

In this issue of Blood, identify huntingtin associated protein 1 (HAP1) loss as a new marker of l-asparaginase resistance in acute lymphoblastic leukemia (ALL) and provide evidence for the pathway involved. They discovered that HAP1 is essential for the formation of the ternary complex with huntingtin (Htt) and inositol 1,4,5-triphosphate receptor (InsP3R) and that its loss impairs the l-asparaginase–mediated increase of cytosolic Ca2+ needed for triggering apoptosis (see figure). Their data were confirmed by specific knockdown of HAP1 in SEM cells and by measurement of both endoplasmic reticulum–released Ca2+ and external Ca2+ influx.1
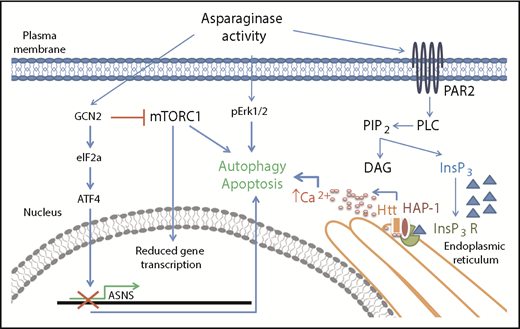
Asparaginase removes asparagine supply from the extracellular environment creating an asparagine-starved environment. At the cellular level, asparagine starvation causes the activation of the amino acid response mechanism triggered by the presence of uncharged transfer RNAs and initiated by the general control nonrepressible-2 (GCN2) kinase. GCN2 activation has a double downstream effect: it induces expression of stress response proteins by activating the eukaryotic initiation factor 2 (eIF2a)-activating transcription factor 4 (ATF4) transduction axis and it represses general protein synthesis by repressing Akt/mTORC1 activity. Failure in expression of stress response proteins such as asparagine synthetase (ASNS) causes activation of proautophagy mechanisms and, eventually, apoptosis. Lee et al described a new route for asparaginase proapoptotic signaling that involves the activation of the protease-activated receptor 2 (PAR2)/InsP3R Ca2+-mediated pathway. Failure in its activation because of HAP1 loss has a relevant effect on the capability of asparagine starvation to trigger apoptosis in leukemia cells.
Asparaginase removes asparagine supply from the extracellular environment creating an asparagine-starved environment. At the cellular level, asparagine starvation causes the activation of the amino acid response mechanism triggered by the presence of uncharged transfer RNAs and initiated by the general control nonrepressible-2 (GCN2) kinase. GCN2 activation has a double downstream effect: it induces expression of stress response proteins by activating the eukaryotic initiation factor 2 (eIF2a)-activating transcription factor 4 (ATF4) transduction axis and it represses general protein synthesis by repressing Akt/mTORC1 activity. Failure in expression of stress response proteins such as asparagine synthetase (ASNS) causes activation of proautophagy mechanisms and, eventually, apoptosis. Lee et al described a new route for asparaginase proapoptotic signaling that involves the activation of the protease-activated receptor 2 (PAR2)/InsP3R Ca2+-mediated pathway. Failure in its activation because of HAP1 loss has a relevant effect on the capability of asparagine starvation to trigger apoptosis in leukemia cells.
ALL is a tumor of lymphoblasts and is characterized by great heterogeneity at the molecular level.2 ALL mostly affects children (more than 50% of total cases) with an overall 5-year survival rate of 68%. The rate increases to 80% if only pediatric patients are included and drops to 40% in the adult population (National Institutes of Health, National Cancer Institute and GLOBOCAN 2018). In addition to the influence of age, ALL subtype is also important for prognosis. In all subtypes, there is an ongoing quest for therapeutic alternatives for the treatment of refractory leukemia.
Therapy for ALL in both pediatric and adult patients usually includes early administration of l-asparaginase, a bacteria-derived protein drug used with vincristine and glucocorticoids. Currently, about 20% of patients treated with standard therapeutic protocols will relapse within 5 years from diagnosis because leukemia cells develop resistance to standard drugs. ALL treatment follows the very successful Berlin-Frankfurt-Münster (BFM) scheme, introduced in 1995. The dosage and schedule have been optimized over the years with no changes in the BFM components.3 ALL risk stratification at diagnosis is routinely accomplished by considering several factors, among which are blast cell counts and cytogenetic and molecular abnormalities. However, risk stratification serves purely to determine leukemia prognosis and relapse probability, because high-risk leukemia (HRL) patients are assigned to a treatment regimen similar to that of low-risk leukemia patients, with modest changes in drug administration schedule and dosage.3,4
Identifying specific molecular profiles for targeted therapy in HRL patients would be useful, as demonstrated by the improvements in survival in Philadelphia chromosome–positive ALL.5 Currently, some potential markers of resistance to standard chemotherapy have been identified and characterized at the bench level.6 The best validated marker of l-asparaginase resistance so far is ASNS. In fact, previous studies have shown that high levels of ASNS are linked to l-asparaginase resistance. However, not all subtypes of ALL that are resistant to l-asparaginase are ASNS positive.7 Therefore, ASNS alone is not enough to explain all cases of observed resistance. The opioid receptor μ1 has been detected as a possible mediator of ALL resistance to Erwinia chrysanthemi–derived l-asparaginase (Erwinase),7 and recent evidence from our laboratory suggests that growth factors receptors might be another (M.M., unpublished data).
Lee et al have performed an unbiased RNA interference screening (24 000 short hairpin RNAs) in a cell line (SEM) derived from a relapsed 5-year-old ALL patient while looking for a single or multiple gene combination whose loss could confer l-asparaginase resistance. They found that HAP1 loss significantly increased the cell line resistance to l-asparaginase cytotoxicity. HAP1 is a marker of positive prognosis for pancreatic cancer, and its overexpression has been associated with reduced aggressiveness and spread of breast cancer.8
Lee et al explored the pathway through which HAP1 rescues l-asparaginase sensitivity by evaluating apoptosis of SEM cells in the presence or absence of HAP1 overexpression. Specifically, they analyzed the l-asparaginase–related pathway activated by PAR2 described by Peng et al,9 which represents an addition to the classical amino acid response mechanism (see figure). The authors also demonstrated that the activated pathway involved the calpain 1-Bid-caspase 3/12 cascade. Next, they inversely correlated HAP1 expression levels with in vitro l-asparaginase resistance in patient-derived ALL cells. To our knowledge, this is the first time a direct correlation between l-asparaginase activity and HAP1-Htt-InsP3R complex proapoptotic activity has been shown.
The main implications of the study are twofold. First, HAP1 loss can be used as a marker of l-asparaginase resistance and can easily be identified in the clinical laboratory by standard techniques, although threshold values must be defined. Second, the pathway that includes HAP1 can be used as a target for designing new drugs: for example, a specific biological therapy could compensate for HAP1 loss in ALL cells, making them l-asparaginase sensitive. Moreover, the molecular data on the proapoptotic correlation between l-asparaginase activity and HAP1 expression can be used to address pancreatic damage that often causes moderate-to-severe pancreatitis in patients treated with l-asparaginase.9
What needs to be done next? This is an exciting contribution to the field of ALL research, and it will be interesting to see its implications on treatment and drug design. This model could also be tested in other l-asparaginase–resistant cell lines and cell lines from non-ALL patients. Finally, in addition to the work by Lee et al, there are more pieces that still need to be added to the puzzle of l-asparaginase resistance.
Conflict-of-interest disclosure: C.S. and M.M. are inventors of several asparaginase-related patents owned by the University of Pavia. C.S. is a shareholder of Ardis S.r.l., a spin-off of the University of Pavia.

