Key Points
Extended ibrutinib treatment showed sustained PFS in previously treated patients with CLL, including those with high-risk cytogenetics.
Overall survival outcomes were sustained and no long-term safety signals have emerged with 4 years of follow-up on ibrutinib treatment.

Abstract
Ibrutinib, a once-daily oral inhibitor of Bruton tyrosine kinase, has greatly improved outcomes for patients with chronic lymphocytic leukemia (CLL). The phase 3 RESONATE trial, which compared single-agent ibrutinib to ofatumumab in high-risk, relapsed patients with CLL, provided support for approval of ibrutinib in the United States and Europe. We describe long-term follow-up of patients treated in RESONATE, where continued superiority of progression-free survival (PFS) (hazard ratio [HR], 0.133; 95% confidence interval [CI], 0.099-0.178) was observed. Overall survival benefit continues (HR, 0.591; 95% CI, 0.378-0.926), although with decreased magnitude relative to that seen before crossover to ibrutinib was implemented for patients on ofatumumab (HR, 0.426; 95% CI, 0.220-0.823). Notably, overall response to ibrutinib increased over time, with 91% of patients attaining a response. The PFS benefit with ibrutinib was independent of baseline risk factors, although patients with ≥2 prior therapies had shorter PFS than those with <2 prior therapies, and the presence of TP53 or SF3B1 mutations showed a trend toward shorter PFS vs without these factors. Median duration of ibrutinib was 41 months, with 46% remaining on treatment at a median follow-up of 44 months. Grade ≥3 adverse events generally decreased over time, causing only a small proportion of patients to cease therapy. Ibrutinib was discontinued due to progressive disease in 27% of patients. This long-term study provides support for sustained efficacy and safety of ibrutinib in relapsed/refractory CLL and consideration of study provisions that allow crossover to investigational therapy when benefit has been clearly demonstrated. This trial was registered at www.clinicaltrials.gov as #NCT01578707.
Introduction
Chronic lymphocytic leukemia (CLL) is characterized by increasing frequency in individuals aged >60 years and has a variable natural history predicted in part by clinical and genomic features.1-4 CLL is typically diagnosed at an early stage and is monitored without therapy until symptoms develop.1 At the time of therapy initiation, a patient’s age, comorbidities, and select genotype features, such as presence or absence of del(17)(p13.1), del(11)(q22.3), TP53 mutations, or immunoglobulin heavy chain variable region gene (IGHV) mutational status are important in treatment choice.1 For initial therapy of CLL, treatment approaches have evolved from monotherapy with alkylating agents to chemoimmunotherapy including a CD20 antibody such as rituximab or obinutuzumab.5-8 Although a subset of patients with CLL requiring treatment are young or fit with IGHV-mutated disease in which durable remissions can be appreciated with fludarabine, cyclophosphamide, and rituximab (FCR), the majority of patients are not and typically relapse within 3 to 5 years or earlier.3,9,10 At relapse, treatment often includes different or similar chemoimmunotherapy or targeted agents such as B-cell receptor signaling pathway inhibitors or BCL-2 inhibitors.11,12
The introduction of ibrutinib, a first-in-class, once-daily, orally bioavailable, covalent inhibitor of Bruton tyrosine kinase approved for treatment of CLL, has greatly changed how this disease is treated.13 Bruton tyrosine kinase is an enzyme whose expression and activity are essential for B-cell receptor signaling, cellular homing, and adhesion.14-17 Ibrutinib was initially approved for treatment of CLL for patients who had received ≥1 prior therapy, based on high response rates and durability of response demonstrated in a single-arm phase 2 study.18 Most patients on the pivotal phase 2 study (PCYC-1102) derived benefit from single-agent ibrutinib therapy. However, the size and single-arm design of this initial trial limited the ability to fully understand safety and efficacy with prolonged ibrutinib treatment, including identification of pretreatment features associated with shorter progression-free survival (PFS).18,19 This randomized phase 3 study (PCYC-1112, RESONATE) was initiated to compare once-daily, oral, single-agent ibrutinib with ofatumumab, an approved single-agent therapy, in patients with relapsed or refractory CLL. The primary analysis (median follow-up, 9.4 months) was reported previously20 and provided justification for full regulatory approval of ibrutinib in previously treated CLL.21 The large population of the RESONATE study (N = 391) with its extended follow-up provides an opportunity to examine pretreatment genomic features and long-term efficacy and safety of ibrutinib treatment in patients with CLL.22 Additionally, midstudy modification of the study design to permit crossover from control treatment to ibrutinib allows analyses toward understanding the impact of crossover on survival.
Methods
Patients
Detailed eligibility criteria have previously been described.20 Eligible patients with CLL or small lymphocytic lymphoma (SLL) had received ≥1 line of prior therapy and met International Workshop on Chronic Lymphocytic Leukemia (IWCLL) 2008 criteria for requiring therapy but were considered inappropriate candidates for purine analog treatment (see supplemental Methods, available on the Blood Web site). All patients provided written informed consent. The study was approved by the institutional review board or independent ethics committee at each participating institution and was conducted in accordance with the Declaration of Helsinki and International Conference on Harmonization Guidelines for Good Clinical Practice. The study was sponsored by Pharmacyclics LLC, an AbbVie company, and Janssen Research and Development, LLC.
Study design
The RESONATE study was an international, open-label, randomized, phase 3 study designed to compare the efficacy and safety of ibrutinib with ofatumumab (registered at www.clinicaltrials.gov as #NCT01578707). An independent review committee (IRC), blinded to treatment and lymphocyte data, assessed disease progression and response. Patients were enrolled between June 2012 and April 2013 and were randomized 1:1 to receive oral ibrutinib 420 mg once daily until disease progression or unacceptable toxicity or IV ofatumumab administered for up to 24 weeks at an initial dose of 300 mg at week 1, followed by 2000 mg weekly for 7 weeks then every 4 weeks for 16 weeks. Patients were stratified according to (1) disease refractory to purine analog-anti-CD20 chemoimmunotherapy regimen (defined as no response or relapse within 12 months of the last purine analog dose) and (2) presence of del(17)(p13.1). During the conduct of this study, continued promising durable responses in the phase 2 study led investigators and the RESONATE steering committee to request, and the independent data monitoring committee (DMC) to endorse, crossover of patients from ofatumumab to ibrutinib.18 This was supported by the sponsor, and health authorities were informed. A protocol amendment was released ∼4 months after the last patient was randomized that allowed ofatumumab-randomized patients with IRC-confirmed disease progression to receive ibrutinib. Subsequently, based on results from the interim analysis, the DMC recommended allowing crossover to the ibrutinib arm for all remaining patients on the ofatumumab arm.
Study assessments and follow-up
The primary end point was PFS assessed by the IRC per IWCLL 2008 criteria with 2012 clarification for treatment-related lymphocytosis not to be considered progressive disease (PD).23 Key secondary end points included overall survival (OS) and overall response rate (ORR) as defined by IWCLL criteria.24 Monitoring of patients for the first 18 months has been previously described.20 Thereafter, patients were seen every 3 months for a disease and toxicity assessment that included history, physical examination, and laboratory monitoring (complete blood count, chemistry, and liver function tests). Further details on study assessments, including laboratory studies, are described in supplemental Methods.
Statistical analysis
The intent-to-treat population was assessed for long-term efficacy. Long-term safety was assessed in treatment-emergent periods for each randomized therapy. In this updated analysis, PFS and ORR were determined by investigator assessment, as IRC assessment was not performed beyond the primary analysis. The proportion of patients with sustained hematologic improvement was compared using a Fisher exact test. All patients who received ≥1 treatment dose were included in the safety analysis. At interim analysis (median 9 months of follow-up), the DMC declared superiority of ibrutinib vs ofatumumab for PFS and OS and recommended early unblinding of the study. The interim analysis was thus considered the primary analysis. An additional OS sensitivity analysis was performed per published guidelines to address the bias introduced by extensive crossover from ofatumumab to ibrutinib. The rank-preserving structural failure time model was used to account for crossover and represents a randomization-based method for estimating counterfactual survival times that would have been observed in the absence of crossover. Multivariate analysis for PFS in the ibrutinib arm was conducted with the following potential covariates included in the model: age, Rai stage, Eastern Cooperative Oncology Group (ECOG) status, number of prior lines of therapy, del(17)(p13.1) status, del(11)(q22.3) status, B2M, lactate dehydrogenase, and refractory status to purine analogs. Covariates in the final model were identified using forward, backward, and stepwise selection.
Results
Patient features
A total of 391 patients were enrolled (supplemental Figure 1). Baseline characteristics, including genomic and mutational features, are shown in Table 1. A large proportion of patients had high-risk features, including del(17)(p13.1) (32% in ibrutinib arm; 33% in ofatumumab arm) or TP53 mutation (51% and 46%, respectively); 25% and 22%, respectively, had complex karyotype. In post hoc analysis of available baseline data, large proportions of patients had high-risk (24% in the ibrutinib arm; 27% in the ofatumumab arm) or very-high-risk CLL-IPI scores (36% and 25%, respectively); 35% and 40% in the ibrutinib and ofatumumab arms, respectively, could not be scored (Table 1). Baseline bulky disease (lymph node ≥ 5 cm) and unmutated IGHV were more frequently observed in the del(11)(q22.3) subgroup (supplemental Table 1). Median follow-up for patients assigned initially to ibrutinib was 44 months (range, 0.33-53.16) and median time on ibrutinib was 41 months (range, 0.2-50.1 months), with 69% receiving >2 years of ibrutinib. At time of analysis, 90/195 patients (46%) remain on initially assigned ibrutinib therapy (supplemental Table 2). The most common reasons for discontinuation of ibrutinib were PD (53/195 patients; 27%) and adverse events (AEs) (23/195; 12%). Of 53 patients for whom PD was the primary reason for discontinuation of ibrutinib, 14 patients discontinued due to Richter transformation (diffuse large B-cell lymphoma, n = 9; Hodgkin disease, n = 3; prolymphocytic lymphoma, n = 2); median time to transformation in these 14 patients was 13.6 months (range, 1.7-27.8). Sequencing data for somatic mutations in TP53, SF3B1, NOTCH1, XPO1, and BIRC3 genes were available from baseline in 10 of 14 Richter transformation patients. Mutations were found to be present in TP53 (8/10 patients), SF3B1 (5/10 patients), NOTCH1 (4/10 patients), XPO1 (1/10 patients), and BIRC3 (1/10 patients). These gene mutations, with the exception of TP53 mutations, were found to occur concurrently with mutations in ≥1 of the other assayed genes. Of 8 patients with TP53 mutations, half (4/8 patients) also had mutations in ≥1 of these other negative prognostic genes, while half only had mutations in TP53. Of 23 patients who discontinued ibrutinib due to AEs, the most frequent AEs were pneumonia (n = 3) and anemia, thrombocytopenia, diarrhea, and anal incontinence (n = 2 each).
Median follow-up time for patients initially assigned to ofatumumab who crossed over to ibrutinib at time of progression (n = 133) was 43.6 months (range, 7.2+ to 50.0); 41% of these patients were still receiving ibrutinib at the time of analysis.
PFS
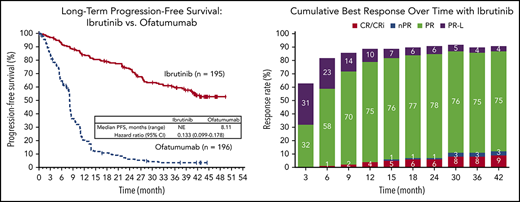
At a median follow-up of 44 months (range, 0.33-53 months) for ibrutinib, PFS remains significantly longer for ibrutinib than for ofatumumab, with a HR of 0.133 (95% CI, 0.099-0.178; P < .0001) (Figure 1). Median PFS was not reached (NR) with ibrutinib and 8.1 months for ofatumumab. Of those patients assigned to ofatumumab, 178 out of 196 (91%) had experienced progression or death at the time of analysis. The 3-year PFS rate was 59% with ibrutinib vs 3% with ofatumumab.
![Figure 1. PFS at a median follow-up of 44 months (intention-to-treat [ITT] population). NE, not evaluated.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/133/19/10.1182_blood-2018-08-870238/3/m_blood870238f1.png?Expires=1769863343&Signature=yghyvHqZFkp2LAolkZKmj~nprsAiFrJVoYZ0R-hR8sdYZNj9paxY3jKO5kU90Cup-GTgNNEi9lcaKvrcDE8EtomVkfKC2sDRIpAPiZk6SMYUB2-CTwb-j~2IHYqBv--DFF-79KzYxhKIHmM5nlu6DADO4kx1qAqS7XRkwhxFptlLHv4EbvYTNUDb24S3Z5rqdwJkq31KUUn1GNOYcDkzi6yPquukJL28tmx-ZSFyn-p-A9O5AJf3ochn4IGr8EACyk-nRnAZA~HnPMHlkGM3ncqjpnPblBmH794s1YdOgkYL3y9iEg0vLcIyQ-3~-OmUw43SpXgKbQLJ2UzT-oRDaA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
PFS at a median follow-up of 44 months (intention-to-treat [ITT] population). NE, not evaluated.
PFS at a median follow-up of 44 months (intention-to-treat [ITT] population). NE, not evaluated.
The benefit of ibrutinib vs ofatumumab on PFS was consistent across patient subgroups analyzed according to baseline clinical characteristics or molecular features, with HRs ranging from 0.049 to 0.183 (Figure 2). Consistent with PFS results for the overall population, similar PFS benefit with ibrutinib treatment was also observed in exploratory post hoc analyses of patient subgroups with high- and very-high-risk CLL-IPI scores. HRs for PFS events with ibrutinib vs ofatumumab by CLL-IPI risk groups were 0.064 (95% CI, 0.008-0.502) for intermediate-risk, 0.115 (95% CI, 0.059-0.222) for high-risk, and 0.118 (95% CI, 0.067-0.207) for very-high-risk CLL-IPI groups. PFS was also examined within the ibrutinib arm. PFS was longer in patients who received ≤2 prior lines of therapies (62% [57/92] had 2 prior) compared with more heavily pretreated patients with >2 prior lines (median PFS NR vs 35.1 months) (Figure 3A). Of the genomic factors examined, PFS for the del(11)(q22.3) subgroup trended to have the most favorable outcome; however, PFS was not statistically different for patients with del(17)(p13.1) (median 40.1 months) or del(11)(q22.3) (median NR) or without these fluorescence in situ hybridization abnormalities (median NR) (Figure 3B). Median PFS among patients with complex karyotype was 40.8 months vs NR for patients without complex karyotype (Figure 3C; HR 1.292; 95% CI, 0.770-2.168). Among patients with complex karyotype, median PFS was 41.2 vs 31.7 months (HR, 0.795; 95% CI 0.329-1.921) in patients with del(17)(p13.1) vs without del(17)(p13.1). No significant differences were observed in PFS with ibrutinib between patients with unmutated and mutated IGHV status; median PFS was NR in both subgroups and 3-year PFS was 63% vs 66%, respectively (Figure 4A). Similarly, no significant differences in PFS with ibrutinib treatment were seen in patients with or without mutations in NOTCH1, TP53, SF3B1, BIRC3, or XPO1. Median PFS was NR for patients with or without mutated NOTCH1 status (Figure 4B). PFS in patients with TP53 mutations had a trend toward less favorable outcomes vs those without TP53 mutations (median PFS 40.7 months vs NR), although this difference was not statistically significant (Figure 4C). Median PFS was 40.6 months in patients with mutated SF3B1 gene but NR for patients with nonmutated SF3B1 status (Figure 4D). Median PFS was NR in patients with or without mutated BIRC3 (Figure 4E). Median PFS was 38.5 months in patients with mutated XPO1 compared with NR for patients with nonmutated XPO1 (Figure 4F).
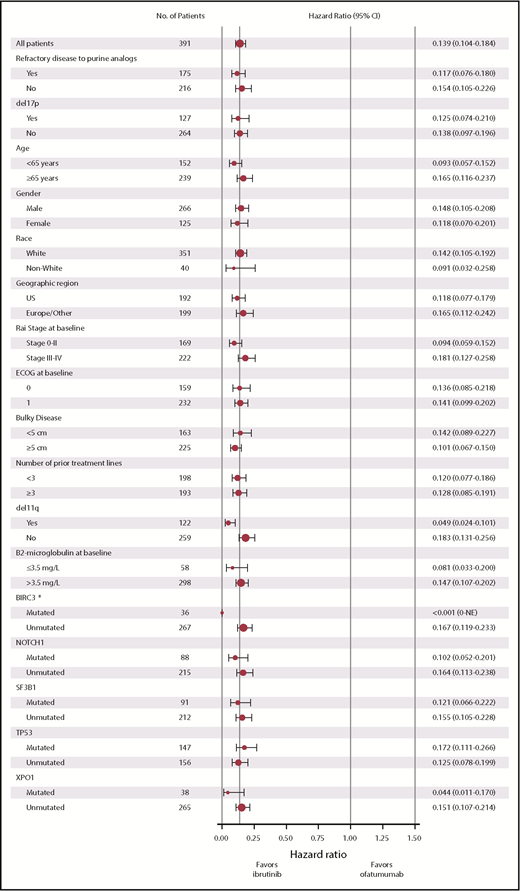
Forest plot of HRs for PFS by baseline subgroups (ITT population). *All 15 patients with mutated BIRC3 in the ofatumumab arm had events.
Forest plot of HRs for PFS by baseline subgroups (ITT population). *All 15 patients with mutated BIRC3 in the ofatumumab arm had events.
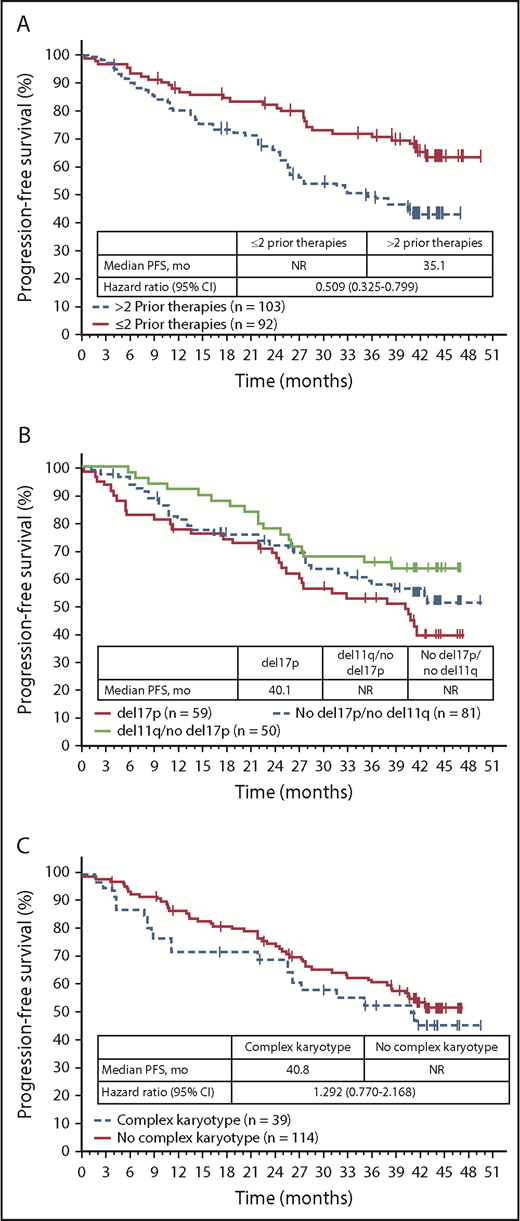
PFS by number of prior therapies and cytogenetic subgroups (ITT population randomized to ibrutinib). (A) Analysis by number of prior therapies. (B) Analysis by del(17)(p13.1) and del(11)(q22.3). (C) Analysis by complex karyotype.
PFS by number of prior therapies and cytogenetic subgroups (ITT population randomized to ibrutinib). (A) Analysis by number of prior therapies. (B) Analysis by del(17)(p13.1) and del(11)(q22.3). (C) Analysis by complex karyotype.

PFS by genomic subgroups (ITT population randomized to ibrutinib). (A) Analysis by IGHV. (B) Analysis by NOTCH1. (C) Analysis by TP53. (D) Analysis by SF3B1. (E) Analysis by BIRC3. (F) Analysis by XPO1.
PFS by genomic subgroups (ITT population randomized to ibrutinib). (A) Analysis by IGHV. (B) Analysis by NOTCH1. (C) Analysis by TP53. (D) Analysis by SF3B1. (E) Analysis by BIRC3. (F) Analysis by XPO1.
In a multivariate analysis of PFS with ibrutinib, independent factors associated with decreased PFS included greater number of prior lines of therapy (HR [95% CI] for ≤2 vs >2 prior therapies, 0.53 [0.333-0.840]; P = .007) and elevated B2M (HR [95% CI] for ≤3.5 vs >3.5 mg/L, 0.45 [0.206-0.977]; P = .044).
OS
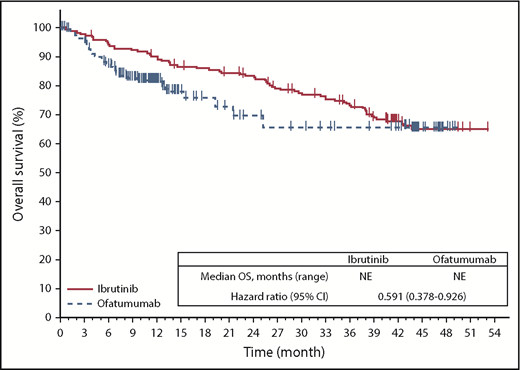
With extended follow-up and despite most patients in the ofatumumab arm (133/196 [68%]) crossing over to ibrutinib as of this updated analysis, OS continues to trend longer for ibrutinib vs ofatumumab (median NR for either arm), with 74% of patients on the ibrutinib arm and 65% on the ofatumumab arm (irrespective of crossover to ibrutinib) alive at 3 years of follow-up after randomization. Censored for crossover, OS was better in patients randomized to ibrutinib (HR, 0.591; 95% CI, 0.378-0.926; P = .0208); Figure 5). A sensitivity analysis of OS adjusting for crossover based on the rank-preserving structural failure time method also showed continued OS benefit with ibrutinib vs ofatumumab (HR, 0.371; 95% CI, 0.223-0.618).
Response and hematologic improvement
The cumulative ORR for ibrutinib was 91%, and the proportion of patients with a best response of complete remission/complete remission with incomplete marrow recovery (9% with current follow-up) increased over time (Figure 6). Among patients with cytopenias at baseline, sustained hematologic improvement in absolute neutrophil count, hemoglobin, and platelet count were noted in 76%, 80%, and 85% of patients treated with ibrutinib, respectively (supplemental Table 3).
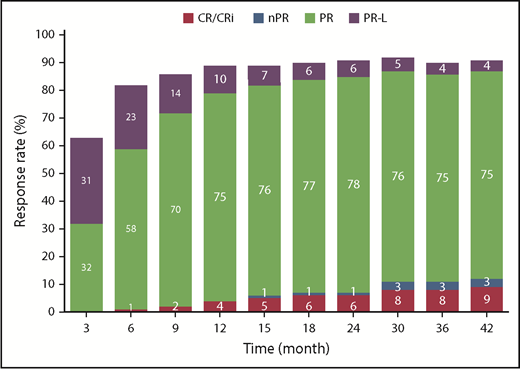
Cumulative best response over time per investigator assessment (ITT population). CR, complete response; nPR, nodular partial response; PR, partial response; PR-L, partial response with lymphocytosis.
Cumulative best response over time per investigator assessment (ITT population). CR, complete response; nPR, nodular partial response; PR, partial response; PR-L, partial response with lymphocytosis.
Patient-reported outcomes
The average (standard deviation) Functional Assessment of Cancer Therapy-Fatigue (FACIT-F) score at baseline was 36.1 (12.3) for ibrutinib and 35.6 (11.9) for ofatumumab, and a greater proportion of patients achieved clinically meaningful improvement in FACIT-F with ibrutinib than with ofatumumab (64% vs 50%).
At baseline, mean (standard deviation) EuroQol 5-Dimensions 5-Level (EQ-5D-5L EuroQol Research Foundation; EQ-5D is a trademark of the EuroQol Research Foundation) Visual Analog Scale scores were 66.4 (21.4) for ibrutinib and 67.6 (19.7) for ofatumumab, and similar proportions (65% vs 45%) of ibrutinib and ofatumumab patients achieved clinically meaningful improvement. Improvements in FACIT-F and EQ-5D-5L Visual Analog Scale scores were largely maintained over time with ibrutinib (supplemental Figure 2A-B).
AEs
The most common AEs of any grade (occurring in ≥20% of patients) were consistent with prior reports with ibrutinib (supplemental Figure 3). The most common grade ≥3 hematologic AEs over the 41-month treatment period included neutropenia (23%), anemia (9%), and thrombocytopenia (8%). The most common grade ≥3 nonhematologic AEs included pneumonia (17%), hypertension (8%), urinary tract infection (6%), atrial fibrillation (6%), and diarrhea (6%). Over the long-term follow-up on the ibrutinib arm, major hemorrhage had occurred in 12 patients (6%). Second primary malignancies included nonmelanoma skin cancers in 29 patients (15%), non-skin cancers in 12 patients (6%), and melanoma in 1 patient (<1%). Figure 7 demonstrates that the prevalence of most reported grade ≥3 AEs of interest decreased over time for patients remaining on treatment. The prevalence of grade ≥3 atrial fibrillation was 4%, 0%, 2%, and 1% over years 0 to 1, 1 to 2, 2 to 3, and 3 to 4, respectively (Figure 7), while atrial fibrillation of any grade was reported in 22 patients (11%) over the course of follow-up. The prevalence of grade ≥3 hypertension was 4%, 9%, 7%, and 6% over years 0 to 1, 1 to 2, 2 to 3, and 3 to 4, respectively. The prevalence of AEs leading to discontinuation generally decreased over time from 11% in year 0 to 1 to 3% in year 3 to 4. The prevalence of AEs leading to dose reduction remained consistent over time (6%, 9%, 4%, and 7% over years 0 to 1, 1 to 2, 2 to 3, and 3 to 4, respectively). Of 26 patients who had dose reductions due to AEs, 6 patients re-escalated ibrutinib for ≥2 cycles (range, 61-272 days) at doses of 280 mg (n = 1) and 420 mg (n = 5); these patients continued ibrutinib for median 931 days after dose reduction, and 4 patients subsequently discontinued treatment. The remaining 20 patients who did not re-escalate dose received ibrutinib for median 686 days after dose reduction, and 11 patients subsequently discontinued.
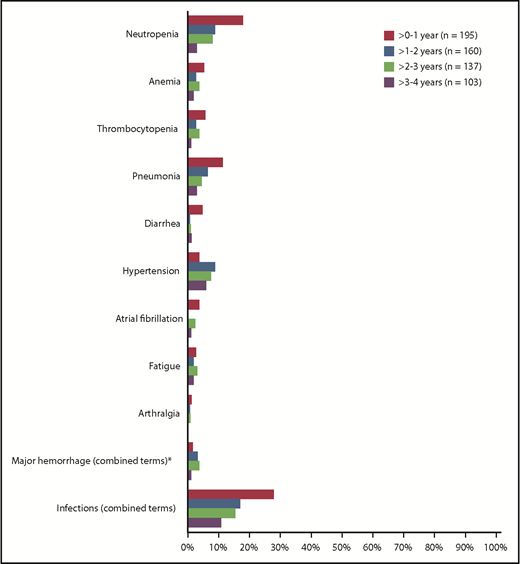
Prevalence of grade ≥3 AEs of clinical interest over time (ITT population). Prevalence was determined by the proportion of patients with a given AE (which can be an existing event or new onset of an event) during each yearly interval. Multiple onsets of the same AE term within a given yearly interval were counted once, and the same AE term continuing across several yearly intervals were counted in each of the intervals. *Defined as any hemorrhagic event grade ≥3 or in severity or that results in one of the following: intraocular bleeding causing vision loss, need for a transfusion of ≥2 U red blood cells or equivalent, hospitalization, or prolongation of hospitalization.
Prevalence of grade ≥3 AEs of clinical interest over time (ITT population). Prevalence was determined by the proportion of patients with a given AE (which can be an existing event or new onset of an event) during each yearly interval. Multiple onsets of the same AE term within a given yearly interval were counted once, and the same AE term continuing across several yearly intervals were counted in each of the intervals. *Defined as any hemorrhagic event grade ≥3 or in severity or that results in one of the following: intraocular bleeding causing vision loss, need for a transfusion of ≥2 U red blood cells or equivalent, hospitalization, or prolongation of hospitalization.
Impact of crossover on OS
Considerable debate existed at trial initiation regarding equipoise of the treatment arms due to durable remissions observed with ibrutinib as part of the initial phase 1 and 2 trials. To understand for future trials how decisions to allow vs prohibit crossover in phase 3 studies influence outcomes, we examined the OS prior to the time at which crossover to ibrutinib treatment was allowed for patients randomized to ofatumumab treatment. Indeed, relative risk of dying trended higher for patients on the ofatumumab arm prior to institution of crossover (HR, 0.426; 95% CI, 0.220-0.823). The reduced risk of dying currently observed for patients randomized to the control arm illustrates the patient benefits of crossover that were subsequently observed in this study.
Discussion
We have described extended follow-up of the randomized phase 3 trial of ibrutinib vs ofatumumab in patients with relapsed CLL/SLL. Primary analysis at median follow-up of 9.7 months demonstrated superiority of ibrutinib over ofatumumab in PFS, OS, and overall response. With extended follow-up of median 44 months, these same results persist; a plateau of PFS has not yet been reached in this long-term follow-up. We also observe very durable remissions among patients of all genomic groups, including those with del(17)(p13.1), del(11)(q22.3), or unmutated IGHV, who are traditionally considered high-risk populations. This contrasts with the inferior outcomes generally observed with chemoimmunotherapy in patients with del(17)(p13.1)/del(11)(q22.3) or unmutated IGHV. A variety of genomic mutations commonly observed in CLL, including BIRC3, SF3B1, NOTCH1, and XPO1, did not affect ibrutinib-conferred PFS, in contrast to the inferior outcomes generally observed with these mutations. Following the pattern observed with most CLL therapies, patients with del(17)(p13.1) or TP53 mutations tended to have a shorter PFS than those without these genomic features, but no significant differences were seen.25-28 While del(17)(p13.1) was identified as an independent prognostic factor for PFS in patients with relapsed/refractory CLL treated with ibrutinib in the PCYC-1102/1103 study, it should be noted that patients in that study had received a greater number of prior regimens and more frequently had complex karyotype than patients in RESONATE.29 In a multivariate analysis examining features associated with shortened PFS in ibrutinib-treated patients in RESONATE, only number of prior therapies (1-2 vs >2) and baseline B2M levels (≤3.5 vs >3.5 mg/L) were predictive. As observed in earlier studies of ibrutinib,19,29 response rates and depth of response have improved over time in RESONATE, with 91% of patients currently responding (9% complete remission/complete remission with incomplete marrow recovery). Although new cases of AEs continue to occur with extended follow-up on ibrutinib, the burden of toxicities (prevalence by yearly intervals), including atrial fibrillation, diarrhea, and infections appeared to plateau or decrease while hypertension was observed more frequently in later years. Discontinuation or dose reductions due to toxicities with ibrutinib were relatively uncommon over the median 41 months of therapy, suggesting that ibrutinib is generally well tolerated over long-term therapy. We acknowledge that patients in this study were relatively young (median age 67 years) and fit (all had ECOG status 0-1), which may not fully represent patients in the real-world setting. Nevertheless, this long-term follow-up study provides evidence that ibrutinib represents a highly efficacious and acceptably tolerated therapy for long-term use in relapsed/refractory patients with CLL.
One aspect of this study that emerged at onset of initial patient enrollment was the clinical equipoise of the 2 treatment arms. Data from a single-arm phase 2 study in heavily pretreated patients with CLL demonstrated remissions in a large proportion of patients treated when this trial was initially planned.18 Given that durability of response to ibrutinib therapy was not known at the time of study conception and ibrutinib was not licensed in any region, a crossover to experimental therapy was not initially included in the study design. During the extended initiation period of this trial, further evidence of durability of response emerged that prompted investigators and patients to advocate for such crossover. In this report, we have provided a retrospective analysis demonstrating a higher risk of death for patients who did not have access to the investigational agent ibrutinib. Future trials in CLL and other cancers where effective therapies are not available may consider phase 3 trial designs that prespecify outcome findings from initial phase 2 studies that would lead to the immediate ability to incorporate crossover to effective therapies. Such design should have prospective agreement by regulatory agencies and sponsors on select surrogate end points acceptable for definitive registration to avoid abrogating the overarching purpose of performing the trial. These considerations would enhance benefit to patients who contribute as control subjects in randomized phase 3 trials.
The evolution of treatment options from intermittent chemoimmunotherapy to include continuous oral administration of targeted therapy raises several important questions related to long-term toxicities. Treatment with chemoimmunotherapy can result in cytopenias and life-threatening infections due to treatment-related myelosuppression.30-32 In previously untreated patients receiving FCR, recurrent late cytopenia episodes occurred in 28% of patients, with actuarial 1- and 6-year incidences of 18% and 23%, respectively, after a median follow-up of 6 years.33 A subset of these cytopenic episodes (8%) were associated with infection. In previously treated patients with CLL, the REACH study of FCR had an 18% frequency of grade 3 to 4 infections; the COMPLEMENT-2 study of ofatumumab, fludarabine, and cyclophosphamide had a 19% frequency of grade ≥3 infections; and the MURANO study of venetoclax and rituximab, although not a chemoimmunotherapy regimen, had a frequency of grade 3 to 4 infections of 18%.34-36 Although direct comparisons cannot be made, collective long-term experience of ibrutinib shows delayed cytopenias are not typically observed and ibrutinib is associated with improved hematologic function in a large majority of patients with baseline cytopenias. Grade 3 infection risk with ibrutinib was similar to that observed with chemoimmunotherapy in the relapsed setting, but with seemingly higher efficacy.37 Additionally, while grade ≥3 atrial fibrillation and major hemorrhage were noted in a minority of patients receiving ibrutinib, incidences were not amplified with extended exposure. The rate of atrial fibrillation in this study (11%) is generally consistent with rates observed in real-world studies of ibrutinib in relapsed/refractory CLL (5% to 12% over follow-up durations of 10-24 months)38-43 and in prior clinical studies (5% to 15% over follow-up of 17-60 months).19,29,44-49 Discontinuation of ibrutinib due to AEs occurred in only 12% of patients over 3.5 years of treatment; although 26 patients (13%) required dose reduction, 11 out of 26 patients were continuing ibrutinib at the time of this analysis, and 6 out of 26 patients re-escalated dose. Ultimately, extended follow-up of randomized trials comparing ibrutinib to chemoimmunotherapy and other targeted agents in previously untreated patients with CLL will be required to adequately compare toxicity, as most relapsed studies reported to date afford only short-term comparative follow-up due to imbalance in efficacy.
After extended follow-up of ibrutinib-treated patients, the most common cause for treatment cessation was PD (n = 53). The most common progression histology was CLL (n = 39); however, 14 patients had transformation to large cell lymphoma (n = 9), Hodgkin disease (n = 3), and prolymphocytic leukemia (n = 2), with many of these patients progressing fairly early (<1 year) after randomization. Survival for study patients in the setting of relapsed disease remained modest and better with immediate vs delayed ibrutinib treatment (after crossover), indicating the need for strategies to prevent and treat ibrutinib-relapsed disease. In this study, we identified decreased PFS among patients receiving ibrutinib beyond second-line therapy compared with those receiving it in second line. In other diseases, such as chronic myeloid leukemia, earlier application of targeted therapy yielded a lower frequency of disease progression.50-52 Studies applying ibrutinib in first-line therapy have observed similar findings.29,44,53 Multiple studies are ongoing to investigate ibrutinib earlier in the course of CLL therapy, including phase 3 studies of first-line ibrutinib (or ibrutinib combined with anti-CD20 therapy) compared with standard chemoimmunotherapy regimens (Alliance 041202 study, NCT01886872; PCYC-1130 study, NCT02264574; ECOG 1912 study, NCT02048813).
In conclusion, this study comprises a large cohort of patients with relapsed CLL/SLL receiving single-agent ibrutinib and provides further evidence for efficacy and safety with prolonged treatment across multiple high-risk genomic and clinical disease features and with increasing depth of response. These findings also support the earlier application of ibrutinib in the treatment of patients with CLL/SLL.
Individual participant data from this clinical study are not available. Pharmacyclics LLC, an AbbVie Company, is currently developing a data sharing plan.
Presented in abstract form at the annual meeting of the American Society of Clinical Oncology, Chicago, IL, 2017.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the patients who participated in this study and their supportive families, as well as the investigators, subinvestigators, and coordinators at each of the study sites. The authors thank Joris Diels of Janssen and Suzy Van Sanden of HEMAR, Janssen-Cilag Ltd (Beerse, Belgium) for their work in generating the crossover-adjusted OS analysis using the rank-preserving structural failure time method. Editorial support was provided by Melanie Sweetlove and was funded by Pharmacyclics LLC, an AbbVie Company.
This study was sponsored by Pharmacyclics LLC, an AbbVie Company, and Janssen Pharmaceuticals. J. C. Byrd is supported by the Four Winds Foundation, D. Warren Brown Foundation, Judy and Michael Thomas, the Sullivan CLL Research Foundation, and the National Institutes of Health, National Cancer Institute (grants R35 CA197734 and R01 CA177292).
Authorship
Contribution: J. C. Byrd, P.H., J.R.B., and D.F.J. designed the study; J. C. Byrd, P.H., S.O., J. C. Barrientos, N.M.R., S.C., C.S.T., S.P.M., U.J., P.M.B., R.R.F., T.J.K., P.T., J.M.P, J.A.B., J.A.W., and J.R.B. collected data; S.D., R.V., and D.F.J. confirmed the accuracy of the data, interpreted the data, and compiled it for analysis; S.D. performed statistical analysis; all authors had access to the data, were involved in the interpretation of data, contributed to the manuscript preparation, and approved the final version of the manuscript for submission; and J. C. Byrd wrote the first draft of the manuscript.
Conflict-of-interest disclosure: J. C. Byrd received research funding and/or honoraria for a consultancy/advisory role from Genentech, Acerta, Verastem, Jazz, Pharmacyclics LLC, an AbbVie Company, and Janssen. P.H. received honoraria and research funding from and reports a consultancy/advisory role for AbbVie, Pharmacyclics LLC, an AbbVie Company, and Janssen and travel expenses from AbbVie and Janssen. S.O. received honoraria from and reports a consultancy/advisory role AbbVie, Janssen, and Pharmacyclics LLC, an AbbVie Company and received research funding from Pharmacyclics LLC, an AbbVie Company. J. C. Barrientos reports a consultancy/advisory role for Pharmacyclics LLC, an AbbVie Company, AbbVie, Gilead, and Janssen and received research funding from Pharmacyclics LLC, an AbbVie Company, AbbVie, and Gilead. N.M.R. reports a consultancy/advisory role for Celgene, AbbVie, Gilead, and BMS and received research funding from BMS. S.C. reports a consultancy/advisory role for Beigene, Abbvie, and Janssen and received research funding from AbbVie, Gilead, Pharmacyclics LLC, an AbbVie Company, Janssen, and Acerta and honoraria from Pharmacyclics LLC, an AbbVie Company, and Janssen. C.S.T. received honoraria from Janssen and Pharmacyclics LLC, an AbbVie Company, and research funding from Janssen. S.P.M. received honoraria from and reports a consultancy/advisory role for Roche, AbbVie, Janssen, Gilead, and GSK; received research funding from Roche, AbbVie, and Janssen; and is a member of the speakers bureau for Roche, AbbVie, Janssen, and Gilead. U.J. received honoraria and travel expenses from and reports a consultancy/advisory role for Gilead, Novartis, and AbbVie. P.M.B. reports a consultancy/advisory role for AbbVie, Celgene, Novartis, Seattle Genetics, Genentech, Verastem, Gilead, and Merck. R.R.F. reports a consultancy/advisory role for AbbVie, Acerta, Genentech, Gilead, Incyte, Loxo Oncology, Janssen, Pharmacyclics LLC, an AbbVie Company, Sunesis, TG Therapeutics, and Verastem and reports another relationship with Incyte DSMB. T.J.K. reports a consultancy/advisory role for AbbVie, Genentech-Roche, Gilead, Celgene, and Pharmacyclics LLC, an AbbVie Company, and receives and research funding from AbbVie, Genentech-Roche, Oncternal, and Pharmacyclics LLC, an AbbVie Company. P.T. received honoraria and travel expenses from Roche and reports a consultancy/advisory role for Janssen and AbbVie. C.M. reports a consultancy/advisory role for Janssen, AbbVie, and Pharmacyclics LLC, an AbbVie Company. M.M. received honoraria from Roche, Gilead, Janssen, and AbbVie; reports a consultancy/advisory role for AbbVie, Gilead, and Janssen; received travel expenses from Gilead and Janssen; is a member of the speakers bureau for AbbVie, Janssen, Gilead, and Roche; and received research funding from Roche. J.M.P. reports a consultancy/advisory role for Pharmacyclics and Gilead. J.A.B. received honoraria and travel expenses from Janssen; reports a consultancy/advisory role for Gilead, Pharmacyclics LLC, an AbbVie Company, and Janssen; and received research funding for Pharmacyclics LLC, an AbbVie Company. S.D. and R.V. are employees of Pharmacyclics LLC, an AbbVie Company, and have stock or other ownership of AbbVie. D.F.J. is an employee of AbbVie and Pharmacyclics LLC, an AbbVie Company (her husband is an employee of AbbVie) and has stock or other ownership with AbbVie (self and husband) and patents, royalties, and other intellectual property with AbbVie. J.R.B. reports a consultancy/advisory role for Janssen, Gilead, Sun Biopharma, AbbVie, Pfizer, AstraZeneca, Astellas, RedX, Pharmacyclics LLC, an AbbVie Company, and TG Therapeutics and received research funding for Gilead and Sun. J.A.W. declares no competing financial interests.
Correspondence: John C. Byrd, B302 Starling-Loving Hall, 320 West 10th Ave, Columbus, OH 43210; e-mail:, john.byrd@osumc.edu.