Abstract
Dysregulation of the B-cell leukemia/lymphoma-2 (BCL-2) family of proteins of the intrinsic apoptotic pathway is fundamental to the pathophysiology of many hematologic malignancies. The BCL-2 family consists of regulatory proteins that either induce apoptosis (proapoptotic) or inhibit it (prosurvival). BCL-2, myeloid cell leukemia-1, and B-cell lymphoma–extra large are prosurvival proteins that are prime targets for anticancer therapy, and molecules targeting each are in various stages of preclinical and clinical development. The US Food and Drug Administration (FDA)-approved BCL-2 inhibitor venetoclax was first proven to be highly effective in chronic lymphocytic leukemia and some B-cell non-Hodgkin lymphoma subtypes. Subsequently, venetoclax was found to be active clinically against a diverse array of hematologic malignancies including multiple myeloma, acute myeloid leukemia, myelodysplastic syndrome, acute lymphoblastic leukemia, and others. Here, we give a brief introduction to BCL-2 family biology and the mechanism of action of BCL-2 Homology 3 (BH3) mimetics, and provide an overview of the clinical data for therapeutically targeting prosurvival proteins in hematologic malignancies, with a focus on BCL-2 inhibition. To prioritize novel agent combinations and predict responders, we discuss the utility of functional assays such as BH3 profiling. Finally, we provide a perspective on how therapies targeting BCL-2 family proteins may be optimally implemented into future therapeutic regimens for hematologic malignancies.
Introduction
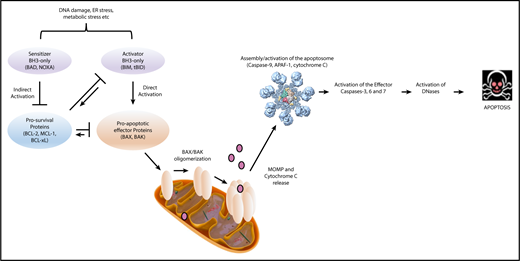
Apoptosis is a form of programmed cell death that is distinct from other forms of cell death such as necroptosis, autophagy, ferroptosis, pyroptosis, netosis, or necrosis.1 During apoptosis, cells undergo morphological changes such as shrinking of the nuclei and mitochondria. Blebbing, in which parts of the plasma membrane tie off cell content, continuously reduces cell size and functionality. These apoptotic bodies are rapidly cleared via phagocytosing cells, avoiding a strong inflammatory response. Apoptosis can be triggered via an extrinsic death receptor pathway, regulated by the tumor necrosis factor (TNF) and first apoptosis signal death receptor super families, or by an intrinsic B-cell leukemia/lymphoma-2 (BCL-2) family-regulated mitochondrial pathway. Both pathways converge at the level of mitochondria via the protein truncated BH3-interacting domain (tBID). In this review, we focus on the intrinsic apoptotic pathway, which has been effectively targeted therapeutically in hematologic malignancies.
Since the discovery of the first protein to prevent cell death, BCL-2,2-4 apoptosis mediated through the intrinsic pathway has become one of the most extensively studied areas of cancer research. Indeed, apoptosis is one of the major hurdles that a nascent neoplastic cell must overcome to transform into a malignant cell.5,6 The BCL-2 gene was initially discovered as the “driver” of follicular lymphoma (FL) pathogenesis due to its involvement in the t(14;18)(q32;q21) chromosomal translocation, the genetic hallmark of FL.4 Later, BCL-2 was also found to contribute to the pathophysiology of chronic lymphocytic leukemia (CLL), mantle cell lymphoma (MCL), and Waldenström macroglobulinemia (WM). In other hematological malignancies such as multiple myeloma (MM),7 diffuse large B-cell lymphoma (DLBCL),8 acute lymphoblastic leukemia (ALL), acute myeloid leukemia (AML), and chronic myeloid leukemia (CML), the dependence of the malignant cell on BCL-2 relative to other prosurvival BCL-2 family proteins is more variable. Unlike most oncogenic proteins, BCL-2 dysregulation does not confer an advantage in terms of cell growth or proliferation, but rather it enables cells that would normally undergo apoptosis to survive.
Another key prosurvival protein, myeloid cell leukemia-1 (MCL-1), was initially discovered as an important regulator of myeloid cell differentiation.9 MCL-1 has since been identified as a crucial survival factor in MM, CML, AML, ALL, and DLBCL.10-14 A third key prosurvival protein, B-cell lymphoma-extra large (BCL-xL; Bcl2l1),15 is essential for megakaryocyte survival, and loss or inhibition of BCL-xL leads to thrombocytopenia.16,17 BCL-xL dysfunction may also contribute to the pathogenesis of several hematologic disorders such as myeloproliferative disorders including polycythemia vera.18
BCL-2 family biology
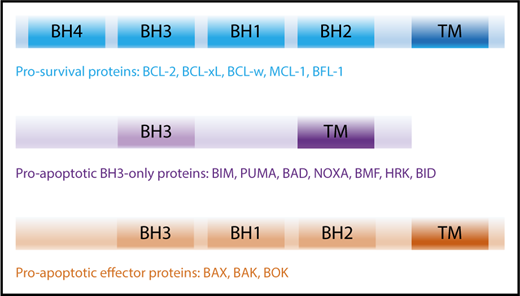
The intrinsic apoptotic pathway is regulated by the BCL-2 family and consists of 2 major groups of proteins: (1) the prosurvival proteins with its founding member BCL-2,3,4 MCL-1,9 BCL-xL,15 B-cell lymphoma-w (BCL-w),19 and BCL-2–related gene expressed in fetal liver-1 (BFL-1),20 all of which inhibit cell death and (2) the proapoptotic proteins, which can be further subdivided into (a) BH3-only proteins: BCL-2–interacting mediator of cell death (BIM),21 p53-upregulated modulator of apoptosis (PUMA; BBC2),22,23 NOXA (phorbol-12 myristate-13-acetate–induced protein 1; PMAIP1),24 BCL-2–associated death promotor (BAD), BH3-interacting domain (BID),25 BCL-2–interacting killer (BIK),26 BCL-2–modifying factor (BMF),27 Harakiri (HRK)28 and (b) the BCL-2–associated X protein (BAX)29 /BCL-2 homologous antagonist killer (BOK)30 -like proteins (including BCL-2–related ovarian killer [BOK]).31 All BCL-2 family proteins share the common BCL-2 homology (BH) domains based on their sequence similarity.32 Prosurvival and BAX/BAK-like proteins contain 4 and 3 BH domains, respectively, whereas BH3-only proteins only share the BH3 domain with the wider family (Figure 1).
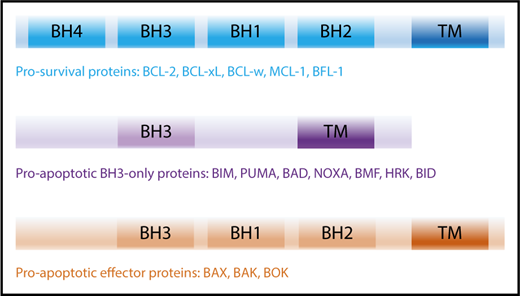
BCL-2 family members and their activation. The BCL-2 family of proteins contain functionally conserved BH domains and can be subdivided into prosurvival, proapoptotic BH3-only and proapoptotic effector proteins. TM, transmembrane.
BCL-2 family members and their activation. The BCL-2 family of proteins contain functionally conserved BH domains and can be subdivided into prosurvival, proapoptotic BH3-only and proapoptotic effector proteins. TM, transmembrane.
Activation of the apoptotic pathway
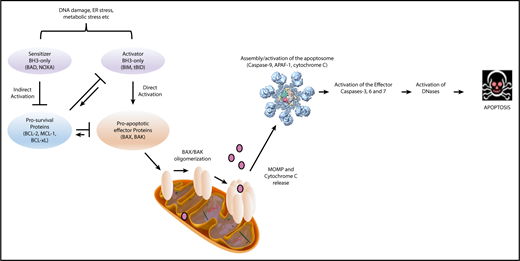
Apoptosis is composed of 3 distinct phases: (1) initiation, (2) commitment, and (3) execution. This process is induced upon stress stimuli such as nutrient deprivation or DNA damage (Figure 2). BH3-only proteins are activated as a death stimulus in a temporal and cell-type–specific manner, and displace/activate the BAX/BAK-like proteins in the initiating step.33 During the commitment phase, BAX and BAK form homodimers and heterodimers, which further assemble to pore-like structures in the outer mitochondrial membrane and cause mitochondrial outer membrane permeabilization (MOMP).34-38 This subsequently leads to the loss of mitochondrial transmembrane potential and oxidative phosphorylation, and the release of cytochrome C from the mitochondrial intermembrane space into the cytoplasm.39 In the execution step, cytochrome C together with APAF-1 and procaspase-9 form the apoptosome, which mediates cleavage of procaspase-9 to its active form caspase-9 and consequently promotes the activation of the effector caspases-3, 6 and 7, which induce DNases to cleave DNA into fragments. It is important to note that the “point of no return” generally occurs at the level of BAX/BAK activation,37 at which point the cell commits to undergo programmed cell death. Due to structural differences, BH3-only proteins have varying affinities for particular prosurvival proteins (Figure 3).40 Some BH3-only proteins such as BIM and PUMA bind promiscuously to all prosurvival proteins.41,42 Other BH3-only proteins bind more selectively, such as BAD, which binds BCL-2, BCL-xL and BCL-W, and NOXA, which binds mainly to MCL-1.40
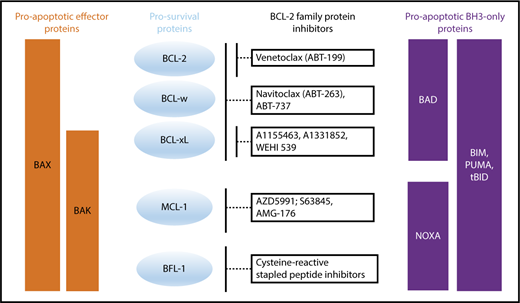
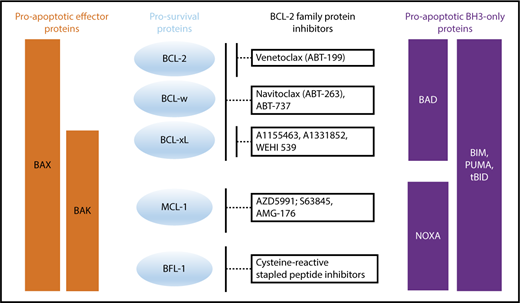
Binding profile of proapoptotic BH3-only and effector proteins to prosurvival proteins and how prosurvival proteins are targeted by BCL-2 family protein inhibitors.
Binding profile of proapoptotic BH3-only and effector proteins to prosurvival proteins and how prosurvival proteins are targeted by BCL-2 family protein inhibitors.
Two main forms of apoptosis induction have been proposed. (1) In the direct activation model, BH3-only proteins act either as activators (eg, BIM, Bid/tBid, PUMA) or sensitizers (eg, BAD, NOXA). Activators bind to both prosurvival and BAX/BAK-like proteins, leading to integration into the outer mitochondrial membrane. Prosurvival proteins such as BCL-xL can sequester BH3-only proteins and thereby prevent BAX activation. Sensitizers bind directly to the prosurvival protein and mediate the release of activators. (2) The indirect (displacement) model assumes that BAX and BAK are constitutively active but are bound to prosurvival proteins to prevent apoptosis. BH3-only proteins displace BAX and BAK upon death stimulus. Because BH3-only proteins have different binding affinities to each prosurvival protein, apoptosis initiation requires the activation of several BH3-only proteins to displace sufficient amounts of BAX and BAK to cause MOMP. Over the last decade, it became apparent that these 2 models likely coexist and operate in a context-specific manner, leading to a unified model in which prosurvival proteins sequester both BH3-only proteins and also activated BAX and BAK (Figure 2).38 The fact that BH3-only proteins have different binding modalities for each prosurvival protein provides an opportunity to target the prosurvival proteins with pharmacologically selective inhibitors with great therapeutic potential in hematologic malignancies.40
Targeting the intrinsic apoptotic pathway in the clinic
Some cancer cells lie close to the apoptotic threshold but are held back from death by prosurvival BCL-2 family proteins. Others depend for survival on loss or reduction of proapoptotic proteins in the setting of increased proliferation, cell growth, and changes in dependence on the microenvironment. Both scenarios create a vulnerability in cancer cells which allows for a therapeutic window for agents targeting this pathway.
The mechanism by which prosurvival protein dependency increases is variable across different hematologic malignancies. In CLL, malignant cells harboring del(13q) express high BCL-2 protein levels due to loss of repression by microRNA (miRNA) 15/16,43 whereas MCL and MM cells are thought to express high BCL-2 due to dysregulation of cyclin proteins.44,45 In both FL and double-hit DLBCL, t(14;18) juxtaposes the BCL-2 gene to the immunoglobulin heavy chain (IgH) locus, leading to high BCL-2 protein levels.46,47 In addition to these internal abnormalities, microenvironmental factors also increase BCL-xL and/or MCL-1 expression in CLL, FL, and MM.48-51 The BCL-2 family lies downstream of tumor protein 53 (TP53), which is often dysfunctional in hematological malignancies, leading to resistance to cytotoxic chemotherapy. This makes the BCL-2 family an attractive therapeutic target in hematologic malignancies.
Several early efforts were made to target the BCL-2 pathway therapeutically, including antisense oligonucleotides, peptide inhibitors, and small molecule inhibitors (Table 1). The first major breakthroughs in targeting the apoptotic machinery directly were the dual BCL-2/BCL-xL inhibitor ABT-73752,53 and its orally bioavailable equivalent ABT-263 (navitoclax) (Figure 3).40,54 Although navitoclax showed remarkable efficacy in preclinical studies,52,55 thrombocytopenia due to inhibition of BCL-xL (an important prosurvival factor for megakaryocytes/platelets56,57 ) was a dose-limiting toxicity in the clinic.58
The first selective and highly potent BCL-2 inhibitor, venetoclax (VEN; formerly ABT-199/GDC-0199), was found to be active in preclinical testing of non-Hodgkin lymphoma (NHL) and AML cell lines as well as in primary CLL samples, irrespective of TP53 mutation status. In xenograft models, VEN induced significantly less thrombocytopenia than navitoclax.59,60 Given that CLL was known to depend on BCL-2 for cell survival,61 the first in human phase 1 study with VEN as monotherapy was conducted in patients with CLL and NHL.62 Table 2 provides an overview of results from selected venetoclax trials to date.
Clinical results of BCL-2 inhibition
Chronic lymphocytic leukemia
A detailed review on targeting BCL-2 in B-cell lymphomas including CLL was recently published.63 Hence, for this review, we summarize key points and review recent clinical updates.
In the phase 1 study, VEN monotherapy was highly active in relapsed/refractory (R/R) CLL.62 The most critical safety issue was tumor lysis syndrome (TLS), resulting in 1 death and an episode of acute renal failure requiring dialysis. To mitigate TLS risk, the initial VEN dose was lowered, TLS monitoring and management were made more stringent, and a 5-week gradual ramp-up period was explored (starting at 20 mg per day, followed by weekly ramp-up to 50, 100, 200, and 400 mg per day). Using this approach, no clinical significant TLS has been observed across the early-phase VEN development program in CLL.64 Other common toxicities were mild gastrointestinal side effects and grade 3/4 neutropenia.2 Similar results were observed in a landmark phase 2 trial65 and led to the accelerated approval of VEN for patients with R/R CLL with del(17p) by the US Food and Drug Administration (FDA) in April 2016 and by the European Medicines Agency (EMA) in December 2016.
Although VEN was efficacious in CLL, the CR rate with monotherapy was low, mainly due to residual lymphadenopathy. As such, combination strategies were subsequently explored. A phase 1b study of VEN in combination with rituximab (R) in R/R CLL showed remarkable efficacy.66 Building on this promising experience, the phase 3 registrational MURANO trial of VEN plus R (VR) vs bendamustine (B) plus R (BR) was conducted. The ORR and CR/CRi (CR with incomplete blood count recovery) rates in the VR arm were 93.3% and 26.8% vs 67.7% and 8.2% in the BR arm, with 62.4% achieving blood minimal residual disease (MRD) negativity with VR vs 13.3% with BR. Despite the elevated risk for grade 3/4 neutropenia in the VR group, no increase in grade 3/4 infections was observed. A small increase in the occurrence of laboratory TLS was observed in the VR arm vs the BR arm.67 The results of this study recently led to the full FDA approval of VEN in R/R CLL in June 2018.
The GP28331 phase 1b study of VEN in combination with the newer type II CD20 monoclonal antibody obinutuzumab (G, for GA-101) in previously untreated patients with CLL found 74% of patients achieving marrow MRD negativity.68 Building on this experience, the randomized, phase 3 CLL14 trial of VEN plus G vs Chl plus G was launched and has now completed accrual, with very early safety and efficacy data looking encouraging.69
Another promising strategy is to combine VEN with inhibitors of Bruton tyrosine kinase (BTK) such as ibrutinib (Ibr). BTK inhibitors increases BCL-2 dependence both in vitro and in vivo, thereby sensitizing CLL cells to VEN.70,71 The CLARITY trial testing the combination of 8 weeks of Ibr monotherapy followed by combination with VEN in patients with R/R CLL reported a CR/CRi rate of 60%, an ORR of 100% and a MRD negativity rate of 28% in BM in an interim analysis.72 A similar study assessed the combination of 12 weeks of Ibr followed by combination with VEN with encouraging efficacy. Adverse events (AEs) in both studies were consistent with the known toxicities of the individual agents.73 A 3 drug combination VEN, Ibr, and G is also currently being tested in patients with TN and R/R CLL with promising preliminary results.74
Non-Hodgkin lymphoma
In the phase 1 trial, VEN monotherapy was active in multiple NHL subtypes.75 The highest response rates were observed in patients with MCL (ORR, 75%; CR, 21%) and FL (ORR, 38%; CR, 14%). All 4 patients with WM responded, and VEN monotherapy is now being evaluated in a phase 2 study in R/R WM (NCT02677324).75-77
Given the modest efficacy of VEN as a single agent in NHL compared with CLL, the importance of exploring combination approaches is crucial. One such combination study is evaluating VEN plus BR in R/R NHL. Preliminary results in patients with MZL (ORR, 100%; 50% CR) and FL (ORR of 75%, 34.4% CR) do suggest the VEN may augment the activity expected with BR alone,78 though larger comparative studies are needed to confirm this. Another promising combination is VEN plus Ibr in R/R and TN MCL, where a phase 2 study found that after 16 weeks, the ORR was 71% with a CR of 62%, and 93% of evaluated CRs being MRD negative by flow cytometry. The 18-month estimates of progression-free survival (PFS) and overall survival (OS) were 57% and 74%, respectively.79 Similar regimens are being tested in R/R FL (NCT02956382).
Several studies are also testing the capacity of VEN as a chemo-sensitizing agent in combination with already established chemo regimens, such as rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) and G-CHOP in R/R NHL. A preliminary analysis found not only a high CR rate, but also a high incidence of grade 3/4 neutropenia and thrombocytopenia, especially in the VEN plus G-CHOP arm. This led to a change in dosing schedule in VEN from a continuous administration to an abbreviated course to mitigate neutropenia and thrombocytopenia.80 A similar approach is being evaluated in 2 studies of VEN and dose-adjusted rituximab plus etoposide, prednisone, vincristine, cyclophosphamide, and doxorubicin (R-EPOCH), 1 in patients with aggressive B-cell lymphomas such as double-hit lymphoma (NCT03036904) and the other in patients with CLL with Richter syndrome DLBCL (NCT03054896).
Multiple myeloma
Human MM cell lines and primary MM samples are highly sensitive to VEN in vitro, particularly MM cells harboring the t(11:14)(q13:q32) translocation, which is present in 15% to 20% of all MM patients.44 This was later verified in a phase 1 study of VEN monotherapy, where the ORR was significantly higher (24%) in patients harboring t(11:14) vs 4% in patients without.81 Patients with t(11:14) expressed a high BCL2:MCL1 ratio in their MM cells, which may have made their disease more VEN sensitive.50
Dexamethasone (DEX) plus VEN was found to induce greater BIM release from BCL-2 than VEN alone, resulting in greater activation of BAX/BAK.82 A phase 1 study evaluating the combination of DEX plus VEN in patients with t(11:14) R/R MM showed an ORR of 65% (13 of 20) with 7 very good partial responses (VGPR) and 6 PRs, and greater ORR was observed in the patients refractory to bortezomib (82%, 9 of 11) and lenalidomide (71%, 12 of 17). Grade 3/4 AEs included lymphopenia, hypophosphatemia, and hyperuricemia.83
Proteasome inhibitors such as bortezomib induce apoptosis by upregulation of NOXA and cleavage of MCL-1,50,84 hence there is a strong rationale for combining VEN and bortezomib in MM patients. In a parallel phase 1b study, patients with R/R MM were treated with VEN in combination with bortezomib and DEX. The ORR was 67% with 43% achieving VGPR or better. Grade 3/4 AEs observed were thrombocytopenia, anemia, and neutropenia. In patients who were not refractory to bortezomib and had 1 to 3 prior therapies, the ORR was 97% and 73% achieved VGPR. Patients with high BCL-2 expression assessed by droplet digital polymerase chain reaction (ddPCR) had a high ORR of 94%, while patients with low BCL-2 expression had an ORR of 59%.85 The combination of bortezomib and DEX with or without VEN is being explored in an ongoing phase 3 trial (NCT02755597). New trials examining VEN combinations (including cobimetinib, VEN with or without atezolizumab [NCT03312530], daratuzumab, VEN, DEX, with or without bortezomib [NCT03314181] and carfilzomib, VEN and DEX [NCT02899052]) are also under way.
Acute myeloid leukemia
Promising preclinical data on VEN in AML cell lines, primary patient samples, and murine primary xenografts found that the drug killed in the low nanomolar range.86 VEN monotherapy at 800 mg daily was assessed in a phase 2 study of 32 patients with R/R or TN AML, who were deemed unfit for intensive induction chemotherapy. The CR/CRi was 19% and antileukemic activity was seen in another 19% of patients not meeting International Working Group (IWG) criteria. In the group of patients with isocitrate dehydrogenase (IDH1/2) mutations, a higher CR/CRi rate of 33% was achieved, whereas no TLS was observed.87
In preclinical studies on AML cells, hypomethylating agents such as DNA methyltransferase inhibitor 5-azacytidine (5-AZA) decrease MCL-1 expression, providing a strong rationale for testing this combination clinically.88,89 A phase 1b study was conducted in older patients with TN AML with VEN plus azacitidine, VEN plus decitabine, or VEN plus decitabine and the CYP3A inhibitor posaconazole to explore whether this would boost the activity of VEN, which is a CYP3A substrate. Sixty-one percent of patients achieved CR or CR with incomplete BM recovery (CRi), which compares favorably with historical results with single agent hypomethylating agents (25.6% and 27.8% with either decitabine or azacitidine alone).90-92 The combination was a successful bridging strategy to allogenic stem cell transplantation for patients unfit for induction chemotherapy.
The efficacy and safety of VEN with low-dose cytarabine have been established in elderly patients with TN AML ineligible for induction therapy with CR/CRi of 62% and a median OS of 11.4 months.93 Another promising approach is combining VEN with MEK and MDM2 inhibition to decrease MCL-1 expression.94,95 An ongoing phase 1 study is evaluating VEN in combination with MEK inhibitor Cobimetinib (Cobi) or MDM2 inhibitor Idasanutlin (Idasa) in R/R AML. In a preliminary analysis of VEN plus Cobi or VEN plus Idasa, an CR/CRi/CRp (CR with incomplete platelet recovery) of 18% and 15% were observed, respectively.96 Based on these promising studies, VEN received its third breakthrough therapy designation from FDA for TN AML patients unfit for chemotherapy. The potential of VEN as a chemo-sensitizing agent will soon be tested in combination with chemotherapy regimens such as 7+3 and FLAG-IDA (NCT03214562).
Myelodysplastic syndrome
Higher levels of BCL-2 protein are observed in high-risk myelodysplastic syndrome (MDS) compared with low-risk MDS.97,98 Furthermore, the expression ratio between BAX/BAD:BCL-2/BCL-xL inversely correlates with both the International Prognostic Scoring System (IPSS) score and the cytogenetic risk group.99 BCL-2 inhibition by navitoclax and VEN was effective in primary bone marrow samples from high-risk MDS patients, whereas samples from low-risk MDS patients were less sensitive.98 Two phase 1 studies are currently evaluating the efficacy of VEN in combination with AZA in TN high-risk MDS (NCT02942290) and VEN alone or in combination with AZA in high-risk MDS patients after hypomethylating agent failure (NCT02966782).
Acute lymphoblastic leukemia
BCL-2 dependence has been demonstrated in both ALL cell lines and patient samples,100 and high BCL-2 expression is associated with slow response to therapy.101 ALL cells express higher levels of BCL-2 and BCL-xL than normal B cells, and therefore dual inhibition of BCL-2 and BCL-xL may be beneficial. Mixed lineage leukemia–rearranged infant ALL appears to be an exception because the activity of VEN and navitoclax are comparable.102 New trials have recently been launched to explore the activity of VEN in ALL patients. These include a study of VEN plus navitoclax as chemo-sensitizing agents in pediatric and adult patients with R/R ALL (NCT03181126) and VEN with or without chemotherapy in pediatric and young adult patients with R/R malignancies including ALL (NCT03236857). Furthermore, a study of VEN plus chemotherapy as frontline therapy is under way for older patients with ALL (NCT03319901).
Other hematologic malignancies
BCL-2 protein expression increases during CML blast crisis.103 Interestingly, tyrosine kinase inhibitors (TKIs) used to treat CML decrease MCL-1 expression, and the combination of BCL-2 and TKI inhibition eradicates CML progenitor stem cells.104 Therefore, dual BCL-2/TKI inhibition may represent a strategy to allow TKI discontinuation, though to our knowledge this approach has not yet entered clinical trials. Recently, blastic plasmacytoid dendritic cell neoplasm (BPDCN) cells and xenografts were found to be BCL-2 dependent and sensitive to VEN. Furthermore, clinical response was seen in 2 patients with R/R BPDCN receiving VEN off label.105 BCL-2 inhibition is also being explored in immunoglobulin light chain (AL) amyloidosis as a phase 1 trial (NCT03000660), as it has been shown that AL bone marrow plasma cells (BMPCs) express high levels of BCL-2.106
Resistance to BCL-2 inhibition and strategies to overcome it
Although VEN has efficacy across a range of hematologic malignancies, many patients do not respond or do respond but subsequently progress. Therefore, understanding mechanisms of VEN resistance is crucial for optimizing therapy. One hypothesis is that VEN monotherapy induces a shift in dependence on prosurvival proteins. In a model assessing changes in protein expression, BCL-xL and BFL-1 modulation appears to be 1 mechanism underlying the development of VEN resistance in primary CLL cells.107 Another possible mechanism relates to the observation that CD40 and IL-4 stimulation resembling T-cell stimulation in vitro increases BFL-1, BCL-xL, and MCL-1 expression in CLL cells and can be reversed by anti-CD20 monoclonal antibodies (mAbs) and BCR pathway kinase inhibitors.51 Similarly, continuous BCR stimulation has been found to increase MCL-1 expression and decrease BIMEL, HRK, and BMF, which can be counteracted by BCR pathway kinase inhibitors.108 Resistance mechanisms may differ depending on compartment; for example, the lymph node microenvironment suppression of NOXA may contribute to the persistence of CLL cells in that site.109 These studies provide important preclinical evidence that combining VEN with BCR pathway inhibitors in CLL may help overcome resistance to VEN monotherapy. In NHL, cell lines exposed to long-term VEN have increased AKT activation and expression of MCL-1 and BCL-xL, suggesting another possible resistance mechanism.110
Although the combination of VEN with Ibr has been active across a range of B-cell NHL, resistance mechanisms to this combination regimen have also already been described. For example, microenvironmental agonists may generate VEN plus Ibr resistance through activation of NF-kB signaling in MCL and CLL cells, thus leading to enhanced expression of MCL-1, BCL-xL and survivin.111 The fact that resistance mechanisms have already been described for dual blockade of BCL-2 and BTK suggests that there is still much that we do not know about this complex biology, and highlights that correlative studies from patients on such combination trials will be critical to help identify new targetable vulnerabilities in resistant cancer cells.
Beyond shifting prosurvival protein dependence, other potential resistance mechanisms for BCL-2 therapy may include genetic changes such as in residue F101 which blocks VEN from binding112 or copy number increases,113 post-translational modification such as ubiquitination114 or other functional alterations, such as activation of aberrant signaling. For example, mutation in the BCL-2 BH3 domain can abolish VEN binding, and mutation in BAX transmembrane domain can prevent mitochondrial translocation.112 Others have reported mutations in BTG1 and homozygous deletions of CDKN2A/B after relapse on VEN.115 Altered phosphorylation of BCL-2 family might be another functional resistance mechanism.116 AML cells resistant to ABT-737 have increased levels of phospho-BCL-2, and phospho-ERK and phospo-BIM have been demonstrated in VEN resistant FL cell lines.117,118
Selectively targeting MCL-1
MCL-1 dysregulation likely contributes to drug-resistance in a range of hematologic malignancies. Recent advances have been made in generating molecules that target MCL-1 either directly or indirectly (Figure 3). One of the first MCL-1 inhibitors developed, maritoclax, causes reduction in MCL-1 protein by targeting the protein for proteasomal degradation and activates cell death in a caspase-dependent manner. It was shown to induce cell death within a micromolar range in primary cells from patients with AML and MM. However, follow-up studies showed that manitoclax could induce cell death even in cells lacking MCL-1, suggesting multiple mechanisms of cell death induction and a lack of specificity. Other compounds that target MCL1 include UMI-77,119 ML311,120 A-1210477,121 and S63845, with these later 2 discovered by using fragment based NMR screening.122,123 One derivate of S63845, S64315/MIK655, is now in clinical trials in AML and MDS (NCT02979366) as well as R/R MM and lymphoma (NCT02992483). Other MCL-1 inhibitors such as AZD-5991 and AMG-176 have recently entered clinical trials (NCT03218683/NCT02675452).
In addition to direct MCL-1 inhibition, agents that indirectly lead to decreased MCL-1 protein levels such as CDK inhibitors like flavopiridol and recently more selective CDK9 inhibitors such as voruciclib124 may have potential in overcoming MCL-1–mediated resistance to BCL-2 inhibition in the clinic. Due to its short half-life, MCL-1 protein levels also decrease rapidly after treatment with several other compounds including MEK inhibitors,94 splicing modulators,125,126 SYK inhibitors,108 and XPO1-selective inhibitors.127
Selectively targeting BCL-xL
In CLL and MZL, BCL-xL protein upregulation has been observed in response to VEN monotherapy128,129 Therefore, targeting BCL-xL has clinical potential if the prior issues with BCL-xL-mediated thrombocytopenia seen with navitoclax can be mitigated. Intermittent scheduling of drug dosing may allow safer use of BCL-xL inhibitors. BCL-xl inhibitors do not inhibit granulopoiesis and should therefore not cause neutropenia, one of the most common toxicities of VEN.130 The first selective BCL-xL inhibitor, WEHI-539, was discovered via high-throughput screening and induces BAK-dependent apoptosis (Figure 3).131 Drugs such as A1155463 and the next generation compound A1331852 were developed using fragment-based approaches and have shown promising results in preclinical studies.132 However, these compounds have not yet entered clinical trials.
BH3 profiling
Assays that aid us in better understanding resistance mechanisms and in predicting clinical response to agents targeting the BCL-2 family would be helpful to optimize therapy. BH3 profiling is one such test developed as a functional assay to interrogate the interactions of BCL-2 family members.41 The profile determines the proximity of a cell to the apoptosis threshold, known as ‘mitochondrial priming’, and also identifies the specific prosurvival proteins on which a cell depends for its survival.133,134 BH3 profiling on blood samples from CLL patients on the phase 1 study of VEN found that pretreatment CLL cells were uniformly dependent on BCL-2, and that mitochondrial priming was associated with the depth of disease reduction in BM and PB.135 In contrast to CLL, primary AML and MM cells are more heterogeneous, with survival related to BCL-2, BCL-xL, MCL-1, or a mixture of these, depending on the sample.45
A new technique known as dynamic BH3 profiling (DBP) measures the early net proapoptotic signaling induced by a drug treatment (Figure 4). DBP takes advantage of the fact that alterations in mitochondrial priming are evident well before cancer cells undergo apoptosis in response to drug treatment.136 DBP results have been found to correlate with response to treatment in patients with a variety of hematologic malignancies.105 Furthermore, DBP has proven to be a useful method for predicting promising novel agent combination regimens such as VEN plus Ibr in CLL.71 This assay has the potential to become a useful tool for developing personalized patient treatments and schedules.
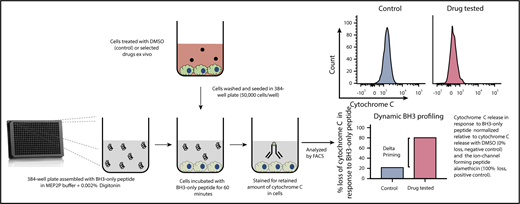
Dynamic iBH3 profiling. A single-cell suspension is obtained from a cell line or primary cell sample. These cells are exposed to apoptosis-inducing drugs of interest for a short period of time. Cells are then gently permeabilized using digitonin to allow BH3 peptide uptake. Apoptotic priming is measured via cytochrome C retention using flow cytometry. By comparing the retained cytochrome C in nontreated vs treated cells, drug efficacy can be determined. The higher the release of cytochrome C the more effective the drug. This assay can be further used to measure potential shifts in prosurvival protein dependency.
Dynamic iBH3 profiling. A single-cell suspension is obtained from a cell line or primary cell sample. These cells are exposed to apoptosis-inducing drugs of interest for a short period of time. Cells are then gently permeabilized using digitonin to allow BH3 peptide uptake. Apoptotic priming is measured via cytochrome C retention using flow cytometry. By comparing the retained cytochrome C in nontreated vs treated cells, drug efficacy can be determined. The higher the release of cytochrome C the more effective the drug. This assay can be further used to measure potential shifts in prosurvival protein dependency.
Future directions
After decades of painstaking research on the BCL-2 family, the approval of VEN in CLL heralded a new era in targeting the intrinsic pathway of apoptosis therapeutically. This important development is only the first step in what is likely to be a powerful new approach to cancer therapeutics that spans across hematologic malignancies and possibly even solid tumors. Eventually, clinicians will likely have access to a toolkit of specific inhibitors of BCL-2, MCL-1, BCL-xL and possibly BFL-1. A major challenge will be selecting the best regimen for an individual, particularly since the optimal regimen may evolve over time, even in an individual patient. A model for how to approach this challenge is highlighted in Figure 5. Genomic and/or functional assays such as BH3 profiling may be used to help select an initial therapy to target the prosurvival proteins on which an individual’s cancer depend at baseline, and testing can be repeated in patients with residual disease or early progression, with treatment adapted to account for changes in prosurvival protein dependencies by either switching agents or adding in new agents. This approach has the potential to maximize therapeutic efficacy for patients whose tumors may require multidrug therapy, while minimizing the toxicities associated with combination approaches in patients whose tumors are sensitive to single-agent therapy. Given the fundamental nature of the intrinsic pathway of apoptosis for cellular survival, this approach may help to overcome resistance to therapies targeting other important pathways that lie upstream of the mitochondria, and would thereby have potential to improve outcomes substantially for patients with hematologic malignancies.
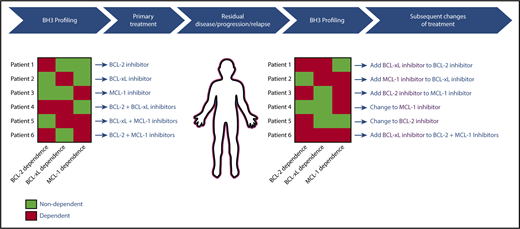
Theoretical model for how BH3 profiling could be used to guide clinical treatments with BCL-2 family protein inhibitors.
Theoretical model for how BH3 profiling could be used to guide clinical treatments with BCL-2 family protein inhibitors.
Authorship
Contribution: R.V., S.G., and M.S.D. wrote the paper.
Conflict-of-interest disclosure: R.V. has received travel support from Abbvie and Roche. M.S.D. has received institutional research support from Genentech, Pharmacyclics, TG Therapeutics, Bristol-Myers Squibb, MEI Pharma, and Surface Oncology, and has received consulting fees from Abbvie, Roche, Genentech, Pharmacyclics, Janssen, TG Therapeutics, Gilead, Astra-Zeneca, Merck, InCyte, Verastem, and MEI Pharma. S.G. declares no competing financial interests.
Correspondence: Matthew S. Davids, Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, MA 02215; e-mail: matthew_davids@dfci.harvard.edu.