and replenish all lineages of blood cells throughout life. BCAS2 (breast carcinoma amplified sequence 2) is a component of the spliceosome and is involved in multiple biological processes. However, its role in hematopoiesis remains unknown. We established a bcas2 knockout zebrafish model by using transcription activator–like effector nucleases. The bcas2−/− zebrafish showed severe impairment of HSPCs and their derivatives during definitive hematopoiesis. We also observed significant signs of HSPC apoptosis in the caudal hematopoietic tissue of bcas2−/− zebrafish, which may be rescued by suppression of p53. Furthermore, we show that the bcas2 deletion induces an abnormal alternative splicing of Mdm4 that predisposes cells to undergo p53-mediated apoptosis, which provides a mechanistic explanation of the deficiency observed in HSPCs. Our findings revealed a novel and vital role for BCAS2 during HSPC maintenance in zebrafish.
Introduction
Hematopoiesis is an orderly but complex dynamic process that includes differentiation, development, and maturation of hematopoietic stem and progenitor cells (HSPCs) controlled by many factors.1,2 HSPCs are characterized by their remarkable ability to self-renew and differentiate into multiple lineages of mature blood cells, including erythrocytes, megakaryocytes, myelocytes (monocytes and granulocytes), and lymphocytes.3,4 Impaired HSPC differentiation and peripheral blood cytopenias are features of several diseases, including acute leukemia and congenital blood disorders.5,6 It is important to have a clear understanding of the mechanisms involved in the specification of HSPCs.
The specification and maintenance of the HSPC program are regulated by many transcription factors and epigenetic interactions.7 Several studies using whole-exome sequencing revealed that mutations in splicing factor genes have been associated with hematopoietic malignancies, including myelodysplastic syndrome (MDS) and secondary acute myeloid leukemia (sAML).8-11 Although many spliceosome mutants characterized to date have hematopoietic perturbations, they are not entirely overlapping, even with regard to factors that are within the same complex.12 Notably, the most frequent novel recurrent mutations occurred in genes that were engaged in the initial steps of RNA splicing, such as U2AF35 (also known as U2AF1), ZRSR2, SRSF2 (SC35), and SF3B1.8.13,14 However, the functional link between other components in pre–messenger RNA (pre-mRNA) splicing and the dysplastic phenotypes of MDS or sAML is poorly defined.
BCAS2 (breast carcinoma amplified sequence 2) is a 26-kDa protein that contains 2 coiled-coil domains in which the C-terminal coiled-coil domain binds directly to CDC5L and recruits hPrp19(PSO4)/PLRG1 to form a core complex in pre-mRNA splicing in mammalian cells.15 BCAS2 is widely expressed during embryonic development in mammals.16 Initial attempts to identify the function of BCAS2 have shed light on the splicing regulation by BCAS2 in developmental processes. Knockout of bcas2 in Drosophila led to larval lethality, and tissue-specific knockdown of bcas2 in Drosophila caused notum and wing deformity, impaired pre-mRNA splicing, and increased apoptosis in wing discs.17 Recent work also showed that BCAS2 regulates the activity of Δ-Notch signaling via pre-mRNA splicing during Drosophila wing development. Deprivation of bcas2 could diminish Δ-Notch activity, a process involved in cell fate determination and development of multiple tissues.18 Depletion of bcas2 in mice resulted in activation of the p53 and CHK1 pathways in response to DNA damage.16 These data reveal the importance of BCAS2 in regulating cell survival and developmental processes, yet its importance in vivo during developmental hematopoiesis has not been reported.
In this study, we provide new insights on the role of BCAS2 during zebrafish HSPC development. Loss of function of BCAS2 in the bcas2−/− homozygous mutants affected the definitive, but not the primitive, stage of hematopoiesis in zebrafish. The defect of hematopoiesis resulted from apoptosis and could be rescued by the deletion of p53. We further identified that bcas2 deletion induces an abnormal alternative splicing of Mdm4 that predisposes cells to undergo p53-mediated apoptosis in HSPCs. Our findings provide evidence, for the first time, that BCAS2, a component of the spliceosome, plays a key role in embryonic HSPC differentiation and development.
Materials and methods
Zebrafish
Zebrafish (Danio rerio) were raised and maintained on a 14-hours-light/10-hours-dark cycle at 28.5°C. Wild-type and injected embryos were maintained in E3 medium (5 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl2, 0.33 mM MgSO4). The bcas2-knockout line was generated from the standard wild-type (AB) laboratory strain. AB wild-type, Tg (cmyb: enhanced green fluorescent protein [EGFP]), and Tg (flk1: EGFP) cell lines were used in this study. All zebrafish maintenance procedures and experiments were in accordance with guidelines approved by the Ethics Committee of the College of Life Science and Technology, Huazhong University of Science and Technology.
Generation of bcas2-mutant zebrafish by TALENs
To generate site-specific mutations in zebrafish, a pair of customized transcription activator–like effector nucleases (TALENs) were designed and constructed using the FastTALE TALEN kit (Sidansai Biotechnology, Shanghai, People's Republic of China). Generation of TALEN arm mRNAs was performed as described previously.19 A 405-bp DNA fragment containing the bcas2 targeting site was amplified from the injected or uninjected control embryos by polymerase chain reaction (PCR) (5′-TGGAGGAAGAGACGAGACGA-3′, 5′-TATGCAAGTAGCCCCAGACC-3′). The PCR products were purified from 1% agarose gel and sequenced. The bcas2−/− homozygous mutant embryos were generated from normal crosses between identified bcas2+/− heterozygotes.
Quantitative real-time PCR analysis
Total RNA was isolated from 3 days postfertilization (dpf) dissected tails that included caudal hematopoietic tissue (CHT) regions of zebrafish by using Trizol (Life Technologies) according to the manufacturer’s protocol; it was reverse-transcribed into complementary DNA by TransScript All-in-One First-Strand cDNA Synthesis SuperMix for quantitative PCR (qPCR) (One-Step gDNA Removal, TransGen Biotech Co., Ltd). The qPCR analysis was performed with AceQ qPCR SYBR Green Master Mix (Vazyme Biotech Co., Ltd) in a StepOnePlus real-time PCR machine (Life Technologies) and analyzed with GraphPad 5.1 software. Data are represented as mean ± standard deviation (SD), and significance was determined by two-tailed Student t tests between control and experimental groups. The PCR primers used are listed in supplemental Table 1 (available on the Blood Web site).
Whole-mount in situ hybridization
Whole-mount in situ hybridization (WISH) was performed as described previously.20 The images were captured by using a fluorescence microscope (Nikon Ti i80). After imaging, DNA was extracted from the embryos for genotyping. Generally, the embryos were immersed in 50 μL of 50 mM NaOH and incubated at 95°C for 20 to 30 minutes. After vortexing, 5 μL of 1 M tris(hydroxymethyl)aminomethane-HCl (pH 8.0) was added to the solution.
Protein extraction and immunoblotting analysis
To obtain fish protein from the CHT region, the heads of embryos were used for genotyping assay, and tails that included the CHT region of embryos with similar genotypes were lysed with radio immunoprecipitation assay lysis buffer containing protease inhibitor cocktail (Sigma). Western blot was performed as described previously.21 The following primary antibodies were used in this study: anti-BCAS2 antibody (GeneTex, Cat# GTX115390), anti-GAPDH antibody (Dia-an, Cat# 2058), anti-p53 antibody (GeneTex, Cat# GTX128135), and anti-γH2AX antibody (Cell Signaling Technology, Cat# 2577). The images were captured by using a ChemiDoc XRS+ gel imaging system (Bio-Rad).
TUNEL immunostaining
The 3- and 4-dpf bcas2−/−/Tg (cmyb: EGFP) zebrafish embryos were fixed in 4% paraformaldehyde overnight at 4°C and dehydrated in 100% methanol at −20°C over 30 minutes. After rehydration, embryos were digested with proteinase K (20 µg/mL) at room temperature (RT) for 40 minutes and then washed with phosphate-buffered saline (PBS)/Tween. The apoptotic cells were labeled with the terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling (TUNEL) BrightRed Apoptosis Detection Kit (Vazyme Biotech Co., Ltd). After blocking in buffer (PBS + 1% dimethyl sulfoxide [DMSO] + 0.1% Triton X-100 + 1% bovine serum albumin) for 2 hours at RT, the embryos were incubated with anti-green fluorescent protein antibody (Dia-an, Cat# 2057) followed by goat-Alexa Fluor 488-conjugated anti-mouse secondary antibody (Invitrogen) incubation. The images were captured by using a fluorescence microscope (Nikon Ti i80).
EdU cell proliferation assay
Zebrafish embryos were incubated in egg-water containing 50 mM 5-ethynyl-2′-deoxyuridine (EdU)/5% DMSO for 30 minutes at RT, fixed in 4% paraformaldehyde at 4°C overnight, and dehydrated in methanol at −20°C for 30 minutes. After digestion with proteinase K (20 µg/mL) at RT for 40 minutes, the embryos were blocked with blocking buffer (PBS + 1% DMSO + 0.1% Triton X-100 + 1% bovine serum albumin) for 2 hours at RT and incubated with anti-green fluorescent protein antibody followed by goat-Alexa Fluor 488-conjugated anti-mouse secondary antibody incubation. After that, the embryos were stained with Cell-Light EdU Cell Proliferation Detection. The images were captured by using a fluorescence microscope (Nikon Ti i80).
Results
bcas2 gene is expressed in the sites of hematopoiesis in zebrafish
To investigate the role of BCAS2 during hematopoiesis, we examined its spatiotemporal expression pattern in both wild-type embryos (supplemental Figure 1A-J) and bcas2−/−-mutant embryos (supplemental Figure 1K-T) by using WISH; bcas2 showed a dynamic expression pattern throughout embryo development (supplemental Figure 1A-J). It was maternally expressed in 1-cell-stage embryos (supplemental Figure 1A) and ubiquitously expressed through the 6-somite stage (supplemental Figure 1B-C). At 18 hours postfertilization (hpf), bcas2 was expressed in the posterior intermediate cell mass (ICM), the region overlying the yolk tube and extending caudally (supplemental Figure 1D). At 24 hpf, bcas2 expression extended to the nascent posterior ICM-derived posterior blood island (PBI) region (supplemental Figure 1E), suggesting its potential involvement in vasculogenesis or hematopoiesis. From 24 hpf onward, the expression of bcas2 in the PBI decreased significantly. By 2 dpf, only a few cells remained in the CHT, a transient secondary site of hematopoiesis (supplemental Figure 1F-G). By 3 dpf, WISH staining was almost undetectable in the CHT. Instead, the expression of bcas2 was mostly anterior, including in the eyes and tectum as well as the kidney and thymus, both being the ultimate sites of definitive hematopoiesis (supplemental Figure 1H). Notably, the expression of bcas2 was detectable again in the CHT at 4 dpf (supplemental Figure 1I) and disappeared at 5 dpf (supplemental Figure 1J). The expression pattern in kidney and thymus was largely maintained up to 5 dpf (supplemental Figure 1I-J).
Generation of bcas2−/−-mutant zebrafish with TALENs
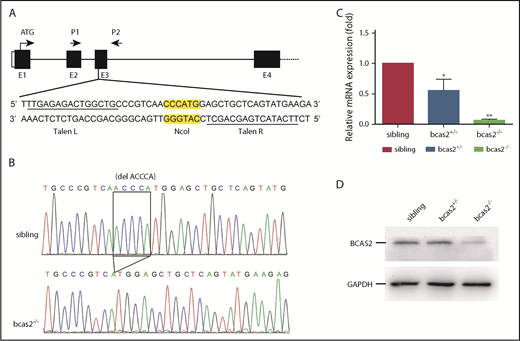
To examine the physiological role of BCAS2 in vivo, we generated bcas2-mutant zebrafish using TALENs. TALEN sites and a 15-bp spacer DNA containing an NcoI digest site were designed at exon 3 in the bcas2 gene (Figure 1A). The 5-base deletion genotype of mutants used in this study (Figure 1B) was predicted to cause a frameshift with premature translation termination, resulting in an abnormal p.Gln77Hisfs*85 BCAS2 protein. Real-time PCR analysis showed that bcas2 mRNA expression was decreased by 50% in heterozygous bcas2+/− embryos and was almost undetectable in homozygous bcas2−/− embryos at 3 dpf (Figure 1C). In addition, WISH results showed that bcas2 was still detected in bcas2−/− embryos and displayed maternal expression at the 1- to 4-cell stage (supplemental Figure 1K-L). However, from 12 hpf onward, bcas2 mRNA levels were severely decreased in bcas2−/− embryos, likely as a result of nonsense-mediated RNA decay, whereas maternally provided bcas2 mRNA was no longer present in bcas2−/− embryos after 12 hpf (supplemental Figure 1M-T). Western blot analysis also showed that the expression of bcas2 was markedly decreased in mutant lines at 3 dpf (Figure 1D). Moreover, anti-BCAS2 staining of sibling/Tg (cmyb: EGFP) showed that BCAS2 co-localized with cmyb-expressed cells, indicative of HSPCs, at 4 dpf in the CHT. However, in bcas2−/−/Tg (cmyb: EGFP) embryos, almost no BCAS2 signals were present (supplemental Figure 1U).

TALEN-mediated targeted mutagenesis of bcas2. (A) A schematic diagram showing the genomic organization of bcas2 with the first 4 exons (E1-E4) and connecting lines depicting introns and the location of the TALEN pair used to generate bcas2 mutants. P1 and P2 show the primer sites for amplification. The NcoI restriction site in the spacer region is used for mutation detection. (B) DNA sequencing identified a deletion of 5 bases in the third exon of bcas2, which forms a premature stop code in the coding region of bcas2 in the mutant line. (C) Real-time PCR analysis shows a 50% decrease in bcas2 mRNA expression in heterozygous embryos and almost undetectable bcas2 mRNA in bcas2−/− mutants at 3 dpf. (D) Western blot analysis shows similar expression of BCAS2 in siblings and heterozygous embryos but greatly decreased expression in bcas2−/− mutants at 3 dpf.
TALEN-mediated targeted mutagenesis of bcas2. (A) A schematic diagram showing the genomic organization of bcas2 with the first 4 exons (E1-E4) and connecting lines depicting introns and the location of the TALEN pair used to generate bcas2 mutants. P1 and P2 show the primer sites for amplification. The NcoI restriction site in the spacer region is used for mutation detection. (B) DNA sequencing identified a deletion of 5 bases in the third exon of bcas2, which forms a premature stop code in the coding region of bcas2 in the mutant line. (C) Real-time PCR analysis shows a 50% decrease in bcas2 mRNA expression in heterozygous embryos and almost undetectable bcas2 mRNA in bcas2−/− mutants at 3 dpf. (D) Western blot analysis shows similar expression of BCAS2 in siblings and heterozygous embryos but greatly decreased expression in bcas2−/− mutants at 3 dpf.
Loss of BCAS2 does not affect primitive hematopoiesis or EMP formation
Our WISH results indicated that bcas2 was detected in the ICM in wild-type embryos where primitive erythroid and myeloid cells arise. To determine whether bcas2−/− mutants had any defects in primitive hematopoiesis, we probed for several hematopoietic markers in both bcas2−/− embryos and in siblings. There was no significant difference between bcas2−/− and sibling embryos as stained by the probes for scl and gata1 at 24 hpf and the probe for hbae1 at 22 hpf (supplemental Figure 2A-F). This suggests that the development of erythroid progenitors in bcas2−/− embryos is normal. The expressions of the myeloid cell marker l-plastin at 22 hpf, lyz at 24 hpf, and the granulocyte marker mpx at 24 hpf were also maintained in bcas2−/− embryos, suggesting that the primitive myeloid cells were unaffected (supplemental Figure 2G-L).
Similar to that in the mouse, definitive hematopoiesis is initiated through committed erythromyeloid progenitors (EMPs) in the PBI, and the initiation of the definitive hematopoietic wave is sustained in zebrafish.22 WISH results showed that the expression levels of gata1, l-plastin, and mpx in the PBI in bcas2−/− embryos at 36 hpf were normal, indicating that the EMPs were correctly specified (supplemental Figure 3A-F). Moreover, the primitive erythroblasts that differentiated into erythrocytes seemed to be unaltered because the expression of hbae1 was unaffected in bcas2−/− embryos at 48 hpf (supplemental Figure 3G-H).
Depletion of zebrafish bcas2 impairs definitive hematopoiesis
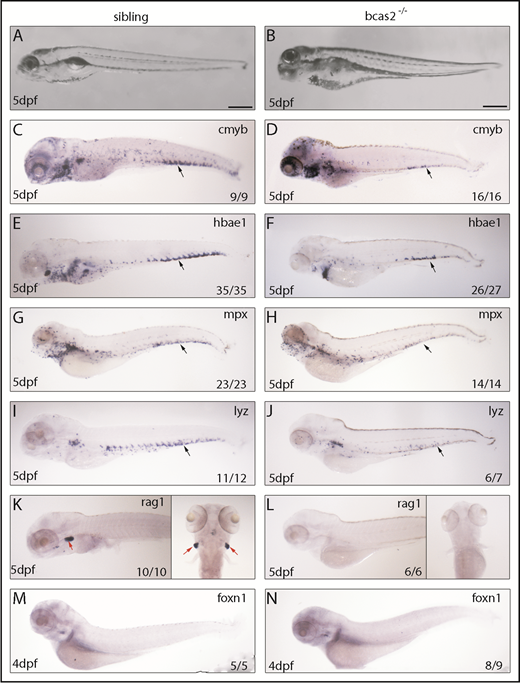
To explore the regulatory role mechanism of BCAS2 in definitive hematopoiesis, we evaluated the presence of definitive blood lineages in bcas2−/− embryos. Bright field images characterized a range of developmental abnormalities, such as a microcephaly-like phenotype, microphthalmia and gradual swelling of the heart, and weak heartbeat from 3 dpf onward in bcas2−/− zebrafish up until 5 dpf (Figure 2A-B). At 5 dpf, WISH results showed that the expression of cmyb was dramatically decreased in the CHT (black arrows) of bcas2−/− embryos compared with siblings (Figure 2C-D). Accordingly, the expression of the erythroid precursor hbae1 was significantly decreased in the CHT and the kidney in bcas2−/− embryos at 5 dpf (Figure 2E-F). We also extended our analysis to include markers of myeloid and lymphoid differentiation and found that at 5 dpf, bcas2−/− embryos also lacked expression of the granulocyte marker mpx and the myeloid marker lyz in the CHT (Figure 2G-J). Accordingly, bcas2−/− mutants lack rag+ T lymphocytes at 4 dpf (supplemental Figure 4A-B) and 5 dpf (Figure 2K-L), despite having normal thymus epithelial cells (Figure 2M-N). Together, our results suggest that BCAS2 is required for the specification of HSPCs.
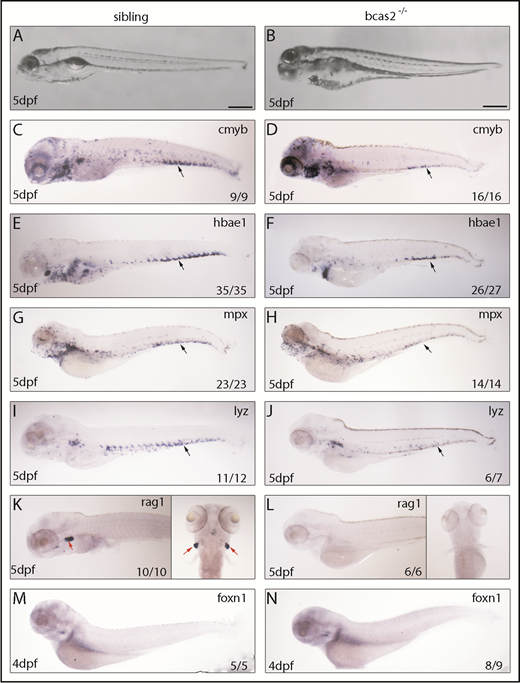
Knockout of bcas2 affects definitive hematopoiesis. (A-B) The bright field images showed developmental defects in bcas2−/− zebrafish at 5 dpf. (C-L) Whole-mount in situ hybridization showed defective definitive hematopoiesis as shown by the alteration of (D) cmyb, (F) hbae1, (H) mpx, (J) lyz, and (L) rag1 in bcas2−/− mutants compared with corresponding sibling embryos (C,E,G,I,K). Thymus epithelial cell marker foxn1 is expressed normally in the thymus at 4 dpf (M-N). Numbers at the bottom right indicate the number of embryos with similar staining patterns among all embryos examined. Black arrows indicate HSPCs in the CHT; red arrows indicate HSPCs in the thymus. Scale bar, 200 μm.
Knockout of bcas2 affects definitive hematopoiesis. (A-B) The bright field images showed developmental defects in bcas2−/− zebrafish at 5 dpf. (C-L) Whole-mount in situ hybridization showed defective definitive hematopoiesis as shown by the alteration of (D) cmyb, (F) hbae1, (H) mpx, (J) lyz, and (L) rag1 in bcas2−/− mutants compared with corresponding sibling embryos (C,E,G,I,K). Thymus epithelial cell marker foxn1 is expressed normally in the thymus at 4 dpf (M-N). Numbers at the bottom right indicate the number of embryos with similar staining patterns among all embryos examined. Black arrows indicate HSPCs in the CHT; red arrows indicate HSPCs in the thymus. Scale bar, 200 μm.
HSPCs originate in arterial vessels, and vasculogenesis and blood flow are essential for HSPC development; disruption of arterial identity could, therefore, cause HSPC defects.23,24 However, WISH results of arterial markers ephrinb2a at 24 hpf (supplemental Figure 5A-B) and flk1 at 36 hpf (supplemental Figure 5C-D) showed that there was no significant difference between bcas2−/− embryos and siblings. In addition, directly observing angiogenesis of bcas2−/− embryos on a Tg (flk1: EGFP) transgenic background from 2 to 5 dpf also showed that vasculature in the CHT seemed normal (supplemental Figure 5E-L), indicating that the defect in HSPC development was not the result of abnormal vasculogenesis.
HSPC defects initiate in the CHT of bcas2−/− embryos
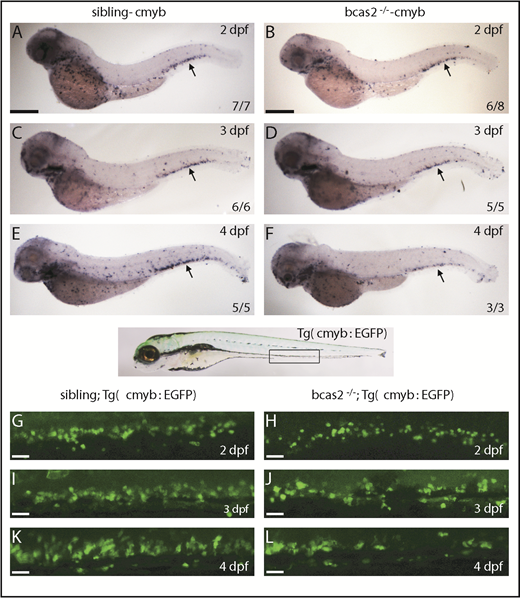
To investigate the initiation of HSPC defects in bcas2−/− embryos, we investigated whether loss of bcas2 affected the onset of definitive hematopoiesis. We performed WISH using cmyb and runx1 probes to evaluate the development of HSPCs. At 36 hpf, the expression of cmyb and runx1 was properly specified and correctly localized in the ventral wall of the aortas of bcas2−/− embryos (supplemental Figure 6A-D). Given the normal expression of cmyb and runx1 in bcas2−/− embryos at 36 hpf, we performed a time course analysis of cmyb expression from 2 to 4 dpf. The WISH results demonstrated that the cmyb expression was still intact at 2 dpf in bcas2−/− embryos (Figure 3A-B). However, the number of HSPCs in the CHT in bcas2−/− embryos was reduced compared with that in siblings at 3 dpf (Figure 3C-D), and the defect was more severe at 4 dpf (Figure 3E-F). This indicated that the maintenance or proliferation of HSPCs was perturbed in the CHT of bcas2−/− embryos. To confirm this discovery, we collected embryos from crossing bcas2+/− zebrafish with Tg (cmyb: EGFP), in which HSPCs could be visualized by EGFP. At 2 dpf, the number of EGFP-positive cells was normal in bcas2−/− embryos compared with that in siblings (Figure 3G-H), whereas gradually decreasing numbers of HSPCs were observed during 3 to 4 dpf (Figure 3I-L). Taken together, our results revealed that neither the initiation of the HSPCs in the aorta-gonad-mesonephros nor their migration to CHT was affected, but that their transitory expansion in the CHT was disturbed in bcas2−/− mutants.
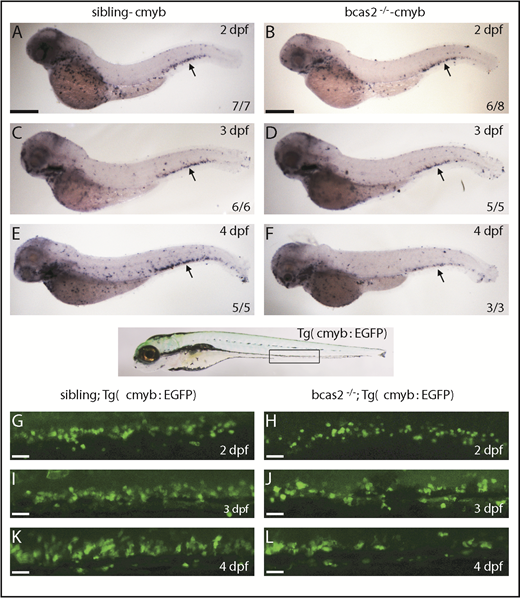
The number of HSPCs was reduced in the CHT of bcas2−/−zebrafish. (A-F) The WISH assay of definitive HSPCs using a cmyb probe from 2 to 4 dpf indicates that the number of HSPCs was decreased in the CHT of bcas2−/− mutants from 3 dpf compared with siblings. Scale bars, 200 μm. (G-L) Live imaging showed that EGFP-positive HSPCs in bcas2−/−/Tg (cmyb: EGFP) zebrafish begin to decrease noticeably in the CHT region from 3 to 4 dpf compared with siblings/Tg (cmyb: EGFP). Scale bars, 50 μm.
The number of HSPCs was reduced in the CHT of bcas2−/−zebrafish. (A-F) The WISH assay of definitive HSPCs using a cmyb probe from 2 to 4 dpf indicates that the number of HSPCs was decreased in the CHT of bcas2−/− mutants from 3 dpf compared with siblings. Scale bars, 200 μm. (G-L) Live imaging showed that EGFP-positive HSPCs in bcas2−/−/Tg (cmyb: EGFP) zebrafish begin to decrease noticeably in the CHT region from 3 to 4 dpf compared with siblings/Tg (cmyb: EGFP). Scale bars, 50 μm.
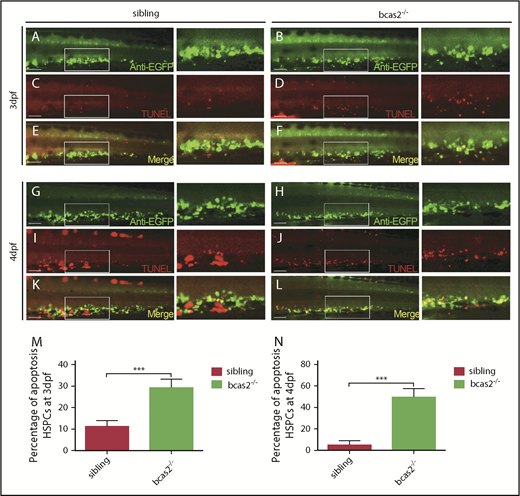
Apoptotic HSPCs are enriched in the CHT of bcas2−/− embryos
To explore how the expansion of HSPCs was disturbed in the CHT in bcas2−/− mutants, we detected the apoptosis of bcas2−/− embryos by using TUNEL assays, which revealed that there is a significant increase in apoptotic HSPC signaling in the CHT region in bcas2−/− embryos compared with that in siblings at 3 dpf (Figure 4A-F,M). In addition, apoptosis spots in the CHT region of bcas2−/− embryos at 4 dpf were more prevalent than those in siblings, whereas the number of HSPCs in the CHT region of bcas2−/− embryos was dramatically decreased (Figure 4G-L,N). EdU assays showed that proliferation signals in the CHT region of mutants were identical to those in siblings at 3 to 4 dpf (supplemental Figure 7). Collectively, these results suggested that the defective definitive hematopoiesis might result from increased apoptosis in HSPCs.

Defective definitive hematopoiesis results from the increased apoptosis of HSPCs of bcas2−/−zebrafish. (A-L) Double immunostaining of cmyb: EGFP and TUNEL at 3 dpf showed an increased apoptosis of HSPCs in the CHT of bcas2−/− mutants that was more prevalent at 4 dpf. Scale bars, 50 μm. (M-N) Quantification of the percentage of the apoptosis of HSPCs in sibling and bcas2−/− mutants at 3 dpf and 4 dpf indicates increased apoptosis of HSPCs in bcas2−/− embryos. For the quantitative analysis, at least 6 embryos were analyzed for each experimental group. Data are represented as mean ± SD. ***P < .001.
Defective definitive hematopoiesis results from the increased apoptosis of HSPCs of bcas2−/−zebrafish. (A-L) Double immunostaining of cmyb: EGFP and TUNEL at 3 dpf showed an increased apoptosis of HSPCs in the CHT of bcas2−/− mutants that was more prevalent at 4 dpf. Scale bars, 50 μm. (M-N) Quantification of the percentage of the apoptosis of HSPCs in sibling and bcas2−/− mutants at 3 dpf and 4 dpf indicates increased apoptosis of HSPCs in bcas2−/− embryos. For the quantitative analysis, at least 6 embryos were analyzed for each experimental group. Data are represented as mean ± SD. ***P < .001.
Apoptosis of HSPCs in bcas2−/− embryos is p53 dependent
To determine how a BCAS2 deficiency triggers apoptosis, we quantified the expression levels of several apoptosis-related genes, including baxa, caspase8, p53, and its target genes in bcas2−/− mutants. Real-time qPCR analysis showed that there was a significantly increased expression of p53 and its target genes p21, mdm2, and puma, as well as apoptosis-related genes baxa and caspase8 in bcas2−/− mutants at 3 dpf. Intriguingly, Mdm4, which has been reported to be 1 of the inhibitors of p53 activity, showed significantly decreased expression (Figure 5A). The activation state of p53 was also confirmed by western blot analysis, and the results indicated that the observed excessive apoptosis may be due to p53 signaling (Figure 5B). To confirm whether the upregulation of p53 expression was responsible for the HSPC defects, we collected embryos for WISH analysis by crossing bcas2+/− mutants with tp53M214K mutants (hereafter p53−/−), which were previously reported to show abolished p53 function in apoptosis.25 WISH results showed that the expression of cmyb in the CHT region was partially rescued in bcas2−/−/p53+/− embryos at 4 dpf, and the rescue was more significant in bcas2−/−/p53−/− embryos (Figure 5C-K). These results imply that the apoptosis of HSPCs in bcas2−/− embryos is p53 dependent.
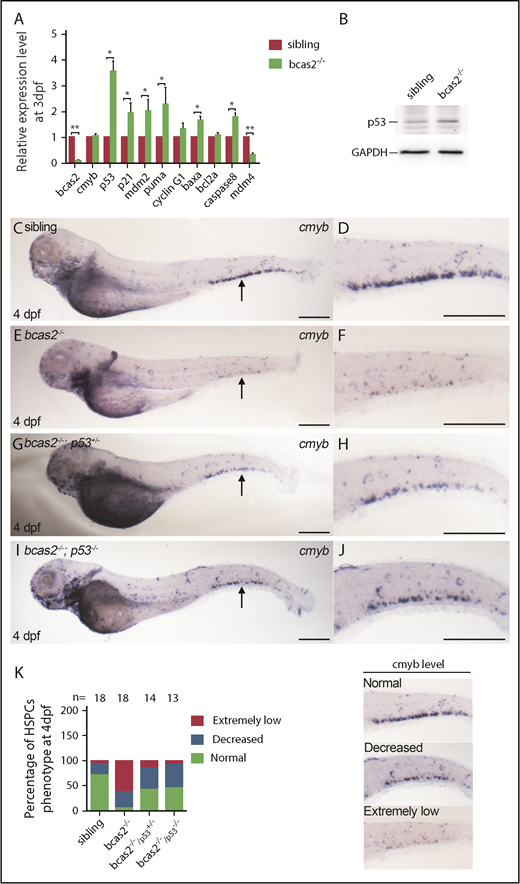
Apoptosis of the HSPCs in bcas2−/−embryos is p53 dependent. (A) Real-time qPCR analysis showed increased expression of all p53-dependent apoptosis-related genes except for cyclin G and bcl2a, after a decrease in Mdm4 expression. (B) Western blot showed increased expression of p53 in bcas2−/− embryos at 3 dpf compared with siblings. (C-F) WISH results showed significantly decreased expression of cmyb in CHT of bcas2−/− embryos at 4 dpf. (G-H) bcas2−/−p53+/− embryos displayed partially rescued cmyb expression at 4 dpf. (I-J) bcas2−/−p53−/− embryos showed well-rescued cmyb expression at 4 dpf. Scale bars, 200 μm. (K) Quantitative analysis of cmyb expression phenotypes in HSPCs, as identified by WISH analysis. The 3 types of cmyb WISH phenotypes in the CHT are shown on the right. The corresponding numbers of embryos are displayed above the columns. Data are represented as mean ± SD. *P < .05; **P < .001.
Apoptosis of the HSPCs in bcas2−/−embryos is p53 dependent. (A) Real-time qPCR analysis showed increased expression of all p53-dependent apoptosis-related genes except for cyclin G and bcl2a, after a decrease in Mdm4 expression. (B) Western blot showed increased expression of p53 in bcas2−/− embryos at 3 dpf compared with siblings. (C-F) WISH results showed significantly decreased expression of cmyb in CHT of bcas2−/− embryos at 4 dpf. (G-H) bcas2−/−p53+/− embryos displayed partially rescued cmyb expression at 4 dpf. (I-J) bcas2−/−p53−/− embryos showed well-rescued cmyb expression at 4 dpf. Scale bars, 200 μm. (K) Quantitative analysis of cmyb expression phenotypes in HSPCs, as identified by WISH analysis. The 3 types of cmyb WISH phenotypes in the CHT are shown on the right. The corresponding numbers of embryos are displayed above the columns. Data are represented as mean ± SD. *P < .05; **P < .001.
bcas2 deletion triggers alternative splicing of Mdm4 without gross changes in DNA damage
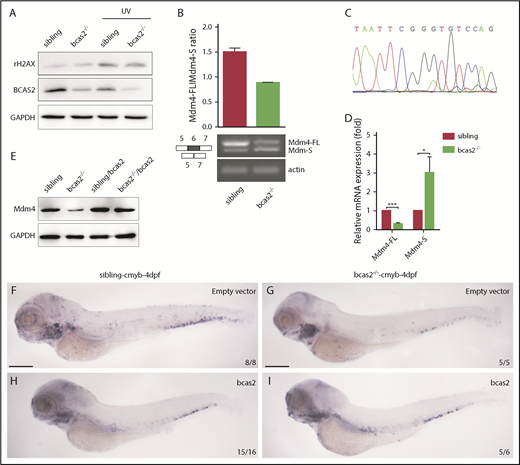
Previous studies have demonstrated that BCAS2 plays conserved roles in DNA damage response.16,26,27 Surprisingly, our results showed that there was no increase in expression of γH2AX in bcas2−/− mutants compared with siblings at 2 dpf, even during UV treatment (Figure 6A). The data suggested that DNA damage may not be the major reason for p53 activation in the context of bcas2 disruption.
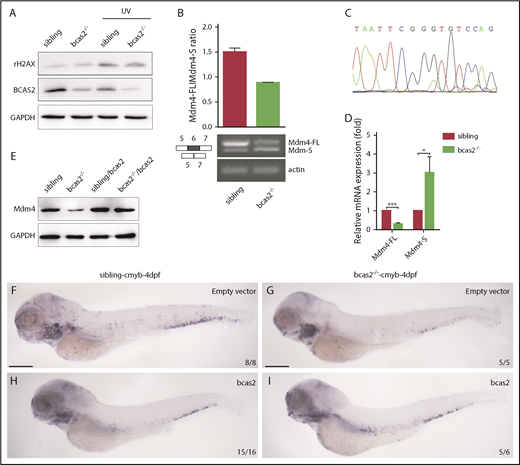
BCAS2 deprivation triggers alternative splicing of Mdm4 without gross changes in DNA damage. (A) The expression of γH2AX in bcas2−/− embryos and siblings showed no difference, even under UV treatment. (B) Semiquantitative PCR analysis and relative quantification of total Mdm4-FL and Mdm4-S isoforms in the CHT region of siblings and bcas2−/− mutants. (C) Sequencing of the shorter product showed a deficiency of exon 6 compared with Mdm4-FL. (D) Real-time PCR analysis of total Mdm4-FL and Mdm4-S isoforms in the CHT region of siblings and bcas2−/− mutants. (E) Mdm4-FL protein levels are reduced in bcas2−/− embryos that can be recovered by re-expression of bcas2. (F-I) Ectopic expression of wild-type bcas2 mRNA can rescue HSPC defects in bcas2−/− mutants at 4 dpf. Scale bars, 200 μm.
BCAS2 deprivation triggers alternative splicing of Mdm4 without gross changes in DNA damage. (A) The expression of γH2AX in bcas2−/− embryos and siblings showed no difference, even under UV treatment. (B) Semiquantitative PCR analysis and relative quantification of total Mdm4-FL and Mdm4-S isoforms in the CHT region of siblings and bcas2−/− mutants. (C) Sequencing of the shorter product showed a deficiency of exon 6 compared with Mdm4-FL. (D) Real-time PCR analysis of total Mdm4-FL and Mdm4-S isoforms in the CHT region of siblings and bcas2−/− mutants. (E) Mdm4-FL protein levels are reduced in bcas2−/− embryos that can be recovered by re-expression of bcas2. (F-I) Ectopic expression of wild-type bcas2 mRNA can rescue HSPC defects in bcas2−/− mutants at 4 dpf. Scale bars, 200 μm.
Mdm4 can inhibit p53 transactivation of target genes by binding to the N-terminal transactivation domain of p53.28 Because BCAS2 participates in splicing reactions and can influence splice site selection,17 we further investigated whether alternative splicing of Mdm4 may be a cause of p53 activation. Alternative splicing of Mdm4 results in the expression of 2 isoforms based on the inclusion of or skipping of exon 6 in humans or exon 7 in mice.29,30 Mdm4 undergoes alternative splicing at exon 6 in bcas2−/− knockout zebrafish, resulting in the production of a shorter MDM4 isoform that has previously been described as Mdm4-S that might attenuate the full-length MDM4 protein (Mdm4-FL) level31 (supplemental Figure 8).
We speculated that disruption of bcas2 might cause defects in constitutive splicing of Mdm4 and increase Mdm4 exon 6 exclusion. As expected, the expression of Mdm4-FL showed a significant decrease in bcas2 mutants, with a concomitant increase in Mdm4-S levels (Figure 6B-D). This results in a lack of Mdm4 protein (Figure 6E), which might contribute to increased p53 activity.
To confirm that bcas2 was responsible for the phenotypes observed, we performed a rescue assay by ectopic expression of wild-type bcas2 in bcas2−/− embryos. After injection of bcas2 mRNA into 1-cell-stage bcas2−/− embryos, ectopic expression of bcas2 mRNA in bcas2−/− embryos increased the Mdm4-FL:Mdm4-S ratio (supplemental Figure 9A), which positively correlated with higher Mdm4 protein expression levels (Figure 6E). In addition, the wild-type bcas2 rescue assay implies that a deficiency of bcas2 was responsible for the hematopoietic phenotypes in the bcas2−/− embryos (Figure 6F-I).
P53 can be activated in response to alternative splicing of Mdm4
To confirm that alternative splicing of Mdm4 was not a consequence of but rather a cause of p53 activation and apoptosis, we measured the Mdm4-FL:Mdm4-S ratio in different p53 genetic backgrounds (Figure 7A). Consistent with our hypothesis, the alternative splicing of Mdm4 was not changed in p53-mutant embryos.
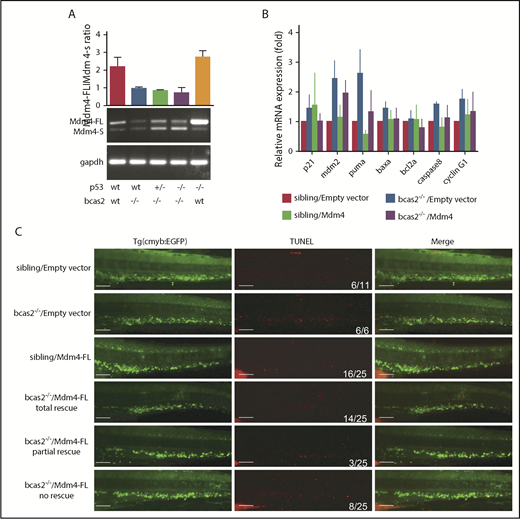
p53 can be activated in response to alternative splicing of Mdm4. (A) PCR analysis of the alternative splicing events on the Mdm4 mRNA upon bcas2 deletion in different p53 genetic backgrounds. (B) Microinjection of Mdm4-FL into bcas2−/− mutants could partially rescue p53 activation. Real-time qPCR of p53 target gene in siblings and bcas2−/− mutants re-expressing Mdm4-FL or empty vector. At least 3 representative experiments were shown as an example. (C) TUNEL staining of siblings and bcas2−/−/Tg (cmyb: EGFP) zebrafish re-expressing Mdm4-FL or empty vector. The last 3 lines represent 3 types of rescue patterns. Scale bars, 50 μm.
p53 can be activated in response to alternative splicing of Mdm4. (A) PCR analysis of the alternative splicing events on the Mdm4 mRNA upon bcas2 deletion in different p53 genetic backgrounds. (B) Microinjection of Mdm4-FL into bcas2−/− mutants could partially rescue p53 activation. Real-time qPCR of p53 target gene in siblings and bcas2−/− mutants re-expressing Mdm4-FL or empty vector. At least 3 representative experiments were shown as an example. (C) TUNEL staining of siblings and bcas2−/−/Tg (cmyb: EGFP) zebrafish re-expressing Mdm4-FL or empty vector. The last 3 lines represent 3 types of rescue patterns. Scale bars, 50 μm.
To further validate whether the Mdm4 decrease is responsible for the increased p53 activity in bcas2−/− mutants, we microinjected full-length Mdm4 mRNA into bcas2−/− mutants. Overexpression of Mdm4 in bcas2 knockout zebrafish could only partially rescue the p53 activation response and the induction of apoptosis (Figure 7B-C). Moreover, the expression of cmyb in bcas2−/− zebrafish was partially rescued, suggesting that Mdm4 exon 6 exclusion was consistently accompanied by a decrease in HSPC survival (supplemental Figure 9B-C).
Discussion
Role of BCAS2 in the maintenance of HSPCs
In this study, we established a novel bcas2−/−-mutant zebrafish line, which retained primitive hematopoiesis but showed a severe defect in definitive hematopoiesis. The reduction of HSPCs is due to increased p53-dependent apoptosis and leads to the embryonic lethality of bcas2−/− embryos at 7 to 10 dpf.
bcas2 transcripts were identified in the zebrafish hematopoietic system, which suggests that there is a conserved role of BCAS2 in blood and immune cell development in vertebrates. WISH analysis revealed a gradual decrease of cmyb expression in bcas2−/− embryos from 3 dpf to 4 dpf (Figure 3), indicating that definitive HSPCs were defective in the BCAS2 loss-of-function embryos. However, we cannot conclude that BCAS2 is not necessary for primitive hematopoiesis or for the specification of definitive HSPCs because it is possible that the maternal BCAS2 could still support the early development of embryos up to 3 dpf in mutants.
Definitive HSPCs emerge from the aortic endothelium transiently, and these cells first migrate to the CHT, a location similar to that of the mammalian fetal liver, where they undergo rapid proliferation and differentiation and then migrate to seed the definitive hematopoietic organs such as the kidney and the thymus.32 Although the number of HSPCs was decreased in bcas2−/− mutants at 3 dpf to 4 dpf (Figure 4), the proliferation of HSPCs still maintained a constant high rate (supplemental Figure 7). Notably, tissues with a high proliferation rate, such as those in hematopoietic organs, usually display hypersensitivity to disadvantageous genetic mutations. In addition, ectopic expression of bcas2 can rescue the failure of fetal hematopoiesis and apoptosis in bcas2−/− mutants. Altogether, our data illustrated a novel and vital role for bcas2 in HSPC maintenance during zebrafish embryogenesis.
Role of BCAS2 in regulation of p53-dependent apoptosis of HSPCs
The role of BCAS2 in development has been previously studied in mice and Drosophila.16-18 Our data showed that BCAS2 has a novel function in activating the p53 pathway during HSPC development in zebrafish. Interestingly, unlike in mice, DNA damage in zebrafish is not a major factor of p53 activation in response to bcas2 deletion. This difference can be attributed to species differences between mice and zebrafish, but it can also be attributed to differences between HSPCs and other cells.
Previous studies suggested that p53 activation is a general consequence of spliceosome defects.33 Although several studies have already identified that p53 is highly expressed in HSPCs and may mediate the quiescence, self-renewal, and apoptosis of HSPCs,34-36 our findings demonstrate that BCAS2 is essential for HSPC survival via regulation of p53 signaling in zebrafish. Importantly, as a component of the spliceosome, BCAS2 has not been reported to be involved during HSPC development, and the underlying mechanism that regulates p53 activation in HSPCs is still unclear. Recently, Mdm4 was reported as a key regulator of p53 activation in several cancer cells.29,37,38 Our results showed that bcas2 mutation results in an accumulation of p53 protein and transcriptional activity accompanied with a reduction in mdm4 mRNA and protein expression, which might provide a mechanism of p53 activation.
Role of BCAS2 in regulating alternative splicing of Mdm4
In our study, the decrease in Mdm4-FL mRNA levels was concomitant with the increase in Mdm4-S mRNA levels. Mdm4-S introduces a premature stop codon into its transcript that potentially encodes a truncated Mdm4-S protein, which might be the reason for decreased Mdm4 protein expression. Ectopic expression of Mdm4-FL partially inhibited p53 transcriptional activation and p53-dependent apoptosis of HSPCs in zebrafish in vivo. However, it does not fully account for p53 activation that results from other mechanisms, including other alternative splicing events induced by the absence of BCAS2.
BCAS2 is a core component of the splicing-related Prp19 complex, which is involved in stable association of U5 and U6 with the spliceosome after U4 dissociation.39 Specific deletion of bcas2 in mouse germ cells affects splicing of several hundred genes.40 In zebrafish, bcas2 mutation might result in defective spliceosome assembly, dysregulated global alternative splicing of genes that play important roles in hematopoiesis, or p53-dependent apoptosis. Further studies may identify the consequences of mutant bcas2-induced splicing alterations for hematopoiesis.
In summary, our results obtained in the bcas2−/− mutants revealed an unexpected role for BCAS2 during definitive hematopoiesis and showed that p53 signaling is involved in this process. The p53 pathway was activated in bcas2−/− mutants as a result of abnormal alternative splicing of Mdm4. Recently, RNA sequencing data revealed significant alterations in gene expression of many splicing factors in sAML, including downregulation of bcas2.11 Notably, the most common clinical feature of MDS is ineffective hematopoiesis in multiple cell lineages with increased apoptosis, which is similar to the phenotype observed in bcas2−/− zebrafish. This study expands our understanding of the special mechanism of BCAS2-mediated pre-mRNA splicing and its effect on HSPC survival during hematopoiesis development, which may lead to new methods of therapeutic interventions for hematopoiesis disorders.
For original data, please contact lium@mail.hust.edu.cn.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Feng Liu (Chinese Academy of Sciences), Luying Liu (Yale University), and James Reilly (Glasgow Caledonian University) for their critical reading and valuable comments.
This work was supported by grants from the National Natural Science Foundation of China (81670099, 31472263, 31471194, 31571303, 31801041, and 81670890).
Authorship
Contribution: M.L. and T.J. developed the study concept; S.Y. and D.L. designed the study; S.Y., D.J., and Y. Han performed most of the experiments in zebrafish and cell lines; D.J. and Y.Z. generated bcas2−/− zebrafish by using TALEN technology; Y. Huang, Z.Q., and Y.L. contributed to vector construction and western blot; J.L., X.H., Z.L., and Y.Q. assisted with cell culture and real-time PCR assays; J.T., S.H., X.L., and S.X. helped prepare zebrafish and the in situ assays; S.Y., M.L., T.J., D.L., Z.T., Q.K.W., and F.L. analyzed the data; S.Y., D.L., and M.L. prepared the draft and final version of the manuscript; and all authors reviewed the results and approved the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Mugen Liu, College of Life Science and Technology, Huazhong University of Science and Technology, 1037 Luoyu Rd, Wuhan, Hubei, 430074, People’s Republic of China; e-mail: lium@mail.hust.edu.cn; and Daji Luo, Department of Genetics, School of Basic Medical Sciences, Wuhan University, 185 Donghu Road, Wuhan, Hubei, 430071, People’s Republic of China; e-mail: luodaji@whu.edu.cn.