Key Points
Cooperative PSGL-1 and CXCR2 signaling in neutrophils increases adhesion and NET release in flow-restricted veins.
Cooperative PSGL-1 and CXCR2 signaling in neutrophils, but not monocytes, promotes DVT.
Inflammation is a major contributor to deep vein thrombosis (DVT). Flow restriction of the inferior vena cava (IVC) in mice induces DVT like that in humans. In this model, P-selectin–dependent adhesion of neutrophils and monocytes leads to release of neutrophil extracellular traps (NETs) and expression of tissue factor. However, it is not known what signals cause myeloid cells to generate these procoagulant effectors. Using ultrasonography and spinning-disk intravital microscopy in genetically engineered mice, we found that engagement of P-selectin glycoprotein ligand-1 (PSGL-1) and the chemokine receptor CXCR2 on rolling neutrophils propagated signals that cooperated to induce β2 integrin–dependent arrest in flow-restricted IVCs. Unlike previous reports, PSGL-1 signaling in neutrophils did not require L-selectin, and it used tyrosine 145 rather than tyrosines 112 and 128 on the adaptor Src homology domain-containing leukocyte phosphoprotein of 76 kDa. PSGL-1 and CXCR2 signaling cooperated to increase the frequency and size of thrombi, in part by stimulating release of NETs. Unlike in neutrophils, blocking PSGL-1 or CXCR2 signaling in monocytes did not affect their recruitment into thrombi or their expression of tissue factor. Our results demonstrate that neutrophils cooperatively signal through PSGL-1 and CXCR2 to promote DVT.

Introduction
Deep vein thrombosis (DVT) and pulmonary embolism, together defined as venous thromboembolism, are major causes of cardiovascular morbidity and mortality.1,2 Reduced venous blood flow without endothelial injury typically precedes DVT.3,4 This frequently occurs in immobilized medical and surgical patients, in pregnant women, and in passengers on long-distance flights. Anticoagulants such as heparin or warfarin are used to prevent or treat DVT. However, they are associated with bleeding, particularly in high-risk surgical patients, and they do not reduce postthrombotic sequelae such as leg swelling and skin ulcerations.5,6 Therefore, drugs that target venous thrombosis without impairing physiological hemostasis are needed.
Inflammation is a major contributor to DVT.4,5 Human venous thrombi contain numerous leukocytes in addition to fibrin, red blood cells, and some platelets. During the latter stages of DVT, inflammatory cells enter the venous wall where they may accelerate fibrosis. Flow restriction of the inferior vena cava (IVC) in mice induces thrombosis that resembles DVT in humans.7,-9 In this model, myeloid cells accumulate on the endothelial cell surface and interact with platelets.8 Neutrophils release DNA- and histone-rich neutrophil extracellular traps (NETs).8,9 Monocytes and, to a lesser degree, neutrophils express tissue factor.8 Both NETs and tissue factor activate thrombin and trigger fibrin formation. However, it is not known what signals cause myeloid cells to inappropriately release these effectors in the vein lumen.
In physiological inflammation, neutrophils roll in venules, arrest, and migrate through endothelial cell junctions into perivascular tissues.10 Signaling events regulate this multistep adhesion cascade.11,-13 Neutrophils rolling on P- or E-selectin on activated endothelial cells transduce signals through P-selectin glycoprotein ligand-1 (PSGL-1).14,,,,,-20 These signals convert integrin αLβ2 from a bent, low-affinity conformation to an extended, intermediate-affinity conformation, which interacts with ICAM-1 to reduce rolling velocities.19 Chemokines such as CXCL1 on endothelial cells engage the G protein–coupled receptor CXCR2 on neutrophils,21,-23 which converts integrin αLβ2 to an extended, high-affinity conformation that causes arrest. Activated integrin αMβ2 on neutrophils mediates intraluminal and perivascular crawling. Migrating neutrophils do not release effectors such as NETs, proteases, and reactive oxygen species until they encounter pathogens or injured tissues. In vitro or with adoptive neutrophil transfers in vivo, selectin signals cooperate with limiting chemokine signals to activate β2 integrins; at higher chemokine levels, integrin activation does not require selectin signals.24 Selectins and chemokines also reportedly cooperate to mobilize endogenous neutrophils to inflamed tissues in vivo.15 In these experiments, however, inhibition of chemokine signaling was likely incomplete, and interventions used to block selectin signaling would also impair integrin outside-in signaling.24 When engaged, both PSGL-1 and β2 integrins trigger a signaling cascade that activates Src family kinases (SFKs), proteins with immunoreceptor tyrosine-activation motifs (ITAMs), spleen tyrosine kinase (Syk), the adaptor Src homology domain-containing leukocyte phosphoprotein of 76 kDa (SLP-76), Tec kinases, p38 MAPK, and other mediators.11,13,25 Thus, it remains unclear whether selectin and chemokine signals cooperate to mobilize endogenous neutrophils in vivo. Whether these signals trigger dysregulated release of NETs or other effectors in inflammatory disorders such as DVT is also unknown.
In this study, we examined whether selectin and/or chemokine signals in neutrophils and monocytes promote DVT in mice. We found that cooperative signaling limited to neutrophils can propagate venous thrombosis.
Methods
Detailed information on reagents and protocols are provided in supplemental Methods (available on the Blood Web site).
Mice
All mice were backcrossed with C57BL/6J mice >10 times. C57BL/6J, Cxcr2−/−, and Itgb2−/− mice were from The Jackson Laboratory. Selplg−/− mice were generated as described.26 Sell−/− mice were made as described,27 with retention of the loxP-flanked neostop cassette to prevent expression of L-selectin. Lcp2f/fLysMCre−, Lcp2f/f LysMCre+, Lcp2Y112/128F, and Lcp2Y145F mice28,29 were gifts from Gary Koretzky.
All mouse protocols were approved by the institutional animal care and use committee of the Oklahoma Medical Research Foundation.
Cells
Flow cytometry
Flow cytometry was performed as described.27
Flow chamber assay
Western blot
Bone marrow leukocytes (1 × 107) were incubated in 6-well plates with P-selectin–immunoglobulin M (IgM), E-selectin–IgM, or control CD45-IgM captured on immobilized anti-human IgM Fc antibody (Ab) on a rotary shaker at 65 rpm for 10 minutes at room temperature. Isolated bone marrow neutrophils (5 × 106) were incubated in plates immobilized with 50 μg/mL F(ab′)2 fragments of control rat IgG, GAME-46, 4RA10, or MEL-14 for 10 minutes.34 Some cells were preincubated with 10 mM methyl-β-cyclodextrin (Sigma-Aldrich) or its inactive analog α-cyclodextrin for 30 minutes at 37°C. After incubation, the cells were lysed and analyzed by western blotting.35
Western blotting of thrombi was performed as described.27
Measurement of NETs
NETs were measured as described.36
DVT
DVT induced by flow restriction of the mouse IVC was performed as described,7,8,27 with minor modifications. At various times after surgery, ultrasonography was performed with a Vevo 2100 system with a 40-MHz mouse scan head (VisualSonics) to confirm flow restriction and to monitor thrombus progression.
In some mice, spinning-disk intravital microscopy of the IVC was performed as described.7 Fluoresbrite Green microspheres (0.5-μm diameter; PolySciences, Inc) were coated with control or anti-CXCL1 monoclonal Ab (mAb).31 One hour before microscopy, 1010 microspheres and 4 μg of phycoerythrin (PE)-conjugated anti-Ly6G mAb or 4 μg of PE-conjugated anti-mouse macrophage colony-stimulating factor receptor (M-CSFR; CD115) mAb in 200 μL of saline were injected through the retro-orbital venous plexus. The Fluoresbrite Green microspheres and PE-labeled Ly6G+ neutrophils or M-CSFR+ monocytes in the IVC were observed 1 mm below the ligation in the experimental group or 1 to 2 mm below the left renal vein in sham-surgery control mice. Rolling cells were defined as cells moving in the direction of the blood flow at slower velocity in the focal plane than free-flowing cells (100-150 m/s). Adherent cells or microspheres were defined as those that remained stationary for at least 30 seconds.
In other experiments, 50 μg of mAb RB40.34, 4RB12, 4RA10, 9A9, or GAME-46 per mouse was injected IV through the retroorbital plexus 1 hour before and 12 hours after the ligation. A total of 1 mg/kg SB225002, dissolved in dimethyl sulfoxide (DMSO) and diluted in saline, was injected intraperitoneally 1 hour before and 12 hours after the ligation.37
The incidence and size of thrombi were monitored by ultrasonography. After 24 hours, mice were killed, and the IVC was excised below the ligation and proximal to the confluence of the common iliac vein. Thrombus formed in the IVC was removed for measurements of weight and length, flow cytometry, and western blots.
Statistical analysis
Statistical differences were analyzed with the unpaired and 2-sided tail Student t test or with 1-way analysis of variance with the post hoc multiple-comparison test. Values were considered significant at P < .05.
Results
PSGL-1 and β2 integrins mediate neutrophil rolling and arrest in flow-restricted IVCs
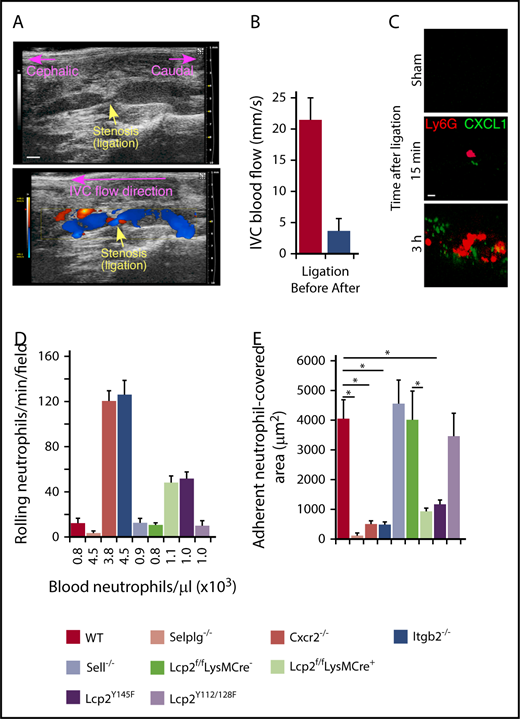
We used a mouse model of DVT to explore the contributions of selectin and chemokine signaling in neutrophils and monocytes.7,8,27 A suture introduced around the IVC reduced blood flow by ∼90%, causing a thrombus to develop upstream of the stenosis in ∼50% of vessels. In this model, P-selectin is mobilized to the endothelial surface and initiates adhesion of neutrophils and monocytes.8,38 We used ultrasound to confirm flow restriction (Figure 1A-B). Spinning-disk intravital microscopy of wild-type (WT) mice documented accumulation of CXCL1 (labeled with anti-CXCL1–coated beads) and neutrophils (labeled with anti-Ly6G) on endothelial cells within 15 minutes after flow restriction (Figure 1C). After 3 hours, we still observed rolling neutrophils (Figure 1D), but most of the endothelial cell surface was covered with firmly adherent cells (Figure 1E). We next examined mice lacking PSGL-1 (Selplg−/−), β2 integrins (Itgb2−/−), or CXCR2 (Cxcr2−/−). These 3 genotypes exhibited an approximately fourfold increase in circulating neutrophils (Figure 1D; supplemental Table 1), consistent with earlier reports.24,26,33 As previously shown in P-selectin–deficient (Selp−/−) mice,8 virtually no neutrophils rolled or arrested in Selplg−/− mice, confirming that P-selectin/PSGL-1 interactions initiate rolling, a prerequisite for arrest (Figure 1D-E). In Itgb2−/− and Cxcr2−/− mice, many neutrophils rolled but few arrested, despite the elevated circulating neutrophils (Figure 1D-E). Thus, rolling neutrophils require chemokine activation of β2 integrins to arrest.
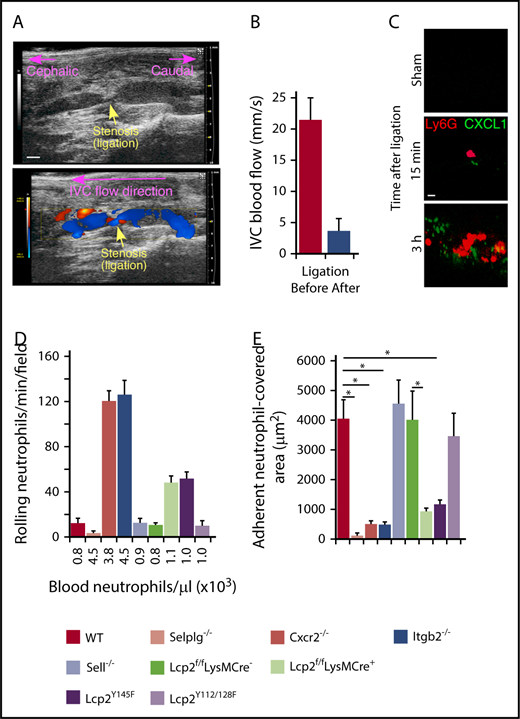
Cooperative PSGL-1 and CXCR2 signaling in neutrophils enhances adhesion in flow-restricted IVC. (A) Top, Sagittal view of B-mode ultrasonography confirms stenosis of the IVC after ligation. The cephalic and caudal directions are marked. Bar, 1 mm. Bottom, Color Doppler mode in B-mode sagittal view reveals the flow direction in the IVC around the stenosis. The blue color indicates blood flowing toward the transducer, from right (caudal) to left (cephalic). The red color indicates blood flowing away from the transducer, from left (cephalic) to right (caudal). (B) Blood flow in the IVC was quantified by pulse Doppler mode before and after ligation. (C) Representative images of CXCL1 expression (green) and adherent Ly6G+ neutrophils (red) in the IVC of WT mice obtained with spinning-disk intravital microscopy. PE-conjugated anti-Ly6G mAb and Fluoresbrite green beads coated with anti-CXCL1 mAb were injected IV into WT mice 1 hour before sham surgery or surgical ligation. Top, Three hours after sham surgery; middle, 15 minutes after ligation; bottom, 3 hours after ligation. Bar, 10 μm. (D) Number of rolling neutrophils per minute per microscopic field 3 hours after ligation (vertical axis). Circulating neutrophil count for each genotype (horizontal axis). (E) Quantification of endothelial surface area covered with firmly adherent neutrophils 3 hours after ligation. The data in the graphs represent the mean ± standard error of the mean (SEM) from 5 mice in each group. *P < .05.
Cooperative PSGL-1 and CXCR2 signaling in neutrophils enhances adhesion in flow-restricted IVC. (A) Top, Sagittal view of B-mode ultrasonography confirms stenosis of the IVC after ligation. The cephalic and caudal directions are marked. Bar, 1 mm. Bottom, Color Doppler mode in B-mode sagittal view reveals the flow direction in the IVC around the stenosis. The blue color indicates blood flowing toward the transducer, from right (caudal) to left (cephalic). The red color indicates blood flowing away from the transducer, from left (cephalic) to right (caudal). (B) Blood flow in the IVC was quantified by pulse Doppler mode before and after ligation. (C) Representative images of CXCL1 expression (green) and adherent Ly6G+ neutrophils (red) in the IVC of WT mice obtained with spinning-disk intravital microscopy. PE-conjugated anti-Ly6G mAb and Fluoresbrite green beads coated with anti-CXCL1 mAb were injected IV into WT mice 1 hour before sham surgery or surgical ligation. Top, Three hours after sham surgery; middle, 15 minutes after ligation; bottom, 3 hours after ligation. Bar, 10 μm. (D) Number of rolling neutrophils per minute per microscopic field 3 hours after ligation (vertical axis). Circulating neutrophil count for each genotype (horizontal axis). (E) Quantification of endothelial surface area covered with firmly adherent neutrophils 3 hours after ligation. The data in the graphs represent the mean ± standard error of the mean (SEM) from 5 mice in each group. *P < .05.
PSGL-1 and integrin signaling pathways in neutrophils require distinct tyrosine residues on SLP-76
In T lymphocytes and platelets, signaling through ITAM receptors activates the adaptor SLP-76 by phosphorylating tyrosine 145, whereas outside signaling through integrins activates SLP-76 by phosphorylating tyrosines 112 and 128.39,40 Phosphorylation of these tyrosines recruits other proteins to the SLP-76 signaling complex. In neutrophils, PSGL-1–triggered signaling through ITAMs was reported to activate SLP-76 by phosphorylating tyrosines 112 and 128 rather than tyrosine 145.20 To determine whether SLP-76–mediated signaling in neutrophils contributes to DVT, we used Lcp2f/fLysMCre+ mice.28 These mice lacked SLP-76 in neutrophils but retained ∼50% levels of SLP-76 in monocytes (supplemental Figure 1A-B), consistent with the inefficiency of LysM-driven Cre recombination in monocytes.41 They expressed normal levels of SLP-76 in platelets and T lymphocytes (supplemental Figure 1A-B). As controls, we used Lcp2f/fLysMCre− mice, which in all experiments yielded the same results as WT mice. We also used Lcp2Y145F and Lcp2Y112/128F mice, which express phosphorylation-defective SLP-76 variants.29,40 We confirmed mutations of codons for specific tyrosine residues on both alleles of Lcp2Y145F and Lcp2Y112/128F mice, respectively (supplemental Figure 1C-E). Rolling neutrophils arrested at much lower frequencies in flow-restricted IVCs of Lcp2f/fLysMCre+ and Lcp2Y145F mice, whereas neutrophils rolled and arrested normally in Lcp2Y112/128F mice (Figure 1D-E). These data suggest that, unlike the previous report,20 PSGL-1–triggered integrin activation in neutrophils requires phosphorylation of tyrosine 145 on SLP-76.
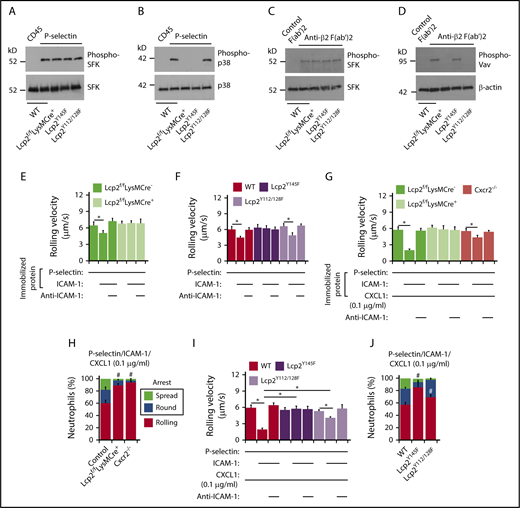
To further address how SLP-76 is activated, we incubated neutrophils on immobilized P-selectin to induce PSGL-1–dependent signaling or on immobilized anti-β2 integrin F(ab′)2 to induce integrin outside-in signaling.25 When incubated on P-selectin, neutrophils from WT, Lcp2f/fLysMCre−, Lcp2f/fLysMCre+, Lcp2Y145F, or Lcp2Y112/128F mice activated SFKs, but only neutrophils from WT and Lcp2Y112/128F mice activated p38 MAPK (Figure 2A-B). When incubated on anti-β2 integrin F(ab′)2, neutrophils of all genotypes activated SFKs, but only neutrophils from WT and Lcp2Y145F mice activated Vav proteins, which trigger Rac- and Rho-dependent actin polymerization that strengthens adhesion (Figure 2C-D). These data confirm that P-selectin–induced phosphorylation of tyrosine 145 on SLP-76 is downstream of SFKs and upstream of p38, whereas integrin-induced phosphorylation of tyrosines 112 and 128 is downstream of SFKs and upstream of Vav proteins. Neutrophils from WT and Lcp2Y112/128F mice rolled slower on coimmobilized P-selectin and ICAM-1 than on P-selectin alone (Figure 2E-F). Anti–ICAM-1 mAb prevented slow rolling, consistent with P-selectin–induced activation of β2 integrins that slowed rolling through interactions with ICAM-1. In contrast, neutrophils from Lcp2f/fLysMCre+ or Lcp2Y145F mice did not undergo P-selectin–induced slow rolling on ICAM-1 (Figure 2E-F). When low-density CXCL1 was coimmobilized with P-selectin and ICAM-1, WT neutrophils, but not Cxcr2−/− neutrophils, rolled even slower (Figure 2G) and frequently arrested and spread (Figure 2H), consistent with an earlier study.24 Neutrophils from Lcp2f/fLysMCre+ and Lcp2Y145F mice failed to undergo integrin-dependent slow rolling (Figure 2G,I) as well as arrest and spreading (Figure 2H,J). This indicates that P-selectin signals must phosphorylate tyrosine 145 on SLP-76 to synergize with chemokine signals to activate β2 integrins. Neutrophils from Lcp2Y112/128F mice were partially defective in slow rolling (Figure 2I) and markedly defective in spreading (Figure 2J). This suggests that adhesion strengthening through integrin outside-in signaling contributes to slow rolling when chemokine density is limiting. For all genotypes, we observed similar results when we immobilized E-selectin instead of P-selectin in flow chambers (supplemental Figure 2), consistent with a common signaling pathway induced by both selectins.14,15
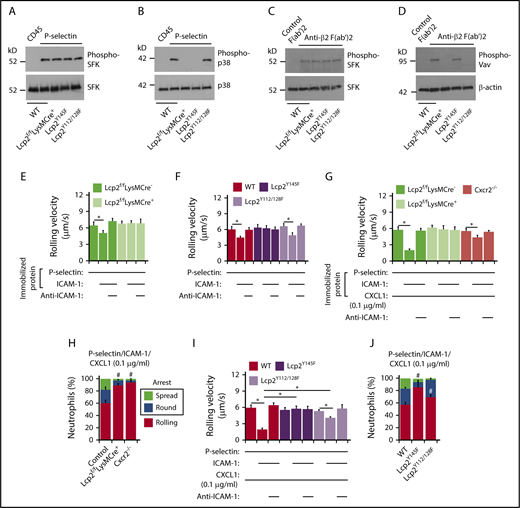
PSGL-1 and integrin signaling pathways in neutrophils require distinct tyrosine residues on SLP-76. (A-D) Bone marrow leukocytes (A-B) of the indicated genotype were incubated on immobilized control CD45-IgM or P-selectin–IgM. Isolated bone marrow neutrophils (C-D) of the indicated genotype were incubated on control F(ab′)2 or anti-β2 integrin F(ab′)2. Lysates were analyzed by immunoblotting with the indicated antibodies. (E-F) Rolling velocities of neutrophils of the indicated genotype on P-selectin with or without coimmobilized ICAM-1 in the presence or absence of anti–ICAM-1 mAb. (G,I) Rolling velocities of neutrophils of the indicated genotype on P-selectin coimmobilized with ICAM-1 and low-dose CXCL1 (0.1 μg/mL) in the presence or absence of anti–ICAM-1 mAb. (H,J) Percentages of neutrophils of the indicated genotype rolling, arrested and round, or arrested and spread on coimmobilized P-selectin, ICAM-1, and low-dose CXCL1. The data in panels A through D are representative of 3 experiments. The data in panels E through J represent the mean ± SEM from 5 experiments, with 5 mice in each experimental group. *P < .05 for rolling velocity; #P < .05 for number of rolling cells.
PSGL-1 and integrin signaling pathways in neutrophils require distinct tyrosine residues on SLP-76. (A-D) Bone marrow leukocytes (A-B) of the indicated genotype were incubated on immobilized control CD45-IgM or P-selectin–IgM. Isolated bone marrow neutrophils (C-D) of the indicated genotype were incubated on control F(ab′)2 or anti-β2 integrin F(ab′)2. Lysates were analyzed by immunoblotting with the indicated antibodies. (E-F) Rolling velocities of neutrophils of the indicated genotype on P-selectin with or without coimmobilized ICAM-1 in the presence or absence of anti–ICAM-1 mAb. (G,I) Rolling velocities of neutrophils of the indicated genotype on P-selectin coimmobilized with ICAM-1 and low-dose CXCL1 (0.1 μg/mL) in the presence or absence of anti–ICAM-1 mAb. (H,J) Percentages of neutrophils of the indicated genotype rolling, arrested and round, or arrested and spread on coimmobilized P-selectin, ICAM-1, and low-dose CXCL1. The data in panels A through D are representative of 3 experiments. The data in panels E through J represent the mean ± SEM from 5 experiments, with 5 mice in each experimental group. *P < .05 for rolling velocity; #P < .05 for number of rolling cells.
P-selectin–triggered signaling through PSGL-1 does not require L-selectin
Signaling through PSGL-1 was reported to require that PSGL-1 associate with L-selectin in the membrane.42 We generated Sell−/− (L-selectin–deficient) mice and confirmed that their neutrophils lack L-selectin but express normal levels of PSGL-1, CD44, and β2 integrins (Figure 3A). Unexpectedly, neutrophils rolled and arrested normally in flow-restricted IVCs of Sell−/− mice (Figure 1D-E).
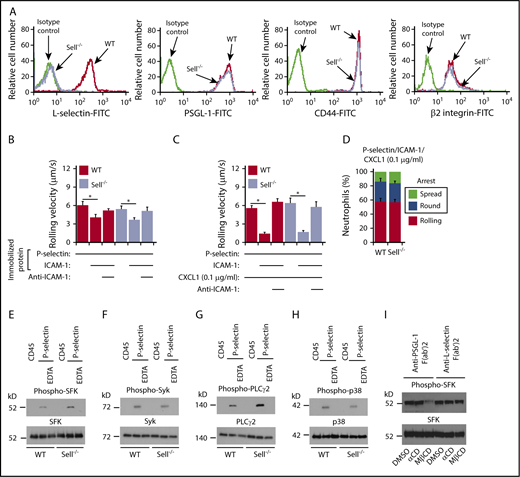
P-selectin–triggered signaling through PSGL-1 does not requireL-selectin. (A) Flow cytometric analysis of expression of L-selectin, PSGL-1, CD44, or β2 integrins on WT or Sell−/− neutrophils. (B) Rolling velocities of WT or Sell−/− neutrophils on P-selectin with or without coimmobilized ICAM-1 in the presence or absence of anti–ICAM-1 mAb. (C) Rolling velocities of WT or Sell−/− neutrophils on P-selectin coimmobilized with ICAM-1 and low-dose CXCL1 (0.1 μg/ml) in the presence or absence of anti–ICAM-1 mAb. (D) Percentages of WT or Sell−/− neutrophils rolling, arrested and round, or arrested and spread on coimmobilized P-selectin, ICAM-1, and low-dose CXCL1. (E-H) Bone marrow leukocytes from WT or Sell−/− mice were incubated on immobilized control CD45-IgM or P-selectin–IgM in the presence or absence of EDTA. Lysates were analyzed by immunoblotting with the indicated antibodies. (I) Isolated bone marrow neutrophils from WT mice were pretreated with DMSO (vehicle control), inactive control α-cyclodextrin (αCD), or methyl-β-cyclodextrin (MβCD). They were then incubated on immobilized F(ab′)2 fragments of anti–PSGL-1 mAb or anti–L-selectin mAb. Lysates were analyzed by immunoblotting with antibody against SFK or phospho-SFK. The data in panels B through D represent the mean ± SEM from 5 experiments, with 5 mice in each experimental group. The data in panels A and E through I are representative of 3 experiments. *P < .05.
P-selectin–triggered signaling through PSGL-1 does not requireL-selectin. (A) Flow cytometric analysis of expression of L-selectin, PSGL-1, CD44, or β2 integrins on WT or Sell−/− neutrophils. (B) Rolling velocities of WT or Sell−/− neutrophils on P-selectin with or without coimmobilized ICAM-1 in the presence or absence of anti–ICAM-1 mAb. (C) Rolling velocities of WT or Sell−/− neutrophils on P-selectin coimmobilized with ICAM-1 and low-dose CXCL1 (0.1 μg/ml) in the presence or absence of anti–ICAM-1 mAb. (D) Percentages of WT or Sell−/− neutrophils rolling, arrested and round, or arrested and spread on coimmobilized P-selectin, ICAM-1, and low-dose CXCL1. (E-H) Bone marrow leukocytes from WT or Sell−/− mice were incubated on immobilized control CD45-IgM or P-selectin–IgM in the presence or absence of EDTA. Lysates were analyzed by immunoblotting with the indicated antibodies. (I) Isolated bone marrow neutrophils from WT mice were pretreated with DMSO (vehicle control), inactive control α-cyclodextrin (αCD), or methyl-β-cyclodextrin (MβCD). They were then incubated on immobilized F(ab′)2 fragments of anti–PSGL-1 mAb or anti–L-selectin mAb. Lysates were analyzed by immunoblotting with antibody against SFK or phospho-SFK. The data in panels B through D represent the mean ± SEM from 5 experiments, with 5 mice in each experimental group. The data in panels A and E through I are representative of 3 experiments. *P < .05.
To further address whether PSGL-1 requires L-selectin to transduce signals, we used flow chamber experiments and signaling assays in vitro. Bone marrow neutrophils from Sell−/− mice had normal, integrin-dependent slow rolling on immobilized P-selectin and ICAM-1 (Figure 3B), and rolled slower, arrested, and spread normally when CXCL1 was coimmobilized (Figure 3C-D). Both WT and Sell−/− neutrophils rapidly activated SFKs, Syk, PLCγ2, and p38 MAPK after incubation on immobilized P-selectin but not on control protein (Figure 3E-H). Addition of EDTA to block Ca2+-dependent selectin interactions prevented signaling, confirming its specificity. We observed similar results when we immobilized E-selectin instead of P-selectin (supplemental Figure 3). Furthermore, incubating WT neutrophils on immobilized anti–PSGL-1 F(ab′)2 activated SFKs; this was prevented by disrupting lipid rafts with methyl-β-cyclodextrin but not with the inactive analog α-cyclodextrin (Figure 3I), confirming earlier studies.14,43 In contrast, methyl-β-cyclodextrin did not block SFK activation when WT neutrophils were incubated on immobilized anti–L-selectin F(ab′)2 (Figure 3H), implying a distinct L-selectin–dependent signaling mechanism. These data demonstrate that PSGL-1–triggered signaling in neutrophils does not require L-selectin.
Cooperative PSGL-1 and CXCR2 signaling in neutrophils promotes DVT
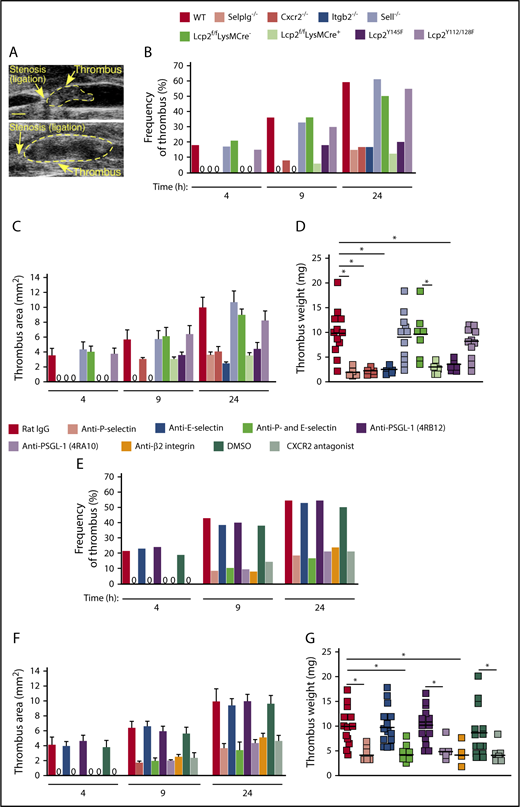
We used ultrasound to monitor the kinetics of thrombus formation and progression in each genotype over the first 24 hours after ligation of the IVC (Figure 4A). Both thrombus formation and thrombus area were reduced in genotypes where neutrophil adhesion was impaired (Selplg−/−, Itgb2−/−, Cxcr2−/−, Lcp2f/fLysMCre+, and Lcp2Y145F) but not in genotypes where adhesion was normal (Sell−/− and Lcp2Y112/128F) (Figure 4B-C). We collected each thrombus 24 hours after ligation. Thrombus weight was markedly lower in each genotype with defective neutrophil adhesion (Figure 4D). The defective thrombus development in Lcp2f/fLysMCre+ and Lcp2Y145F mice provides evidence that PSGL-1–mediated signaling, which requires tyrosine 145 on SLP-76, cooperates with CXCR2-mediated signaling to promote venous thrombosis.

Cooperative PSGL-1 and CXCR2 signaling in neutrophils promotes DVT. (A) Representative thrombi scanned by ultrasonography. Thrombus areas: top, 3.1 mm2; bottom, 11.6 mm2. Bar, 1 mm. (B-C) Kinetics of thrombus development (frequency) and thrombus size (area) in mice of the indicated genotype, measured by ultrasonography at the indicated times after ligation. (D) Thrombus weight in the indicated genotype 24 hours after ligation. Each symbol represents an individual thrombus. Horizontal black bars represent median values. (E-F) Kinetics of thrombus development (frequency) and thrombus size (area) in mice with the indicated treatment, measured by ultrasonography at the indicated times after ligation. (G) Thrombus weight with the indicated treatment 24 hours after ligation. Each symbol represents an individual thrombus. Horizontal black bars represent median values. The data in panels B and E were taken from 10 to 18 mice in each group. The data in panels C and F represent the mean ± SEM from 3 to 14 mice in each group. *P < .05.
Cooperative PSGL-1 and CXCR2 signaling in neutrophils promotes DVT. (A) Representative thrombi scanned by ultrasonography. Thrombus areas: top, 3.1 mm2; bottom, 11.6 mm2. Bar, 1 mm. (B-C) Kinetics of thrombus development (frequency) and thrombus size (area) in mice of the indicated genotype, measured by ultrasonography at the indicated times after ligation. (D) Thrombus weight in the indicated genotype 24 hours after ligation. Each symbol represents an individual thrombus. Horizontal black bars represent median values. (E-F) Kinetics of thrombus development (frequency) and thrombus size (area) in mice with the indicated treatment, measured by ultrasonography at the indicated times after ligation. (G) Thrombus weight with the indicated treatment 24 hours after ligation. Each symbol represents an individual thrombus. Horizontal black bars represent median values. The data in panels B and E were taken from 10 to 18 mice in each group. The data in panels C and F represent the mean ± SEM from 3 to 14 mice in each group. *P < .05.
Injecting WT mice with a CXCR2 antagonist, with a blocking mAb to P-selectin, with a blocking mAb (4RA10) but not a nonblocking mAb (4RB12) to PSGL-1, or with a blocking mAb to β2 integrins also inhibited thrombus formation (Figure 4E), thrombus area (Figure 4F), and thrombus weight (Figure 4G). In marked contrast, injecting a blocking mAb to E-selectin had no effect on thrombus development, and coinjecting anti–E-selectin mAb did not enhance the antithrombotic effects of anti–P-selectin mAb (Figure 4E-G).
Cooperative PSGL-1 and CXCR2 signaling in neutrophils enhances neutrophil recruitment and NET formation in deep vein thrombi
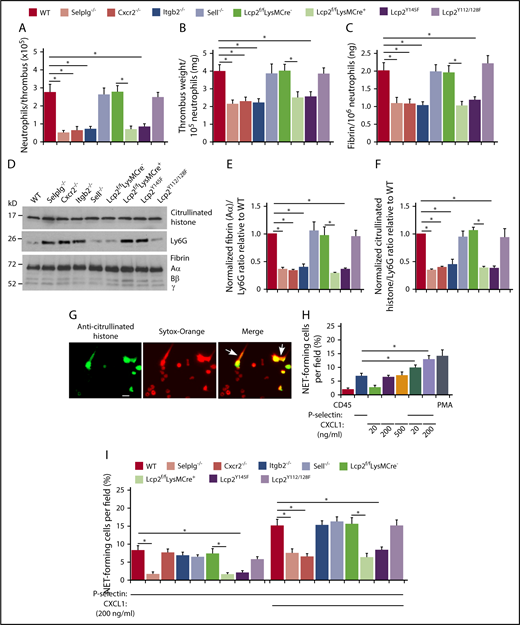
In all genotypes, the weight of each thrombus was proportional to the total amount of fibrin (supplemental Figure 4), and immunoblots revealed a constant ratio of the red blood cell marker TER-119 to fibrin (supplemental Figure 5). The number of neutrophils per thrombus was significantly lower in each genotype with defective neutrophil adhesion, including Cxcr2−/−, Lcp2f/fLysMCre+, and Lcp2Y145F (Figure 5A). Strikingly, these thrombi had lower weight and less fibrin per incorporated neutrophil (Figure 5B-C). Immunoblots also revealed less fibrin and less citrullinated histones, a component of NETs, relative to the neutrophil marker Ly6G (Figure 5D-F). In vitro, dimeric or oligomeric forms of fluid-phase P-selectin cause neutrophils to release NETs.36,44 We observed that phorbol myristate acetate, CXCL1, or an oligomeric P-selectin–IgM chimera36 induced WT neutrophils to citrullinate histones, as measured by immunofluorescence, and to release NETs, as measured by staining of citrullinated histones and DNA outside cells (Figure 5G-H). Combining P-selectin–IgM and CXCL1 further increased NET formation by neutrophils from WT mice, but not from mice with defects in PSGL-1 or CXCR2 signaling (Figure 5H-I). These results demonstrate that cooperative PSGL-1 and CXCR2 signaling in neutrophils enhances procoagulant activity, which accelerates thrombus development.
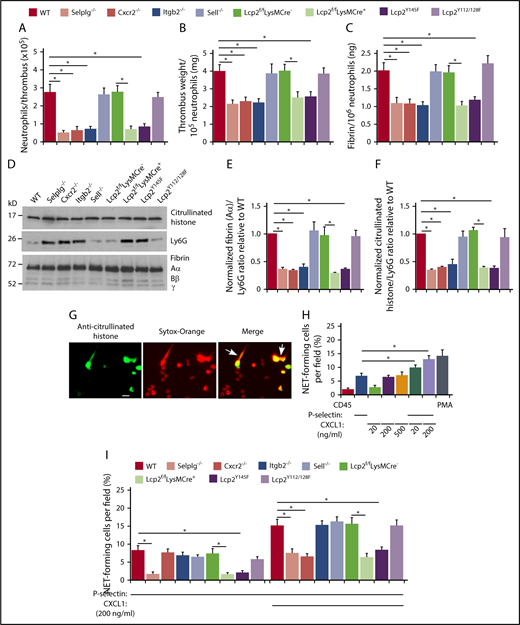
Cooperative PSGL-1 and CXCR2 signaling in neutrophils enhances neutrophil recruitment and NET formation in deep vein thrombi. (A) Number of Ly6G+ neutrophils per thrombus 24 hours after ligation in the indicated genotype, as measured by flow cytometry. (B) Normalized thrombus weight per Ly6G+ neutrophil. (C) Normalized fibrin level per Ly6G+ neutrophil. The data in panels A through C represent the mean ± SEM from 5 to 10 mice in each group. (D) Western blot of thrombus lysates probed with antibodies to fibrin, Ly6G, and citrullinated histone. The Aα, Bβ, and γ chains of fibrin are marked. The data are representative of 3 experiments. (E) Normalized densitometric ratio of fibrin Aα chain to Ly6G. (F) Normalized densitometric ratio of citrullinated histone to Ly6G. The data in panels E and F represent the mean ± SD from 3 mice in each experimental group. (G) Representative fluorescent images of isolated WT bone marrow neutrophils incubated with phorbol myristate acetate (PMA), stained with anti-citrullinated histone IgG followed by Alexa 488–conjugated anti-rabbit IgG (left), with Sytox Orange to label DNA (middle), or merged image (right). The white arrow marks extracellular staining for both citrullinated histones and DNA, indicating NET release. Scale bar, 10 μm. (H-I) Percentage of NET-forming neutrophils treated with the indicated agonist, calculated by dividing the number of cells with both extracellular citrullinated histones and Sytox Orange–positive DNA by the total number of Sytox Orange–positive cells. The data in panel H are from WT mice. The data in panel I are from mice of the indicated genotype. The data in panels H and I represent the mean ± standard deviation (SD) from 3 mice in each experimental group. *P < .05.
Cooperative PSGL-1 and CXCR2 signaling in neutrophils enhances neutrophil recruitment and NET formation in deep vein thrombi. (A) Number of Ly6G+ neutrophils per thrombus 24 hours after ligation in the indicated genotype, as measured by flow cytometry. (B) Normalized thrombus weight per Ly6G+ neutrophil. (C) Normalized fibrin level per Ly6G+ neutrophil. The data in panels A through C represent the mean ± SEM from 5 to 10 mice in each group. (D) Western blot of thrombus lysates probed with antibodies to fibrin, Ly6G, and citrullinated histone. The Aα, Bβ, and γ chains of fibrin are marked. The data are representative of 3 experiments. (E) Normalized densitometric ratio of fibrin Aα chain to Ly6G. (F) Normalized densitometric ratio of citrullinated histone to Ly6G. The data in panels E and F represent the mean ± SD from 3 mice in each experimental group. (G) Representative fluorescent images of isolated WT bone marrow neutrophils incubated with phorbol myristate acetate (PMA), stained with anti-citrullinated histone IgG followed by Alexa 488–conjugated anti-rabbit IgG (left), with Sytox Orange to label DNA (middle), or merged image (right). The white arrow marks extracellular staining for both citrullinated histones and DNA, indicating NET release. Scale bar, 10 μm. (H-I) Percentage of NET-forming neutrophils treated with the indicated agonist, calculated by dividing the number of cells with both extracellular citrullinated histones and Sytox Orange–positive DNA by the total number of Sytox Orange–positive cells. The data in panel H are from WT mice. The data in panel I are from mice of the indicated genotype. The data in panels H and I represent the mean ± standard deviation (SD) from 3 mice in each experimental group. *P < .05.
PSGL-1 and CXCR2 signaling in monocytes does not enhance monocyte recruitment and tissue factor expression in deep vein thrombi
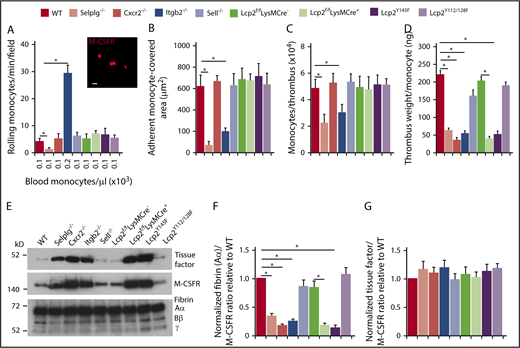
Compared with neutrophils, significantly fewer monocytes circulate in blood (Figure 6A; supplemental Table 1). Nevertheless, we observed rolling and arrest of monocytes (labeled with anti–M-CSFR) in flow-restricted IVCs of WT mice (Figure 6A-B). As with neutrophils, virtually no monocytes rolled or arrested in Selplg−/− mice, whereas many monocytes rolled but few arrested in Itgb2−/− mice (Figure 6A-B). However, we noted normal rolling and arrest of monocytes in the other genotypes (Figure 6A-B). Monocytes from Lcp2f/fLysMCre+ mice retained ∼50% levels of SLP-76 (supplemental Figure 1B) and they also express the related adaptor SLP-65 that may substitute for SLP-76.45 Furthermore, monocytes express CCR2,46 a receptor for the chemokine CCL2 that is upregulated in flow-restricted IVCs.8 Therefore, CCR2 may substitute for CXCR2 in monocytes. These observations enabled us to probe the neutrophil-specific functions of CXCR2 and SLP-76 during DVT.
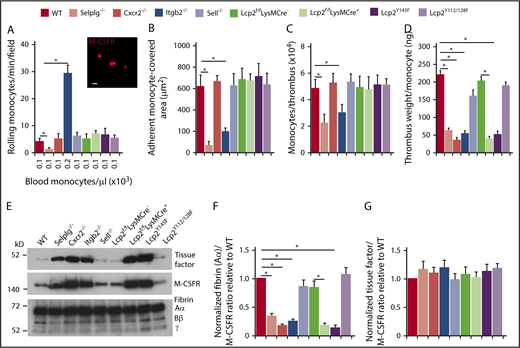
PSGL-1 and CXCR2 signaling in monocytes does not enhance monocyte recruitment and tissue factor expression in deep vein thrombi. (A) Number of rolling M-CSFR (CD115)+ monocytes per minute per microscopic field in the IVC 3 hours after ligation (vertical axis). Circulating monocyte count for each genotype (horizontal axis). Inset, Representative image of adherent M-CSFR+ monocytes (red) in WT mice obtained with spinning-disk intravital microscopy. PE-conjugated anti–M-CSFR mAb was injected IV into WT mice 1 hour before surgery. Bar, 10 μm. (B) Quantification of endothelial surface area covered with firmly adherent monocytes. The data in the graphs of panels A and B represent the mean ± SEM from 5 mice in each group. (C) Number of M-CSFR+ monocytes per thrombus 24 hours after ligation, as measured by flow cytometry. (D) Normalized thrombus weight per M-CSFR+ monocyte. The data in panels C and D represent the mean ± SEM from 5 to 10 mice in each group. (E) Western blot of thrombus lysates probed with antibodies to fibrin, M-CSFR, and tissue factor. The Aα, Bβ, and γ chains of fibrin are marked. The data are representative of 3 experiments. (F) Normalized densitometric ratio of fibrin Aα chain to M-CSFR. (G) Normalized densitometric ratio of tissue factor to M-CSFR. The data in panels F and G represent the mean ± SD from 3 mice in each experimental group. *P < .05.
PSGL-1 and CXCR2 signaling in monocytes does not enhance monocyte recruitment and tissue factor expression in deep vein thrombi. (A) Number of rolling M-CSFR (CD115)+ monocytes per minute per microscopic field in the IVC 3 hours after ligation (vertical axis). Circulating monocyte count for each genotype (horizontal axis). Inset, Representative image of adherent M-CSFR+ monocytes (red) in WT mice obtained with spinning-disk intravital microscopy. PE-conjugated anti–M-CSFR mAb was injected IV into WT mice 1 hour before surgery. Bar, 10 μm. (B) Quantification of endothelial surface area covered with firmly adherent monocytes. The data in the graphs of panels A and B represent the mean ± SEM from 5 mice in each group. (C) Number of M-CSFR+ monocytes per thrombus 24 hours after ligation, as measured by flow cytometry. (D) Normalized thrombus weight per M-CSFR+ monocyte. The data in panels C and D represent the mean ± SEM from 5 to 10 mice in each group. (E) Western blot of thrombus lysates probed with antibodies to fibrin, M-CSFR, and tissue factor. The Aα, Bβ, and γ chains of fibrin are marked. The data are representative of 3 experiments. (F) Normalized densitometric ratio of fibrin Aα chain to M-CSFR. (G) Normalized densitometric ratio of tissue factor to M-CSFR. The data in panels F and G represent the mean ± SD from 3 mice in each experimental group. *P < .05.
The number of monocytes per thrombus was lower only in Selplg−/− and Itgb2−/− mice (Figure 6C), which had defective monocyte arrest in flow-restricted IVCs (Figure 6B). There was lower thrombus weight per monocyte in these mice as well as in Cxcr2−/−, Lcp2f/fLysMCre+, and Lcp2Y145F mice (Figure 5D), in which monocytes arrested normally (Figure 6B). The thrombi of all of these genotypes had less fibrin relative to the monocyte marker M-CSFR than in those of control mice (Figure 6E-F). In marked contrast, the thrombi had the same ratio of tissue factor to M-CSFR as those of control mice (Figure 6E,G). These data indicate that venous thrombosis can be propagated by cooperative PSGL-1 and CXCR2 signals that generate effectors such as NETs in neutrophils but not effectors such as tissue factor in monocytes.
Discussion
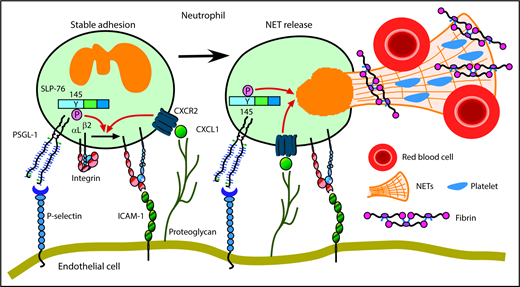
We have shown that neutrophils cooperatively signal through PSGL-1 and CXCR2 to mediate adhesion and to generate prothrombotic effectors in flow-restricted IVCs. PSGL-1 initiated neutrophil rolling by interacting with P-selectin, which was rapidly mobilized to endothelial cell surfaces. CXCL1, the major ligand for CXCR2, was also rapidly mobilized. Signaling through PSGL-1 and CXCR2 activated β2 integrins that enabled neutrophils to decelerate and arrest. PSGL-1–triggered signals, in part through activation of SLP-76, cooperated with CXCR2 signals to generate NETs that propagated venous thrombi (Figure 7). Disrupting these signals in neutrophils, but not in monocytes, inhibited thrombus formation and progression.
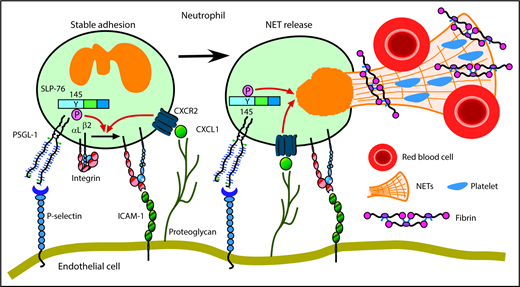
Cooperative PSGL-1 and CXCR signaling in neutrophils promotes DVT. In flow-restricted IVCs, neutrophils rolling on P-selectin signal through PSGL-1, in part through phosphorylation of tyrosine 145 on SLP-76. Rolling neutrophils signal through CXCR2 after engaging CXCL1 immobilized on proteoglycans. These signals cooperatively activate integrin αLβ2, which causes neutrophils to decelerate and arrest on ICAM-1. Cooperative signaling in adherent neutrophils triggers release of procoagulant NETs. See “Discussion” for details.
Cooperative PSGL-1 and CXCR signaling in neutrophils promotes DVT. In flow-restricted IVCs, neutrophils rolling on P-selectin signal through PSGL-1, in part through phosphorylation of tyrosine 145 on SLP-76. Rolling neutrophils signal through CXCR2 after engaging CXCL1 immobilized on proteoglycans. These signals cooperatively activate integrin αLβ2, which causes neutrophils to decelerate and arrest on ICAM-1. Cooperative signaling in adherent neutrophils triggers release of procoagulant NETs. See “Discussion” for details.
When chemokines are limiting, PSGL-1 and CXCR2 signals in neutrophils cooperate to mediate β2 integrin–mediated slow rolling and arrest in vitro or with adoptive neutrophil transfers in vivo.24 Here, we found that SLP-76, a PSGL-1–signaling intermediate, is required for this cooperative function. In vivo, SLP-76 deficiency impaired rolling neutrophils from arresting in flow-restricted IVCs, whereas CXCR2 deficiency prevented arrest. Thus, PSGL-1 and CXCR2 signals cooperated to mediate slow rolling and arrest of endogenous neutrophils in a model that links inflammation to thrombosis.
How does SLP-76 function downstream of PSGL-1? In lymphocytes, mast cells, and platelets, the signaling cascade initiated by ITAM proteins uses the adaptor SLP-76 to recruit Tec kinases, which upon activation, propagate downstream signals.47 SLP-76 must be phosphorylated on tyrosine 145 to enable Tec kinases to be recruited and activated.29,39,40,48 Integrin outside-in signaling also uses SLP-76 to propagate downstream signals. In this pathway, SLP-76 must be phosphorylated on tyrosines 112 and 128 to recruit Vav proteins, which stimulate actin polymerization, cell spreading, and adhesion strengthening.29,39,40,48 The neutrophil Tec kinase Btk is required for selectin-triggered, integrin-dependent slow rolling.14,17 Btk is activated downstream of SFKs, Syk, and SLP-76 but upstream of p38 MAPK. Using Lcp2f/fLysMCre+ mice to delete SLP-76 only in neutrophils, we confirmed that selectin signaling uses SLP-76 downstream of SFKs but upstream of p38 MAPK. We further found that β2 integrin outside-in signaling uses SLP-76 downstream of SFKs but upstream of Vav proteins. We could not reproduce the surprising observation that SLP-76 requires tyrosines 112 and 128 rather than tyrosine 145 to propagate selectin signals in neutrophils.20 Instead, neutrophils expressing the Y145F mutation in SLP-76 failed to undergo selectin-induced, integrin-dependent slow rolling in vitro or in vivo. After adhering to P-selectin or E-selectin, they activated SFKs but not p38 MAPK. Integrin outside-in signaling activated both SFKs and Vav proteins. In contrast, neutrophils expressing the Y112/128F mutations in SLP-76 had normal selectin-induced slow rolling but defective chemokine-induced cell spreading. Integrin outside-in signaling activated SFKs but not Vav proteins. We cannot explain why our results conflict with the previous study.20 Because of the discrepancy, we repeatedly confirmed the genotypes of the Lcp2f/fLysMCre+, Lcp2Y145F, and Lcp2Y112/128F mice in our experimental groups. Our data align with many other reports that SLP-76 requires tyrosine 145 to activate Tec kinases but tyrosines 112 and 128 to activate Vav proteins.29,39,40,48
Upstream of SLP-76, engagement of PSGL-1 activates SFKs, which recruit the ITAM proteins DAP12 and FcRγ and the kinase Syk.14,,,,,-20 Although signaling was reported to require that PSGL-1 associate with L-selectin,42 we observed that neutrophils rolled and arrested normally in flow-restricted IVCs of L-selectin–deficient (Sell−/−) mice. Thrombus incidence and size in Sell−/− mice was normal. Consistent with this observation, injecting blocking anti–L-selectin F(ab′)2 into WT mice does not reduce thrombus incidence or size in flow-restricted IVCs.27 We found that neutrophils from WT and Sell−/− mice exhibited equivalent integrin-dependent slow rolling and arrest in vitro. WT and Sell−/− neutrophils plated on immobilized P-selectin or E-selectin similarly activated SFKs, Syk, PLCγ2, and p38 MAPK. Collectively, our results demonstrate that PSGL-1 signaling in neutrophils does not require L-selectin. We cannot explain the discrepancy with the previous study.42 Our data support other reports that L-selectin is dispensable in models of inflammation where P- and E-selectin have major roles.49,-51 In some circumstances, L-selectin mediates rolling of leukocytes on adherent leukocytes or leukocyte fragments in venules,52,53 but there is no evidence that this requires L-selectin–dependent signaling. PSGL-1 must associate with lipid rafts to signal,14,43 whereas we found that disrupting lipid rafts does not impair signaling through L-selectin.
Cooperative PSGL-1 and CXCR2 signaling promoted thrombosis by facilitating neutrophil adhesion in flow-restricted veins. However, cooperative signaling also augmented production of prothrombotic effectors in adherent neutrophils. Mice with defects in CXCR2 or PSGL-1 signaling generated thrombi with fewer incorporated neutrophils. Furthermore, the thrombi had lower weights and less fibrin per incorporated neutrophil, consistent with impaired neutrophil procoagulant activity. We confirmed NETs as 1 prothrombotic effector. Mice with defects in CXCR2 or PSGL-1 signaling had fewer citrullinated histones per neutrophil incorporated into thrombi. In vitro, cooperative signaling through PSGL-1 and CXCR2 in neutrophils enhanced formation of NETs. Multiple mechanisms to induce NETs in vitro have been described.54 In flow-restricted veins, other mediators may contribute to NETs, but if present, they were much less effective when PSGL-1 or CXCR2 signaling was disrupted.
Defects in neutrophil adhesion reduced thrombus incidence and size in genotypes that retained normal monocyte adhesion in flow-restricted IVCs, consistent with observations that selective depletion of neutrophils prevents thrombus formation.8 Although thrombus formation requires neutrophils, monocytes further augment thrombosis by expressing tissue factor.8 We found that the level of tissue factor per thrombus-incorporated monocyte was unchanged in Selplg−/− and Itgb2−/− mice with defective monocyte adhesion. Indeed, an early study demonstrated that P-selectin–dependent adhesion of activated platelets to monocytes triggers expression of chemokines but not tissue factor.55 This suggests that monocytes generate tissue factor through other means. In the same DVT model, for example, platelet-derived high-mobility group box 1 protein activates monocytes by binding to receptor for advanced glycation end products and Toll-like receptor 2.56 Red blood cells also contribute to DVT through several mechanisms.57
Several mouse models of DVT have been developed, each with advantages and disadvantages.58 A stasis model, in which flow through the IVC is completely blocked, provides valuable information during the later stages of DVT. In this model, however, depleting neutrophils or dissolving NETs does not affect thrombus size.59,60 We used a flow-restriction (stenosis) model that avoids endothelial cell injury and is well suited to explore the early stages of DVT.7,8,27,38 Neutrophil adhesion and signaling were critical events during the first 24 hours. Notably, thrombus development was dependent on P-selectin but not on E-selectin. This suggests that flow restriction did not generate enough cytokines to trigger endothelial cell synthesis of E-selectin.
Our results illustrate how dysregulated neutrophil adhesion and signaling can contribute to pathological inflammation and thrombosis, which may guide new therapeutic strategies.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Dietmar Vestweber and Gary Koretzky for reagents.
This work was supported by National Institutes of Health, National Heart, Lung, and Blood Institute grant HL034363 and National Institutes of Health, National Institute of General Medical Sciences grant GM114731.
Authorship
Contribution: T.Y. and Z.L. performed experiments; J.A. provided expertise in ultrasonography; and T.Y. and R.P.M. conceived the study, interpreted data, and wrote the manuscript, with input from all authors.
Conflict-of-interest disclosure: R.P.M. is a cofounder of Selexys Pharmaceuticals, now part of Novartis AG, and of Tetherex Pharmaceuticals. The remaining authors declare no competing financial interests.
Correspondence: Rodger P. McEver, Cardiovascular Biology Research Program, Oklahoma Medical Research Foundation, 825 NE 13th St, Oklahoma City, OK 73104; e-mail: rodger-mcever@omrf.org.