Key Points
TLT-1 is an independent risk factor for ARDS.
In an ALI model, TLT-1 mediates fibrinogen deposition in the lungs and facilitates platelet-neutrophil release during transmigration.

Abstract
Acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) affect >200 000 individuals yearly with a 40% mortality rate. Although platelets are implicated in the progression of ALI/ARDS, their exact role remains undefined. Triggering receptor expressed in myeloid cells (TREM)–like transcript 1 (TLT-1) is found on platelets, binds fibrinogen, and mediates clot formation. We hypothesized that platelets use TLT-1 to manage the progression of ALI/ARDS. Here we retrospectively measure plasma levels of soluble TLT-1 (sTLT-1) from the ARDS Network clinical trial and show that patients whose sTLT-1 levels were >1200 pg/mL had nearly twice the mortality risk as those with <1200 pg/mL (P < .001). After correcting for confounding factors such as creatinine levels, Acute Physiology And Chronic Health Evaluation III scores, age, platelet counts, and ventilation volume, sTLT-1 remains significant, suggesting that sTLT-1 is an independent prognostic factor (P < .0001). These data point to a role for TLT-1 during the progression of ALI/ARDS. We use a murine lipopolysaccharide-induced ALI model and demonstrate increased alveolar bleeding, aberrant neutrophil transmigration and accumulation associated with decreased fibrinogen deposition, and increased pulmonary tissue damage in the absence of TLT-1. The loss of TLT-1 resulted in an increased proportion of platelet-neutrophil conjugates (43.73 ± 24.75% vs 8.92 ± 2.4% in wild-type mice), which correlated with increased neutrophil death. Infusion of sTLT-1 restores normal fibrinogen deposition and reduces pulmonary hemorrhage by 40% (P ≤ .001) and tissue damage by 25% (P ≤ .001) in vivo. Our findings suggest that TLT-1 uses fibrinogen to govern the transition between inflammation and hemostasis and facilitate controlled leukocyte transmigration during the progression of ARDS.
Introduction
Acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) are inflammatory lung conditions characterized by acute onset and the presence of arterial hypoxia with bilateral pulmonary infiltrates. ALI/ARDS arises as a complication of other primary diagnoses such as sepsis, trauma, pneumonia, aspiration, or multiple transfusions. With an estimated incidence of >200 000 cases annually in the United States, ARDS imposes a substantial burden on the health care system because management requires intensive care and mechanical ventilation.1 During ALI/ARDS progression, inflammatory mediators such as CXCL2 recruit neutrophils to the lungs.2 Neutrophils initiate the epithelial/endothelial damage leading to fluid leakage, neutrophil infiltration, and influx of red blood cells in the alveoli.3 The ensuing inflammatory response mediates the clinical progression from the acute exudative phase to subacute and chronic phases, which may resolve with epithelial and endothelial repair. Unresolved, the condition requires persistent ventilator support, may lead to coagulation or organ failure, and is lethal in ∼40% of the cases.1
The role of platelets in hemostasis is well established, but in recent years a more holistic understanding has emerged of platelets as central effectors of inflammation and tissue repair, as well as coagulation.4 Circumstantial evidence implicates platelets in both the etiology and resolution of ARDS, but the specific mechanisms are not well defined.5 In animal models of ALI, platelets have been shown to exacerbate disease progression through activation of and recruitment of neutrophils to sites of endothelial damage.6 Platelets are an important source of the inflammatory modulators that mediate early stages of ARDS and the anti-inflammatory and surfactant molecules needed for disease resolution.5,7 In a mouse model, it has been shown that necrotic platelets provide membranes that mediate neutrophil macroaggregates.8 In case-control studies, markers of platelet activation and dysregulated coagulation were elevated in Bronchoalveolar lavage (BAL) from patients with ARDS, and multicenter clinical trials have identified correlations between risk of ARDS or ARDS prognosis and thrombocytopenia.9-12 Antiplatelet agents such as aspirin and ketoconozaole had promising results in preclinical models of ALI but have produced mixed results as prophylaxis or treatment of ARDS in clinical settings.5 These studies highlight the fact that there is no clearly defined role for platelets in the progression of ARDS and emphasize an underlying need to better understand how platelets function in ARDS as well as identify reliable platelet biomarkers during ARDS progression.
Our studies of platelet function in sepsis revealed the receptor, triggering receptor expressed on myeloid cells (TREM)–like transcript 1 (TLT-1), as a key mitigating factor during sepsis progression.13 TLT-1 is a type-1 immunoglobulin domain receptor that is stored in the platelet α-granules and, upon platelet activation, translocates to the surface.14 Additionally, some TLT-1, consisting of the extracellular domain from the receptor, is released as a soluble form (sTLT-1) into plasma. Although splice variants, with shortened cytoplasmic tails, and truncated extracellular domains have been identified, sTLT-1 is a platelet-specific product.15-18 Among the most abundant receptors stored in the α-granules, TLT-1 facilitates platelet aggregation and mediates platelet interactions with both endothelial cells and neutrophils.19,20 Fibrinogen is the only known ligand to bind TLT-1 appreciably.13,19
We have demonstrated that patients diagnosed with sepsis have significant increases of sTLT-1 in their blood. In our patients, elevated levels of sTLT-1 strongly correlated with high levels of d-dimers and to disseminated intravascular coagulation (DIC) score, suggesting sTLT-1 could be used as a DIC prognostic factor.13 Subsequent studies using the treml1−/− mouse demonstrated that TLT-1 promotes survival during sepsis.13 We have also shown that sTLT-1 levels are elevated in a small cohort of patients with diagnosed ARDS.21 Based on these observations, we hypothesized that TLT-1 levels may be a prognostic indicator for ARDS and that the protein may play a role in the progression or resolution of ALI/ARDS. To determine whether TLT-1 can be used as a biomarker for ARDS, we retrospectively evaluated the relationships between clinical outcomes and plasma concentrations of sTLT-1 of patients enrolled in the National Heart, Lung, and Blood Institute ARDS Network clinical trials.22,23 Further, we used a mouse model to interrogate the function of TLT-1 in platelet response to lung injury. Our findings indicate that platelets use TLT-1 to minimize tissue damage and that it is indeed an independent prognostic indicator in ARDS.
Materials and methods
For detailed methods, please see supplemental Data (available on the Blood Web site).
Study population
Deidentified samples from patients for this study were obtained from the ARDS Network clinical trial and, after institutional review board approval, were sent to us by the National Institutes of Health biorepository. Further description can be found in supplemental Methods.
Mice
Treml1−/− mice were reported elsewhere.13 All mice were between 8 and 10 weeks of age and weighed 20 to 23 g. Equal numbers of males and females were used. Animal care was provided in accordance with the procedures outlined in Guide for the Care and Use of Laboratory Animals (National Institutes of Health publication no. 85-23, revised 1985). Experimental procedures were approved by the Animal Care and Use Committee.
Acute lung inflammation model
Age- and sex-matched mice were anesthetized intraperitoneally with 100 mg/kg ketamine and 10 mg/kg xylazine and inoculated intranasally with 10 μg Escherichia coli lipopolysaccharide (LPS) (Sigma-Aldrich) or sterile phosphate-buffered saline (PBS) vehicle. BAL was performed by cannulating the trachea with an 18-gauge angiocath. Mice lungs were lavaged 4 times with 0.8 mL cold sterile PBS. Pooled BAL fluid was analyzed by flow cytometry (C6 Accuri, BD Sciences). In parallel experiments, LPS-challenged lungs were dissected and fixed for 24 hours in 4% paraformaldehyde and then placed in 30% sucrose until the tissue sank in the solution. Lung cryosections of 5 µm were stained with propidium iodide (PI) (3 μg/mL) to determine cellular death and damage. Mean fluorescent intensity was analyzed using NIS element viewer 4.20, and total percentage of mean fluorescent intensity per each image was used for quantifications.
Isolation of neutrophils from bone marrow
Femurs and tibias were flushed with 5 mL of Hanks buffered saline solution buffer using a 25G needle. Isolation and bone marrow and subsequent assays are described elsewhere and in supplemental Methods.
Isolation of platelet-rich plasma and washed murine platelets
Platelet-rich plasma was isolated as previously described.13
Flow cytometry
Flow cytometry was completed as previously described.13
Statistics
Descriptive results of continuous variables were expressed as mean ± standard error of the mean. Paired, 2-tailed Student t test analysis available in Prism, version 7.01 (Graph Pad Software) was applied to evaluate statistical differences for 2 groups. P < .05 was considered statistically significant. Detailed description of methods for clinical data are described in supplemental Methods.
TLT-1 copy number
Quantum Simply Cellular anti-mouse immunoglobulin G bead kit (Bangs Laboratories) was used per instructions.
Results
Patient characteristics
The characteristics of the 799 patients included in this analysis have been published22,24 and are shown in supplemental Table 1. The mean age of the patients in our cohort was 52 years, and 42% of the patients were female. The mean Acute Physiology And Chronic Health Evaluation (APACHE) III score was 85. Although the mean platelet count at baseline was within normal range, 57% of the patients in the trial were thrombocytopenic at baseline, and 26% presented with coagulation failure (<80 000 plt/μL). Neither platelet counts nor plasma sTLT-1 concentrations (mean [range], 1831 [0-21 864] pg/mL) varied significantly with age, gender, ventilation volume, drug type, or baseline PaO2/FiO2.
Platelet counts and clinical outcomes
Because one of the goals of our study is to assess the value of TLT-1 as a biomarker for ARDS, we first examined the prognostic value of platelet counts in the ARDS Network trial. Previous studies demonstrated a direct relationship between platelet count and survival in ARDS.10,12 Similar to previous reports, higher platelet counts were associated with improved survival and lower morbidity. Patients who survived had higher baseline platelet counts compared with patients who died (P = .007; Figure 1A). The 28-day survival rate was significantly lower (log rank, P = .003) for patients who presented with coagulation failure (<80 000 plt/μL) compared with patients with platelet counts >80 000 plt/μL at baseline (Figure 1B). Patients presenting with <80 000 plt/μL were also less likely to achieve 48 hours of unassisted breathing than those with >80 000 plt/μL (P = .0027; Figure 1C). Table 1 presents the unadjusted and adjusted risks of mortality and requirement for persistent ventilation for patients presenting with platelet levels <80 000 plt/μL. These patients were 50% more likely to die and were less likely to breath without assistance. Coagulation failure at baseline correlated with higher mean APACHE scores (93 vs 82 for patients with high baseline platelet counts; P < .001), but it lost significance as a predictor of mortality when we controlled for covariates such as age, ventilator volume, creatinine levels, and APACHE score.
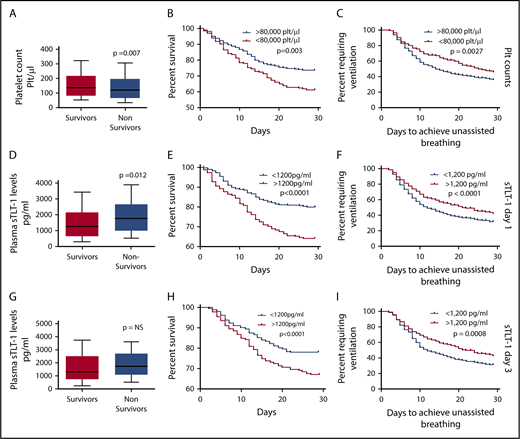
Predictive validation of platelet counts and plasma sTLT-1 in mortality associated with ARDS. (A) Comparison of baseline platelet counts between survivors and nonsurvivors (Student t test, P = .007). Kaplan-Meier survival curves comparing survival (n = 782; log rank, P = .003) (B) and days to achieve unassisted breathing (n = 782; log rank, P = .009) (C) between patients with <80 000 plt/μL and >80 000 plt/μL. Comparison of day 1 (D) and day 3 (G) sTLT-1 levels between survivors and nonsurvivors (day 1: n = 799; Student t test, P = .012; day 3: n = 626; NS, not significant). The whiskers represent the 10th and 90th percentile. Kaplan-Meier survival curves using the day 1 sTLT-1 levels and the 1200 pg/mL cutoff to compare survival (n = 799; log rank, P < .0001) (E) and ventilation-free days (n = 799; log rank, P = .027) (F). Kaplan-Meier survival curves using the day 3 sTLT-1 levels and the 1200 pg/mL cutoff to survival (n = 684; log rank, P < .0001) (H) and ventilation free days (n = 626; log rank, P < .005) (I).
Predictive validation of platelet counts and plasma sTLT-1 in mortality associated with ARDS. (A) Comparison of baseline platelet counts between survivors and nonsurvivors (Student t test, P = .007). Kaplan-Meier survival curves comparing survival (n = 782; log rank, P = .003) (B) and days to achieve unassisted breathing (n = 782; log rank, P = .009) (C) between patients with <80 000 plt/μL and >80 000 plt/μL. Comparison of day 1 (D) and day 3 (G) sTLT-1 levels between survivors and nonsurvivors (day 1: n = 799; Student t test, P = .012; day 3: n = 626; NS, not significant). The whiskers represent the 10th and 90th percentile. Kaplan-Meier survival curves using the day 1 sTLT-1 levels and the 1200 pg/mL cutoff to compare survival (n = 799; log rank, P < .0001) (E) and ventilation-free days (n = 799; log rank, P = .027) (F). Kaplan-Meier survival curves using the day 3 sTLT-1 levels and the 1200 pg/mL cutoff to survival (n = 684; log rank, P < .0001) (H) and ventilation free days (n = 626; log rank, P < .005) (I).
Plasma sTLT-1 concentrations and clinical outcomes
Whereas lower platelet counts were associated with higher risks of mortality, we observed the opposite trend with plasma sTLT-1 concentrations. Plasma concentrations of sTLT-1 measured from samples collected on day 1 were significantly higher (P = .012) in samples of patients who died than in those who survived (Figure 1D). Day 3 sTLT-1 levels were not significantly different between survivors and nonsurvivors (Figure 1G). When stratified by sTLT-1 plasma quartiles, the first quartile (0-783 pg/mL) of day 1 sTLT-1 levels correlated with survival (P < .001; supplemental Table 2). Based on the peak of the frequency distribution of sTLT-1 concentrations among patients, we chose 1200 pg/mL as a single cutoff value to evaluate survival and other clinical outcomes. The 28-day survival time and survival rates are significantly lower for patients with plasma sTLT-1 concentrations exceeding 1200 pg/mL on either day 1 (mean 23.6 days vs 26.8 days) or day 3 (mean 24.7 days vs 26.7 days; P < .0001; Figure 1E,H). Patients with plasma sTLT-1 >1200 pg/mL also had prolonged need for ventilation (log rank, P ≤ .001; Figure 1F,I). Table 1 presents the adjusted risks of mortality and need for persistent ventilation for patients with sTLT-1 levels >1200 pg/mL on days 1 and 3. Statistically significant odds ratios are seen for mortality (P = .001) and persistent ventilation requirement (P < .001). In contrast to coagulation failure, elevated sTLT-1 retained significance as an independent predictor of mortality as well as the need for persistent ventilator support when controlled for age, ventilator volume, creatinine, thrombocytopenia, and APACHE scores (Table 1). Ethnicity, drug type,23 and gender were not found to be significant contributing factors. The ROC suggests that 1200 pg/mL as a single cutoff value is a better predictor of mortality than platelet count or coagulation failure (Table 1).
TLT-1 protects against inflammatory associated hemorrhage in murine ALI model
To gain insights into how platelets and TLT-1 function during the progression of ALI/ARDS, we used LPS to induce ALI in our treml1−/− model. We inoculated wild-type (WT) and treml1−/− mice intranasally with LPS and evaluated the bronchoalveolar lavage fluid (BALF) for bleeding 24 hours postchallenge. Consistent with a role for TLT-1 in ALI, our results demonstrate that LPS treatment led to significant hemorrhage in the BALF of treml1−/− mice compared with WT mice that was confirmed by lung histology (Figure 2A-B). Evaluation of the BALF by flow cytometric analysis demonstrated that treml1−/− mice had 40 times more RBCs than WT mice (2000/µL vs 50/µL, P < .05; Figure 2C). Our results demonstrate that treml1−/− mice fail to maintain hemostasis in ALI where WT mice maintain competency.
![Figure 2. Neutrophil transmigration is delayed in treml1−/− mice. (A) Examples of gross BALF obtained from treml1−/− and WT mice after 24 hours of intranasal PBS or LPS treatment. The reddish BALF in treml1−/− is consistent with increased bleeding. (B) Hematoxylin and eosin histological analysis of the lungs of the WT and treml1−/− mice treated with either PBS or LPS. (C) Flow cytometric based quantification of bleeding in BALF from treml1−/− and WT mice after 24 hours of intranasal PBS or LPS treatment. As shown, BALF from treml1−/− mice had significantly more red blood cells (RBCs) compared with WT (n = 5-7 per group; *P ≤ .05, 1-way analysis of variance [ANOVA]). (D-E) WT and Treml1−/− mice were given LPS or PBS control intranasally. The number of neutrophils in BALF was examined 12 hours (D) and 24 hours (E) after intranasal LPS or PBS treatment (n = 6 mice per group; *P ≤ .05, **P ≤ .001; 1-way ANOVA). All cytometric quantifications were done using BD Accuri C6 software (BD Biosciences).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/132/23/10.1182_blood-2018-03-841593/6/m_blood841593f2.png?Expires=1767816019&Signature=Yl7AmWwvlfP2Ig9voNyqqTwtTAXkS94I-2O5H7dZqKQVUH1aPeOwXP83wwsRZoD-VRMooZHEVOsGeKeVob1Ttdh32UmbZApB0Xy5SAExGYCreAdbut0lsEmy5I3FwM0PRsNWYDlCmjgXFnUMED9r2iArLWBR76luKBYvJ0VpjwSGwxtb7HB4JPpV08bD0n6lwnZzJCye4SZa~rCsiDGnLco4lcevl6pPZofEEtXDsdHm4rL8rwstdcou8o-F9fCn0mlWxiZ1BaIUXK6gCxvx9xt6GQ8pxhSm2JsPDGBrhw9aMd71BkFmLaiao8ANRnGjffQosGKAEVAxhq~Sx3zCxA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Neutrophil transmigration is delayed in treml1−/−mice. (A) Examples of gross BALF obtained from treml1−/− and WT mice after 24 hours of intranasal PBS or LPS treatment. The reddish BALF in treml1−/− is consistent with increased bleeding. (B) Hematoxylin and eosin histological analysis of the lungs of the WT and treml1−/− mice treated with either PBS or LPS. (C) Flow cytometric based quantification of bleeding in BALF from treml1−/− and WT mice after 24 hours of intranasal PBS or LPS treatment. As shown, BALF from treml1−/− mice had significantly more red blood cells (RBCs) compared with WT (n = 5-7 per group; *P ≤ .05, 1-way analysis of variance [ANOVA]). (D-E) WT and Treml1−/− mice were given LPS or PBS control intranasally. The number of neutrophils in BALF was examined 12 hours (D) and 24 hours (E) after intranasal LPS or PBS treatment (n = 6 mice per group; *P ≤ .05, **P ≤ .001; 1-way ANOVA). All cytometric quantifications were done using BD Accuri C6 software (BD Biosciences).
Neutrophil transmigration is delayed in treml1−/−mice. (A) Examples of gross BALF obtained from treml1−/− and WT mice after 24 hours of intranasal PBS or LPS treatment. The reddish BALF in treml1−/− is consistent with increased bleeding. (B) Hematoxylin and eosin histological analysis of the lungs of the WT and treml1−/− mice treated with either PBS or LPS. (C) Flow cytometric based quantification of bleeding in BALF from treml1−/− and WT mice after 24 hours of intranasal PBS or LPS treatment. As shown, BALF from treml1−/− mice had significantly more red blood cells (RBCs) compared with WT (n = 5-7 per group; *P ≤ .05, 1-way analysis of variance [ANOVA]). (D-E) WT and Treml1−/− mice were given LPS or PBS control intranasally. The number of neutrophils in BALF was examined 12 hours (D) and 24 hours (E) after intranasal LPS or PBS treatment (n = 6 mice per group; *P ≤ .05, **P ≤ .001; 1-way ANOVA). All cytometric quantifications were done using BD Accuri C6 software (BD Biosciences).
TLT-1 mediates neutrophil transmigration during inflammation in vivo
Parallel studies in our laboratory using the chemokine CXCL2 (MIP2) to attract neutrophils into the cremaster muscle demonstrate a delay of neutrophil transmigration in treml1−/− mice (supplemental Figure 1). Because CXCL2 and CXCR2 play a significant role in attracting neutrophils to the lungs,2 we investigated if this apparent delay witnessed at the cellular level in the cremaster muscle applies in the lungs. We evaluated neutrophil content in the BALF at 12 and 24 hours after nasal LPS treatment (Figure 2D-E). At 12 hours, the WT mice have on average 56.4% more neutrophils in the BALF than the treml1−/− mice. At 24 hours, the WT mice have resolved the neutrophil influx and exhibit baseline levels of neutrophils. In contrast, the treml1−/− mouse is inundated with neutrophils, demonstrating 25 times more cells than WT, even 3 times more than control mice at 12 hours, supporting the concept that the neutrophils from treml1−/− mice are delayed in transmigration.
In vitro evaluation of TLT-1 effects on neutrophils and neutrophil transmigration
To directly examine TLT-1’s effect on neutrophil transmigration, WT and treml1−/− neutrophils were isolated from the bone marrow and seeded with platelets from WT or treml1−/− mice into the upper chamber of a fibrinogen-coated transwell plate. Without fibrinogen, platelets pass through the membrane freely as single platelets or as conjugates to neutrophils (data not shown). Figure 3A demonstrates that WT and treml1−/− neutrophils incubated with WT platelets migrated equally into the bottom chambers as measured by Gr1-positive events by flow cytometry (Figure 3D-E). WT neutrophils incubated with treml1−/− platelets also migrated, but 43.73 ± 24.75% did so as platelet-leukocyte conjugates compared with only 8.92 ± 2.4% with WT platelets (Figure 3B,D-F). Neutrophils from treml1−/− mice incubated with treml1−/− platelets also migrated as conjugates, and there were significantly fewer cells that migrated (2125 ± 489 vs 4600 ± 684; n = 4/group, P ≤ .001; Figure 3B). As neutrophils have the capability to phagocytize platelets, we examined whether the loss of TLT-1 resulted in altered neutrophil-mediated platelet phagocytosis. There was no difference in platelet phagocytosis between WT and treml1−/− neutrophils, confirming that differences in neutrophil migration were not because of platelet ingestion (supplemental Figure 2). Staining with PI revealed that more treml1−/− neutrophils underwent apparent necrosis in the transmigration process as denoted by a twofold increase in PI staining compared with WT neutrophils (Figure 3C). The twofold difference in cell death raised the possibility that neutrophils may express one of the TLT-1 isoforms. Transcriptome analysis of resting and activated WT, treml1−/−, or human neutrophils did not reveal any treml1−/− derived transcripts (supplemental Figure 3). From these studies, we can conclude that platelet-derived TLT-1 is necessary for platelet modulation of neutrophil transmigration and longevity during inflammation.

TLT-1 promotes neutrophil release from platelets during transmigration. (A-B) In transwell plates, WT or treml1−/− neutrophils were exposed to resting or thrombin-activated (0.05 U/mL) WT platelets (A) or treml1−/− platelets (B), and transmigration of neutrophils was measured by flow cytometry. Shown are representative flow cytometry plots. Neutrophils were measured by Gr1fitc (y-axis), and platelets were identified by Gp1b Alexa 647 (x-axis). (C) Measurement of neutrophil cell death plotted as the number of neutrophils (gated on the red boxes in panels A and B) that stained positive for the cell impermeable PI dye (n = 3; **P ≤ .001). Quantification of the transmigrated neutrophils (D-E) and platelet-neutrophil conjugates (F-G) from panels A and B. (H) Flow cytometric analysis of WT neutrophils incubated with WT platelets in the presence of nonimmune antiserum (i) or anti-TLT-1 (ii). (I) Analysis of treml1−/− neutrophils incubated with treml1−/− platelets in the presence of supernatant from mock transfected cells (i) or supernatant from sTLT-1 transfected cells (ii, n = 4). Quantifications of transmigration from panels H and I of transmigrated WT (J) or treml1−/− (K) neutrophils. Quantifications of platelet-neutrophil aggregates from panels H and I from WT (L) or treml1−/− cells (M). *P ≤ .05, **P ≤ .01; Student t test. All cytometric quantifications were done using BD Accuri C6 software (BD Biosciences).
TLT-1 promotes neutrophil release from platelets during transmigration. (A-B) In transwell plates, WT or treml1−/− neutrophils were exposed to resting or thrombin-activated (0.05 U/mL) WT platelets (A) or treml1−/− platelets (B), and transmigration of neutrophils was measured by flow cytometry. Shown are representative flow cytometry plots. Neutrophils were measured by Gr1fitc (y-axis), and platelets were identified by Gp1b Alexa 647 (x-axis). (C) Measurement of neutrophil cell death plotted as the number of neutrophils (gated on the red boxes in panels A and B) that stained positive for the cell impermeable PI dye (n = 3; **P ≤ .001). Quantification of the transmigrated neutrophils (D-E) and platelet-neutrophil conjugates (F-G) from panels A and B. (H) Flow cytometric analysis of WT neutrophils incubated with WT platelets in the presence of nonimmune antiserum (i) or anti-TLT-1 (ii). (I) Analysis of treml1−/− neutrophils incubated with treml1−/− platelets in the presence of supernatant from mock transfected cells (i) or supernatant from sTLT-1 transfected cells (ii, n = 4). Quantifications of transmigration from panels H and I of transmigrated WT (J) or treml1−/− (K) neutrophils. Quantifications of platelet-neutrophil aggregates from panels H and I from WT (L) or treml1−/− cells (M). *P ≤ .05, **P ≤ .01; Student t test. All cytometric quantifications were done using BD Accuri C6 software (BD Biosciences).
To evaluate the possible therapeutic potential of TLT-1, we used voldemort, a polyclonal anti-TLT-1 antibody with reactivity to amino acids 122 to 139 of TLT-1 produced in our laboratory.25 When we inhibited TLT-1 by adding anti-TLT-1 polyclonal antibodies to the transmigration assays, there was a 3.2 times reduction in transmigration (P = .035) and an associated 47% increase in the abundance of platelet-neutrophil conjugates over assays with control antibodies, supporting a concept that TLT-1 is necessary for platelet release of the neutrophil (P = .0065, n = 4; Figure 3H,J,L). Conversely, the addition of sTLT-1 reduced the platelet-neutrophil conjugate phenotype of treml1−/− neutrophils by 2.8 times (P = .0035) and increased the proportion of neutrophils that transmigrated as single units by 54.6% (P = .046; Figure 3I,K,M). We subsequently asked if sTLT-1 was affecting protease release. The addition of sTLT-1 to neutrophil protease release assays (supplemental Figure 4) demonstrated a relatively small effect on elastase and myeloperoxidase and no impact on cathepsin G release. These results suggest that sTLT-1’s effects on neutrophil transmigration were not because of a direct effect on neutrophils but a secondary consequence of TLT-1 effects on platelets and/or the immediate environment. It seems that platelets are retained on the fibrinogen matrix when TLT-1 is present, releasing neutrophils for transmigration, whereas relative adherence to neutrophils is enhanced and prolonged when TLT-1 is not present.
In vivo modulation of platelet function with exogenous sTLT-1
Next, we examined if we could affect platelet function in vivo with exogenous sTLT-1. Soluble TLT-1 injected into treml1−/− mice is detectable by enzyme-linked immunosorbent assay for 2 days in the serum of null mice (data not shown). To evaluate if sTLT-1 is taken up by platelets and presented at the site of inflammation, we evaluated TLT-1 content in treml1−/− platelets. After injection of sTLT-1, it appears that the sTLT-1 is brought in to the α-granules before translocating to the platelet surface. It seems that at 3 hours more of the sTLT-1 colocalizes with P-selectin than the surface receptor Gp1b (supplemental Figure 5). Immunohistochemistry at 1, 3, and 6 hours postinjection revealed that the sTLT-1 is taken up by the platelets and found on the platelet surface, with levels peaking at 3 hours postinjection (Figure 4A-B). Twenty-four hours after injection, sTLT-1 can be found along the linings of lungs, similar to what is seen in WT mice (Figure 4C). Subsequently, we tested if sTLT-1 could rescue the bleeding phenotype. The addition of sTLT-1 after LPS challenge significantly decreased tissue damage by 25% (P ≤ .001) and reduced RBC influx by 40% (P ≤ .001) in sTLT-1-treated treml1−/− mice compared with nontreated mice (Figures 4D-H). Taken together, these data suggest that platelets not only use TLT-1 to regulate lung damage, but also can resorb and redistribute exogenously administered TLT-1 to sites of disuse.
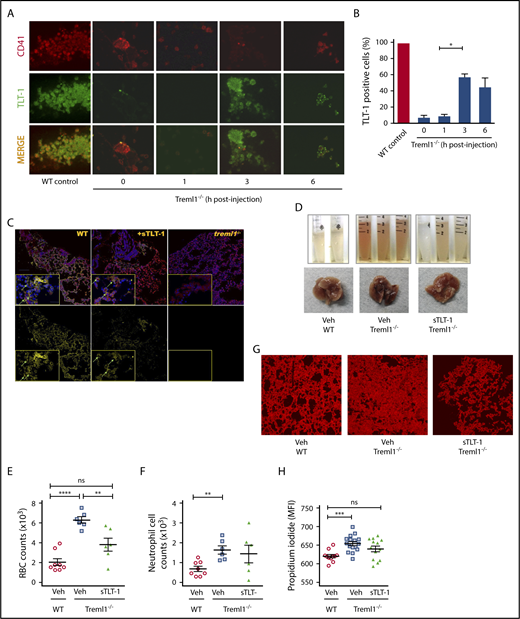
Soluble TLT-1 partially rescues bleeding and tissue damage in ALI model. (A) LPS-treated treml1−/− mice were injected IV with smTLT-1, and platelets were evaluated before injection (time 0), 1, 3, and 6 hours postinjection. Platelets were stained with voldemort and anti-rabbit-fitc secondary, and anti GPIIα IIIα-PE. (B) Quantification of the percentage of TLT-1 positive cells relative to WT (gray column). (C) Immunofluorescent staining of lungs from WT, treml1−/−, or treml1−/− mice treated with sTLT-1, 24 hours after LPS inhalation. CD41, red; TLT-1, yellow; 4′,6-diamidino-2-phenylindole, blue. (D-H) Treml1−/− mice were pretreated with either sTLT-1 or vehicle and then subjected to intranasal LPS challenge and evaluated at 24 hours. (D) Representative images of gross BALF and whole lungs, illustrating reduced bleeding in treml1−/− mice pretreated with sTLT-1. Pretreating treml1−/− mice with sTLT-1 reduced RBC counts (E), neutrophil influx into BALF (F), and PI staining of mouse lungs (G and H). Scale bar represents 100 μM (n = 5-6 mice per group; *P ≤ .05, **P ≤ .01, ***P ≤ .001, ****P ≤ .0001, 1-way ANOVA). Confocal images were processed using Nikon Confocal Microscope and analyzed using NIS element viewer 4.20, magnification ×20. All cytometric quantifications were done using BD Accuri C6 software (BD Biosciences). MFI, mean fluorescent intensity.
Soluble TLT-1 partially rescues bleeding and tissue damage in ALI model. (A) LPS-treated treml1−/− mice were injected IV with smTLT-1, and platelets were evaluated before injection (time 0), 1, 3, and 6 hours postinjection. Platelets were stained with voldemort and anti-rabbit-fitc secondary, and anti GPIIα IIIα-PE. (B) Quantification of the percentage of TLT-1 positive cells relative to WT (gray column). (C) Immunofluorescent staining of lungs from WT, treml1−/−, or treml1−/− mice treated with sTLT-1, 24 hours after LPS inhalation. CD41, red; TLT-1, yellow; 4′,6-diamidino-2-phenylindole, blue. (D-H) Treml1−/− mice were pretreated with either sTLT-1 or vehicle and then subjected to intranasal LPS challenge and evaluated at 24 hours. (D) Representative images of gross BALF and whole lungs, illustrating reduced bleeding in treml1−/− mice pretreated with sTLT-1. Pretreating treml1−/− mice with sTLT-1 reduced RBC counts (E), neutrophil influx into BALF (F), and PI staining of mouse lungs (G and H). Scale bar represents 100 μM (n = 5-6 mice per group; *P ≤ .05, **P ≤ .01, ***P ≤ .001, ****P ≤ .0001, 1-way ANOVA). Confocal images were processed using Nikon Confocal Microscope and analyzed using NIS element viewer 4.20, magnification ×20. All cytometric quantifications were done using BD Accuri C6 software (BD Biosciences). MFI, mean fluorescent intensity.
TLT-1 and fibrin(ogen) interaction
We have previously demonstrated that TLT-1 binds fibrinogen.13 To quantify the avidity of the TLT-1/fibrinogen interaction, we measured the TLT-1 copy number on resting and activated platelets from normal human volunteers. We measured TLT-1 surface expression at baseline and after activation with concentrations of 5 and 20 μM adenosine 5′-diphosphate (ADP). We show a mean of 2089 copies at baseline, 48 835 copies with 5 μM, and 55 778 copies with 20 μM ADP (n = 3; Figure 5A-B). Some of the volunteers displayed a reduction of surface TLT-1 at 20 μM, which is consistent with receptor shedding. Next, we measured TLT-1’s affinity to fibrinogen using a plate bound assay. We demonstrate a concentration-dependent binding of sTLT-1 to fibrinogen. Our results estimate a Kd of 53 nM (Figure 5C). Taken together, the expression of TLT-1 on the surface of cells would strengthen platelet interaction with fibrinogen by increasing both affinity and avidity of platelets for fibrinogen. To test if there were differences in fibrin(ogen) deposition in our mice, we treated treml1−/− mice with exogenous sTLT-1 and compared them with mock-treated treml1−/− or WT mice. We harvested lungs from these mice 24 hours after nasal LPS treatment and stained for fibrinogen. Figure 5D demonstrates that the treml1−/− mice have a significant 29% (P ≤ .001) decrease in fibrinogen deposition compared with WT mice. Introduction of sTLT-1 reestablishes fibrinogen accretion in the treml1−/− mouse (P ≤ .01), clearly demonstrating a role for TLT-1/sTLT-1 in regulation of fibrinogen deposition in the lungs.

TLT-1 copy number and interaction with fibrinogen. (A) Measurement of TLT-1 copy number on resting and ADP-activated platelets (n = 3). (B) Quantification of panel A. (C) Plate bound binding curve of sTLT-1 interaction with fibrinogen predicting a Kd of 53 nM. (D) Confocal images of fibrinogen staining in the lung 24 hours after intranasal LPS treatment. Lungs from WT, treml1−/−, or treml1−/− mice treated with sTLT-1 were stained with fibrinogen (red) and 4′,6-diamidino-2-phenylindole (blue), demonstrating that sTLT-1 increases fibrinogen deposition in the lung. Side panel: quantification of the staining on the left (n = 5 mice per group, 5 slides per lung; **P ≤ .01, ***P ≤ .001). The red line equals 100 μM.
TLT-1 copy number and interaction with fibrinogen. (A) Measurement of TLT-1 copy number on resting and ADP-activated platelets (n = 3). (B) Quantification of panel A. (C) Plate bound binding curve of sTLT-1 interaction with fibrinogen predicting a Kd of 53 nM. (D) Confocal images of fibrinogen staining in the lung 24 hours after intranasal LPS treatment. Lungs from WT, treml1−/−, or treml1−/− mice treated with sTLT-1 were stained with fibrinogen (red) and 4′,6-diamidino-2-phenylindole (blue), demonstrating that sTLT-1 increases fibrinogen deposition in the lung. Side panel: quantification of the staining on the left (n = 5 mice per group, 5 slides per lung; **P ≤ .01, ***P ≤ .001). The red line equals 100 μM.
Discussion
Although platelets are assumed to play a causative role in both ARDS progression and resolution, most of those assumptions are based on the known roles of platelets as mediators of coagulation, inflammation, and tissue remodeling. Animal models have produced some indirect evidence of platelet activity in ARDS; however, supporting data in both human and animal models are lacking. To our knowledge, this is the first detailed study that defines a platelet-specific ARDS biomarker as an independent prognostic factor in ARDS. We examined the relationship between platelet counts and clinical outcome in this group of patients and found an inverse correlation between survival and platelet counts, consistent with reports in other populations.10,12 We subsequently demonstrate that subjects with total sTLT-1 levels >1200 pg/mL have a 91% increased risk of mortality compared with subjects with levels <1200 pg/mL (P < .05).
Prior clinical studies in ALI/ARDS have focused exclusively on either platelet counts or biomarkers of endothelial damage, epithelial damage, or inflammation. Here, we demonstrate that both very low baseline platelet counts (<80 000/µL) and elevated plasma sTLT-1 concentrations (>1200 pg/mL) are predictors of mortality and morbidity. Only sTLT-1 retains significance when we control for other covariates, suggesting that the contents of α-granules released either through platelet activation or platelet destruction may mediate clinical sequalae that lead to organ failure, permanent lung damage, or death. Recent work by Yuan et al demonstrates necrotic platelets are subject to having their membranes removed by neutrophils. The membranes subsequently facilitate neutrophil macroaggregate formation and occlusion of medium-sized vessels in the lung.8 Platelet destruction of this magnitude could account for the higher levels of sTLT-1 seen in patients that do not survive. Although these studies demonstrate that TLT-1 is a platelet specific prognostic factor in ARDS, they also suggest that TLT-1 plays a role in the underlying mechanisms of disease progression, prompting our further investigation.
The restoration of the TLT-1 function by the soluble fragment may seem contradictory to the ARDS data, where high levels are associated with mortality. However, in our mouse model we are rescuing the null phenotype, which strengthens our argument for TLT-1 regulation of fibrinogen deposition and defines a role for sTLT-1. Interestingly, we have cloned a soluble TLT-1 spice variant and have identified using RNA sequencing the transcript that represents this soluble form of TLT-1. It is predicted to represent ∼5% of the transcript present in human platelets (supplemental Figure 6; supplemental Table 3) and further supports the idea of an important role of the soluble form of TLT-1. The reduction of TLT-1 surface expression (Figure 5A) is suggestive of receptor shedding and consistent with the work of Fong et al.26 Because only ∼5% of the TLT-1 transcript is of the soluble form, it more likely that the larger part of what we measure is from cleaved surface receptor. We are, however unable to comment at this time if the cleaved form and the secreted form have the same function.
TLT-1 binds fibrinogen, and we have previously demonstrated that TLT-1 increases fibrinogen deposition and platelet adherence to the endothelium in in vitro assays.13,19 Here, we demonstrate in an in vivo model that TLT-1 is important for fibrinogen deposition and reduction of bleeding in the lung. Moreover, we demonstrate that introduction of sTLT-1 restores fibrinogen deposition and significantly reduces bleeding and tissue damage in an ALI model. As such, our findings not only demonstrate a role for TLT-1 in the progression of ALI/ARDS, but also implicate fibrinogen. Current literature supports a role for fibrinogen in neutrophil trafficking. Several studies have implicated fibrinogen deposition on the endothelium as a neutrophil-honing signal during inflammation27-29 and demonstrated that neutrophil transmigration and neutrophil-mediated clearance of pathogens are mediated by fibrinogen.30-32 TLT-1 interaction with fibrinogen may regulate immune-derived bleeding, a role thought not to be played by the integrin αIIbβ3’s interaction with fibrinogen.33
Our data demonstrate that exogenously administered TLT-1 is absorbed and released, making platelets a potential cache for sTLT-1. This finding, together with the clinical trends noted with the ARDS cohorts21 and sepsis patients,13 suggests that when plasma TLT-1 is high, the cache capacity of platelets is exhausted. We posit that under these conditions, fibrin(ogen)/TLT-1 interaction is no longer confined to sites of inflammation giving greater access to plasmin, leading to classical DIC, consumption of coagulation factors, and mortality. Accordingly, in treml1−/− mice where TLT-1 is not available to regulate fibrin(ogen) deposition, we see higher d-dimer levels during sepsis compared with WT mice.13
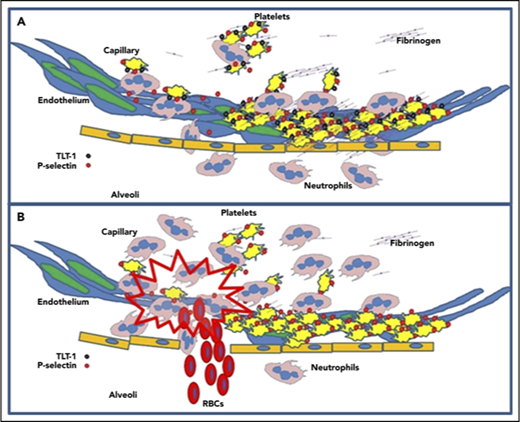
Neutrophil infiltration into lung interstitia is a hallmark of ARDS, and formation of platelet neutrophil conjugates has been shown to be essential in both progression of ARDS and in the resolution of tissue damage.6 When platelets are activated P-selectin is expressed. P-selectin interacting with PSGL-1 initializes34 adhesion between platelets and neutrophils, which is strengthened by the interaction between GP1b and the integrin, αmβ2,35 during acute inflammation. Neutrophils subsequently extravasate into the tissues; platelets on the other hand, are rarely seen in the tissues or extracellular matrix. Logic suggests that a mechanism exists to break the tethered platelet-neutrophil interaction. One interpretation of the data presented here is that TLT-1 mediation of fibrinogen deposition facilitates neutrophil honing for transmigration27 and subsequent platelet release from neutrophils resulting in vessel protection and reduced tissue damage. In the transmigration assays, without fibrinogen in the wells, platelets pass through as single units as well as platelet neutrophil conjugates (data not shown). With the addition of fibrinogen (Figure 3A), only the neutrophils pass to the bottom chamber, suggesting that platelet interaction with fibrinogen modulates platelet-neutrophil interactions.
Our work extends our findings on TLT-1’s role on clot formation and sepsis.13 This is the first in vivo demonstration of TLT-1 regulation of fibrinogen binding. We suggest that TLT-1 interaction with fibrinogen is responsible for control of bronchoalveolar hemorrhage, as well as platelet modulation of neutrophil transmigration, and that elevated levels of TLT-1 in inflammatory conditions are biomarkers of dysregulation of these processes.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Diana Lim for her arduous work with the figures.
This work was supported by grants from the National Heart, Lung, and Blood Institute (R01HL090933, HL126547, F31HL136183, and HL112311), the National Institute of General Medical Sciences (IDeA Networks of Biomedical Research Excellence grant P20GM103475), and the National Institute of Minority Health and Health Disparities (grants 2U54MD007587 and 8G12MD007583), National Institutes of Health.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Authorship
Contribution: J.M.-O. and F.R. were responsible for cremaster studies; V.D., N.L., C.C., R.H., G.M.-M., and A.G. were responsible for ARDS data analysis; J.M.-O. was responsible for the ALI model, transmigration assays, mouse husbandry; J.M.-O. and A.P. were responsible for the mouse neutrophil studies; A.V.W. was responsible for the whole blood flow studies; A.S.J., J.C., and J.L. were responsible for the TLT-1 copy number study; A.G. was responsible for the fibrinogen studies; J.M.-O., F.S., and H.O.-Z. were responsible for the mouse transcriptome studies; J.W.R. was responsible for the identification of the splice variants in human populations; M.T.R. and C.C.Y. were responsible for the human transcriptome studies; J.M.-O., F.R., and B.M. were responsible for the confocal studies; J.M.-O., A.G., M.T.R., and A.V.W. were responsible for writing the manuscript; and A.V.W. was responsible for the overall direction of the project.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: A. Valance Washington, University of Puerto Rico-Rio Piedras, Departamento de Biologia, 17 Ave Universidad Ste 1701, San Juan, Puerto Rico 00925-2537; e-mail: anthony.washington@upr.edu.