

Key Points
Autologous HSCT induces functional renewal of regulatory T cells as well as a strong Treg TCR diversification in autoimmune patients.
Adding regulatory T cells to the graft does not lead to additional clinical improvement but results in delayed donor T-cell reconstitution.
Abstract
Autologous hematopoietic stem cell transplantation (HSCT) is increasingly considered for patients with severe autoimmune diseases whose prognosis is poor with standard treatments. Regulatory T cells (Tregs) are thought to be important for disease remission after HSCT. However, eliciting the role of donor and host Tregs in autologous HSCT is not possible in humans due to the autologous nature of the intervention. Therefore, we investigated their role during immune reconstitution and re-establishment of immune tolerance and their therapeutic potential following congenic bone marrow transplantation (BMT) in a proteoglycan-induced arthritis (PGIA) mouse model. In addition, we determined Treg T-cell receptor (TCR) CDR3 diversity before and after HSCT in patients with juvenile idiopathic arthritis and juvenile dermatomyositis. In the PGIA BMT model, after an initial predominance of host Tregs, graft-derived Tregs started dominating and displayed a more stable phenotype with better suppressive capacity. Patient samples revealed a striking lack of diversity of the Treg repertoire before HSCT. This ameliorated after HSCT, confirming reset of the Treg compartment following HSCT. In the mouse model, a therapeutic approach was initiated by infusing extra Foxp3GFP+ Tregs during BMT. Infusion of Foxp3GFP+ Tregs did not elicit additional clinical improvement but conversely delayed reconstitution of the graft-derived T-cell compartment. These data indicate that HSCT-mediated amelioration of autoimmune disease involves renewal of the Treg pool. In addition, infusion of extra Tregs during BMT results in a delayed reconstitution of T-cell compartments. Therefore, Treg therapy may hamper development of long-term tolerance and should be approached with caution in the clinical autologous setting.
Introduction
Even in the era of biologicals, some patients with autoimmune diseases (AIDs) remain therapy refractory. For these severely ill patients, autologous hematopoietic stem cell transplantation (HSCT) is the only treatment able to induce long-term drug- and symptom-free remission.1-5 Because stem cell therapy is not expected to be a mainstream treatment, studies to explore how stem cell therapy resets the immune balance are pivotal. Insights in this mechanism may yield new therapeutic options that achieve the same goal (disease- and medication-free remission, but with reduced toxicity).
The general concept of autologous HSCT for AID is that immune reconstitution after profound lymphodepletion and immune suppression leads to restoration of the immune balance and immune tolerance.6 How the procedure rewires a faulty immune system is still unknown. For example, it is not clear which cells must be destroyed prior to transplantation, nor is it known which ones keep disease under control afterward.7 Immediately after reinfusion through HSCT, the lymphopenic environment induces selective expansion and activation of the few (potentially autoreactive) T-cell clones that have survived the conditioning regimen or may have been retransferred with the graft.8 Therefore, lymphopenia-induced proliferation and activation of T cells may pose a risk of loss of self-tolerance early after HSCT. The second phase of T-cell reconstitution starts when T cells develop in the thymus and naive T cells are introduced in the periphery,9 contributing to the resetting of the immune system.8 In humans, CD4+ T-cell receptor (TCR) sequencing has confirmed selective expansion of several TCR clones directly after treatment, followed by broadening of the total TCR repertoire during follow-up due to thymus output.10,11 During both reconstitution phases, regulatory T cells (Tregs) may be essential to control T-cell reconstitution and activation, but little is known about the renewal of the Treg compartment.
Tregs are thought to be important players for disease remission in autologous HSCT-treated AID, especially in the lymphopenic reconstitution phase when the delicate immune balance has to be re-established.12,13 In children with refractory juvenile idiopathic arthritis (JIA), it has been shown that prior to HSCT, Treg blood levels are decreased, whereas after HSCT, Treg levels are comparable to healthy controls.14-16 Furthermore, we have shown in an experimental arthritis model that depletion of Tregs after congenic bone marrow transplantation (BMT) results in an improvement of arthritis scores, suggesting a pivotal early role for Tregs in disease remission.17
Expansion of the Treg compartment following HSCT starts with lymphopenia-induced proliferation followed by thymic output of stem cell–derived Tregs.13 What remains to be established is the relative contribution of stem cell graft-derived Tregs vs conditioning-survived host Tregs in controlling T-cell activation and disease remission. This knowledge is of increasing importance after the recent introduction of less toxic nonmyeloablative conditioning regimens that may limit the role of the graft.7 We hypothesized that graft-derived Tregs are the main contributors in the restoration of a functional Treg compartment following autologous HSCT. In the human transplantation setting, it is impossible to study the relative contribution due to the autologous nature of the treatment. Here, we tested our hypothesis, distinguishing “host” and “donor” Treg dynamics and function following BMT in an experimental AID setting by using a congenic marker. To translate our findings to the human setting, we performed Treg TCR β-chain (TCRβ) sequencing in autoimmune patients undergoing autologous HSCT. In addition, we addressed whether the infusion of additional Tregs during BMT led to the suppression of residual potentially harmful effector T cells.
Material and methods
Mice
CBy.PL(B6)-Thy1a.ScrJ (CD90.1, The Jackson Laboratory) mice were used for bone marrow (BM) grafts or served as recipients when indicated. Foxp3-IRES-GFP mice (CD90.2) were obtained for the isolation of Tregs. Female retired breeder BALB/c mice (CD90.2) were acquired from Charles River Laboratory, and offspring of crossed CD90.1 and CD90.2 mice were bred in-house. Both served as recipients. All mice are on a BALB/c background.
Mice were kept in the Utrecht University Animal Facility under regular conditions. After congenic BMT, recipient mice were accommodated under sterile conditions. All experiments were approved by the Animal Experiment Ethical Committee of the University of Utrecht.
Induction and assessment of arthritis.
Both have been described in detail earlier.18,19 In short, 2 and 5 weeks before BMT, arthritis was induced by 2 intraperitoneal injections of proteoglycan together with adjuvant DDA (dimethyldioctadecylammonium bromide). The onset and severity of arthritis were assessed 3 times a week in a blinded fashion using a visual scoring system.17
Treatment protocols
Congenic bone marrow transplantation.
Two weeks after the second proteoglycan/DDA injection, mice were lethally irradiated (7.5 Gy). Within 6 hours of irradiation, mice were injected with 2 × 106 BM cells. As the genetic background of BM was identical except for the congenic T-cell marker, the term “congenic BMT” is used throughout this paper.
BM suspension.
BM was acquired by flushing the tibia and femur bones. BM cells were resuspended in 200 μL 0.2% bovine serum albumin before injection into the tail vein. The mean ± standard deviation percentage of T cells present in BM was 2.05% ± 0.3%.
Infusion of extra Tregs.
After sacrificing Foxp3-IRES-GFP mice, spleens and joint-draining (inguinal and popliteal) lymph nodes (LNs) were harvested. Cells were harvested by pushing them through a cell strainer, after which CD4+ T cells were positively selected via magnetic cell-sorting beads (L3T4, Miltenyi Biotec). Tregs were stained and isolated as TCRβ+CD4+CD25+GFP+ T cells on a FACS Aria II (BD Biosciences).
In vitro assays
Flow cytometry.
A total of 1, 3, 5, and 7 weeks after BMT, thymus, spleen, blood, and joint-draining LN cells were harvested when indicated. In addition, 7 weeks posttransplantation, synovial fluid was obtained by needle aspiration of the knee and ankle joints. Cells were stained with antibodies against TCRβ (clone H57-597), CD25 (clone PC61), CD90.1 (clone OX-7), Ki-67 (clone B56), CD45RB (clone 16A), CD44 (clone IM7) (BD Biosciences, San Jose, CA), CD4 (clone RM4-5), CD90.2 (clone 53-2.1), and Foxp3 (clone FJK-16a) (eBioscience, San Diego, CA)
To calculate absolute T-cell numbers, BD Tru Count Beads were added just before the acquisition of the samples. Data were analyzed with FACS Diva (6.13, BD Biosciences).
Foxp3 demethylation assay.
Seven weeks after BMT, spleen-derived Tregs were isolated with flow cytometry as TCRβ+CD4+CD25+ cells and distinguished for host cells (CD90.2+) or donor cells (CD90.1+). DNA from these cells was isolated using the DNeasy Blood & Tissue kit (Qiagen) and bisulfate converted applying the EpiTect Bisulfate kit (Qiagen Hilden) following the manufacturer’s recommendations. The demethylation assay was performed by Epiontis GmbH (Berlin, Germany).20
Suppression assay.
Seven weeks after BMT, spleen cells were harvested and enriched for CD4+ T cells by CD4 microbead isolation. The negative fraction was used as antigen-presenting cells in the culture. Host and donor Tregs were separated with flow cytometry. Carboxyfluorescein diacetate succinimidyl ester–labeled TCRβ+CD4+CD25− spleen cells from healthy BALB/c mice were used as effector T cells. Tregs were added in different ratios to the effector T cells. Soluble anti-CD3 (1 μg/mL, clone 145-2c11; BD Pharmingen, San Diego, CA) was used as a stimulus. At day 4, the proliferation of effector T cells was analyzed with flow cytometry.
In Figure 5B, host and donor Tregs originated from CD90.1 origin and Foxp3-IRES-GFP Tregs expressed CD90.2.
For the Foxp3 demethylation, suppression, and quantitative polymerase chain reaction (qPCR) assays, only a relatively small amount of material was available. Therefore, mice in the same groups were combined to obtain enough material for the assays.
Real-time qPCR
After RNA extraction from lysed host and donor Tregs, real-time qPCR was performed for Helios, neuropillin-1, interleukin-10 (IL-10), and mGAPDH (for a list of primers, see supplemental Table 1, available on the Blood Web site). Gene expression was calculated as CT ((2−dCT) × 100) with mGAPDH as housekeeping gene.
Next-generation TCRβ sequencing analysis
Frozen PBMCs from autologous-HSCT–treated patients with juvenile dermatomyositis (patients 1 and 2) and JIA (patients 3 and 4) and 4 healthy controls were thawed. Samples were obtained in accordance with the Declaration of Helsinki. Tregs (CD3CD4CD25highCD127low) and non-Tregs (CD3CD4CD25low/mediumCD127medium/high) were sorted (between 9.700 and 48.500 Tregs and between 190.000 and 1 × 106 non-Tregs were obtained) and frozen at −80°C. On average, 74.1% of the sorted patient Tregs expressed FOXP3.
Total RNA was isolated using the RNeasy Mini Kit (Qiagen) for cell fractions >0.2 × 106 cells and the RNeasy Micro Kit (Qiagen) for fractions <0.2 × 106 cells following the manufacturer’s instructions. cDNA was synthesized using the SMARTer RACE cDNA Amplification kit (Clontech). Amplification of the TCRβ VDJ region was performed using previously described primers and amplification protocols.21 PCR products were analyzed with a QIAxcel Advanced System (Qiagen). Upon successful amplification, end repair was performed with the ClaSeek Library Preparation kit, Illumina compatible (Thermo Scientific). Subsequently, TruSeq Barcode adapters (Illumina) were ligated using the ClaSeek Ligation Mix (Thermo Scientific) according the recommendations of the manufacturer. Cleanup of the samples was performed with The Agencourt AMPure XP system (Beckman Coulter). Next-generation sequencing was performed on an Illumina MiSeq system 500 (2 × 250 bp) (Illumina).
Sequencing data were analyzed with the MiTCR program.22 The MiTCR output file was used to calculated the Simpson’s index (D), 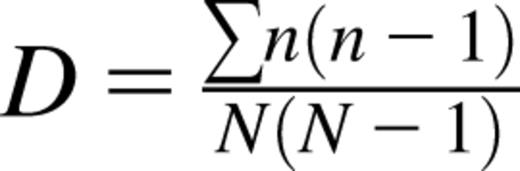 , where n is the total number of specific TCRβ VDJ sequences and N is the total number of all TCRβ sequences. Data are presented as the Simpson’s index of diversity (Di),
, where n is the total number of specific TCRβ VDJ sequences and N is the total number of all TCRβ sequences. Data are presented as the Simpson’s index of diversity (Di),  , where Di = 0 indicates no diversity and Di = 1 represents maximal diversity.
, where Di = 0 indicates no diversity and Di = 1 represents maximal diversity.
Multiplex assay
Spleen cells were obtained 7 weeks after transplantation. Supernatants of a 96-hour culture of 2 × 105 spleen cells with the addition of 1 μg/mL anti-CD3 were analyzed with a mouse cytokine multiplex kit (Bio-Rad). Analysis was performed using the Bio-plex manager software v4.1 (Bio-Rad).
Statistical analysis
To identify differences between BMT-treated proteoglycan-induced arthritis (PGIA) animals and untreated PGIA animals, the Mann-Whitney U test was used. To achieve normal distribution for cytokine data, logarithmic transformation was performed before applying the Mann-Whitney U test. Significant differences between host and donor cells were tested using the Wilcoxon matched-pairs signed rank test. All data are presented as the mean ± standard error of the mean (SEM) values (error bars). P values less than .05 were considered significant. Statistical analysis was performed using IBM SPSS Statistics Version 20.
Results
BMT renews the Treg compartment with donor-derived Tregs that directly home to the site of inflammation
To investigate the reconstitution of T cells after BMT, a congenic marker was used to distinguish host T cells (cells that survived conditioning, CD90.2+) from donor T cells (cells originating from the graft, CD90.1+). One week after BMT, most TCRβ+CD4+CD25+Foxp3+ Tregs present in thymus and spleen were of host origin (Figure 1A left and middle graphs). During the reconstitution phase 3 weeks following transplantation, the thymus harbored more donor-derived Tregs (50.3% ± 9.1) compared with the spleen (37.5% ± 4.8) and LN (38.6 ± 4.1) (Figure 1A). Seven weeks post-BMT, the majority of Tregs were of donor origin in all investigated organs (Figure 1A). As expected, similar reconstitution dynamics in the absolute number of donor-derived Tregs was seen post-BMT, whereas host cells remained relatively stable over time (supplemental Figure 1A).

Congenic BMT for arthritis renews the Treg compartment with donor-derived Tregs. Host T cells (black bar, CD90.2+) were distinguished from donor T cells (white bar, CD90.1+) using a different congenic marker. After transplantation, thymus, spleen, and LNs were analyzed at weeks 1, 3, and 7 for the presence of host and donor Tregs (TCRβ+CD4+CD25+Foxp3+). Results shown are from 2 combined experiments. (A) Percentage of host and donor Tregs. Thymus: 1 week, N = 3; 3 weeks, N = 6; and 7 weeks, N = 8. Spleen: 1 week, N = 2; 3 weeks, N = 6; and 7 weeks, N = 8. LN: 3 weeks, N = 6; and 7 weeks, N = 8. (B) Host and donor Treg distribution in synovial fluid 7 weeks post-BMT (N = 2). All results shown are in percentages (±SEM values). N.D., not determined due to lack of cells.
Congenic BMT for arthritis renews the Treg compartment with donor-derived Tregs. Host T cells (black bar, CD90.2+) were distinguished from donor T cells (white bar, CD90.1+) using a different congenic marker. After transplantation, thymus, spleen, and LNs were analyzed at weeks 1, 3, and 7 for the presence of host and donor Tregs (TCRβ+CD4+CD25+Foxp3+). Results shown are from 2 combined experiments. (A) Percentage of host and donor Tregs. Thymus: 1 week, N = 3; 3 weeks, N = 6; and 7 weeks, N = 8. Spleen: 1 week, N = 2; 3 weeks, N = 6; and 7 weeks, N = 8. LN: 3 weeks, N = 6; and 7 weeks, N = 8. (B) Host and donor Treg distribution in synovial fluid 7 weeks post-BMT (N = 2). All results shown are in percentages (±SEM values). N.D., not determined due to lack of cells.
Comparable results were obtained for total CD4+ T cells (supplemental Figure 1B-C). In thymus and spleen, host CD4+ T cells were most abundant directly after BMT. During reconstitution, the majority of donor CD4+ T cells were first detectable in the thymus, followed by spleen and LN in percentages and absolute numbers. It is important to realize that the absolute CD4+ T-cell number is very low at 1 week posttransplantation and increases over time.
To investigate if donor Tregs could home to the site of inflammation, synovial fluid was obtained of knee and ankle joints. The majority of synovial fluid Tregs were donor derived (Figure 1B).
These results indicate that BMT induces graft-derived renewal of the CD4+ T cell and Treg compartments through thymus-derived donor T cell reconstitution. Furthermore, donor-derived Tregs home to the joint, the site of inflammation.
Donor-derived Tregs have a more naive and functional phenotype than host-derived Tregs
Next, we explored the Treg phenotype following BMT. Three weeks after BMT, the minority of both host- and donor-derived Tregs expressed a naive phenotype (CD45RB+CD44−; Figure 2A and supplemental Figure 2A). Seven weeks posttransplantation, the percentage of naive Tregs increased in both the host and donor compartments, with a slightly higher rise in the donor-derived population (Figure 2A and supplemental Figure 2A). Both Treg populations showed increased proliferation compared with Tregs of PGIA control animals (Figure 2B and supplemental Figure 2B), and this increase was still observed for donor Tregs 7 weeks after BMT. Furthermore, at 7 weeks, Ki-67 expression was significantly higher in donor Tregs than host Tregs.

Post–congenic BMT, donor Tregs show a naive, proliferative, and anti-inflammatory phenotype. Host (circles) and donor (squares) Tregs (TCRβ+CD4+CD25+Foxp3+) were isolated from spleen and LN 3 and 7 weeks post-BMT from BMT-treated mice with arthritis and PGIA controls (pooled data from 3 and 7 weeks). Each Treg set, circle, and square joined together by the line in-between derive from the same mouse. Results shown are from 2 combined experiments. (A) Percentages host vs donor naive Tregs (CD45RBhigh, CD44low). Spleen (left): PGIA control (N = 7) and BMT-treated mice (N = 4) at 3 weeks (N = 5) and 7 weeks post-BMT. LN (right): PGIA control (N = 8) and BMT-treated mice (N = 4) at 3 weeks (N = 3) and 7 weeks. (B) Percentages host vs donor proliferative Tregs (Ki-67+). Spleen (left): PGIA control (N = 3) and BMT-treated mice (N = 4) at 3 weeks (N = 5) and 7 weeks. LN (right): PGIA control (N = 3) and BMT-treated animals (N = 4) at 7 weeks. (C) Host vs donor-derived spleen Tregs from PGIA+BMT-treated animals. Seven weeks after BMT, spleen Tregs (TCRβ+CD4+CD25+) were sorted and relative messenger RNA expression of Helios (left; N = 7, 10 mice) and neuropillin-1 (right; N = 7, 10 mice) was measured by qPCR. (D) Relative expression of the anti-inflammatory cytokine marker IL-10 messenger RNA in spleen Tregs (N = 7, 10 mice). *P < .05 calculated using Wilcoxon rank test. N.D., not determined due to lack of cells.
Post–congenic BMT, donor Tregs show a naive, proliferative, and anti-inflammatory phenotype. Host (circles) and donor (squares) Tregs (TCRβ+CD4+CD25+Foxp3+) were isolated from spleen and LN 3 and 7 weeks post-BMT from BMT-treated mice with arthritis and PGIA controls (pooled data from 3 and 7 weeks). Each Treg set, circle, and square joined together by the line in-between derive from the same mouse. Results shown are from 2 combined experiments. (A) Percentages host vs donor naive Tregs (CD45RBhigh, CD44low). Spleen (left): PGIA control (N = 7) and BMT-treated mice (N = 4) at 3 weeks (N = 5) and 7 weeks post-BMT. LN (right): PGIA control (N = 8) and BMT-treated mice (N = 4) at 3 weeks (N = 3) and 7 weeks. (B) Percentages host vs donor proliferative Tregs (Ki-67+). Spleen (left): PGIA control (N = 3) and BMT-treated mice (N = 4) at 3 weeks (N = 5) and 7 weeks. LN (right): PGIA control (N = 3) and BMT-treated animals (N = 4) at 7 weeks. (C) Host vs donor-derived spleen Tregs from PGIA+BMT-treated animals. Seven weeks after BMT, spleen Tregs (TCRβ+CD4+CD25+) were sorted and relative messenger RNA expression of Helios (left; N = 7, 10 mice) and neuropillin-1 (right; N = 7, 10 mice) was measured by qPCR. (D) Relative expression of the anti-inflammatory cytokine marker IL-10 messenger RNA in spleen Tregs (N = 7, 10 mice). *P < .05 calculated using Wilcoxon rank test. N.D., not determined due to lack of cells.
Helios and neuropilin-1 have been described as markers for thymic-derived Tregs.23-25 The amount of Helios or neuropilin-1 mRNA expression, associated with thymic-derived Tregs (Figure 2C left and right panels), did not differ between donor and host Tregs. In terms of functionality, significantly increased IL-10 mRNA expression was seen in donor-derived Tregs (Figure 2D).
These data indicate that both donor Treg and host Tregs initially expand vigorously after BMT. At a later stage, naive, donor-derived Tregs with a possible suppressive phenotype develop.
Congenic BMT induces a stable and functional donor-derived Treg compartment
The demethylation status from the Treg-specific demethylated region (TSDR),26 a highly conserved element within the first intron of the Foxp3 gene, was investigated to asses stability of the Treg (Figure 3A). The level of Foxp3 TSDR demethylation of donor Tregs was comparable to Tregs of PGIA control mice. However, host-origin Tregs showed less TSDR demethylation compared with donor-origin Tregs, suggesting that donor Tregs are more stable.

Donor-derived Tregs are stable and functional. (A) Demethylation status of the Foxp3 gene (TSDR) was analyzed from spleen host and donor Tregs (TCRβ+CD4+CD25+), isolated 7 weeks post-BMT from PGIA control and BMT-treated animals. PGIA control (N = 4) were pooled in 2 groups and BMT-treated mice (N = 5) in 3 groups (3 PGIA-induced mice, of which 2 were pooled in 1 group, and 2 nonarthritic-induced animals were pooled into 1 group) before measurement. (B) Representative carboxyfluorescein diacetate succinimidyl ester (CFSE) dilution histogram of suppressive capacity of fluorescence-activated cell-sorted host and donor Treg compared with proliferative capacity of effector T cells of healthy mice (left). Summarized suppressive capacity of host and donor Tregs after BMT at different effector T-cell:Treg ratios (1:1, 2:1, 4:1, and 8:1) (right). BMT-treated animals, N = 6 (4 PGIA-induced and 2 noninduced animals). Tregs were paired before the suppression assay. *P < .05, host vs donor (Mann-Whitney U test).
Donor-derived Tregs are stable and functional. (A) Demethylation status of the Foxp3 gene (TSDR) was analyzed from spleen host and donor Tregs (TCRβ+CD4+CD25+), isolated 7 weeks post-BMT from PGIA control and BMT-treated animals. PGIA control (N = 4) were pooled in 2 groups and BMT-treated mice (N = 5) in 3 groups (3 PGIA-induced mice, of which 2 were pooled in 1 group, and 2 nonarthritic-induced animals were pooled into 1 group) before measurement. (B) Representative carboxyfluorescein diacetate succinimidyl ester (CFSE) dilution histogram of suppressive capacity of fluorescence-activated cell-sorted host and donor Treg compared with proliferative capacity of effector T cells of healthy mice (left). Summarized suppressive capacity of host and donor Tregs after BMT at different effector T-cell:Treg ratios (1:1, 2:1, 4:1, and 8:1) (right). BMT-treated animals, N = 6 (4 PGIA-induced and 2 noninduced animals). Tregs were paired before the suppression assay. *P < .05, host vs donor (Mann-Whitney U test).
Subsequently, the suppressive function of host and donor Tregs was determined in a suppression assay. Donor-derived Tregs were efficient in suppressing proliferation of effector T cells (Figure 3B). Although host-derived Tregs were also able to suppress effector T cell proliferation, a significant difference was observed in the suppressive function of donor versus host Tregs, showing that donor Tregs were superior suppressors (Figure 3B).
These results reveal that a stable and functional donor-derived Treg compartment is formed post-BMT.
The oligoclonal Treg receptor repertoire becomes more diverse after successful autologous HSCT in refractory autoimmune patients
To translate our findings on the renewal of the Treg compartment to the human setting, we performed next-generation TCRBV CDR3 sequencing of Tregs and CD4+ non-Tregs in refractory autoimmune patients undergoing HSCT. Following HSCT, patients 1, 2, and 3 had achieved disease remission without medication (longest follow-up, 12 years), whereas patient 4 experienced disease relapse 6 months after transplantation (for patient characteristics, see supplemental Table 2).
Here, strikingly, the number of unique Treg TCR sequences (n) and TCR diversity (Di) prior to HSCT found for patients 1-4 was extremely low, ranging from n = 4 to 19 and Di = 0.114 to 0.687 (Figure 4A-B) compared with healthy controls (characteristics are listed in supplemental Table 3) with n = 225 to 320 and mean DI = 0.983 (range, 0.978-0.988) (Figure 4C). In addition, the non-Tregs showed a higher number of unique TCR sequences and higher Di calculations (Figure 4A-B). After HSCT, the number and diversity of Treg TCR sequences increased over time (patients 1, 2, and 3; Figure 4A-B). The non-Treg compartments showed an increase in TCR sequences as well, although the Di increase was not as pronounced as in the Tregs (Figure 4B). In the patient with disease relapse (patient 4), the Treg TCR repertoire even increased in oligoclonality following transplantation (n = 9, Di = 0.071) (Figure 4A-B). None of the predominant Treg and non-Treg TCR sequences existing prior to transplantation were retraceable in the same population after treatment (cutoff, 2 reads per sequence). Altogether, we conclude that successful autologous HSCT leads to a renewed and more diverse Treg TCR repertoire.

Successful autologous HSCT leads to a renewed and more diverse Treg TCR repertoire. (A) Tregs and non-Tregs from different blood collections of 4 HSCT-treated patients with refractory AID were sorted. These cell samples were used for TCR TCRβ sequencing. Per patient, each time point of blood sampling is shown. Per time point, 2 pie charts show the number and abundance of TCR sequences found for the Tregs and non-Tregs sorted from that cell sample. Color overlap between different pie charts does not represent the same TCR sequence. N represents the number of different TCR sequences found per sample, Di indicates the sample’s diversity (0 = no diversity, 1 = maximal diversity). (B) For patients 1 to 4, the changes in Di prior to HSCT and during follow up is shown in graphs. (C) TCRβ sequencing results of Tregs derived from 4 healthy controls are shown in a similar fashion as in panel A. aSCT, autologous stem cell transplantation; HC, healthy controls.
Successful autologous HSCT leads to a renewed and more diverse Treg TCR repertoire. (A) Tregs and non-Tregs from different blood collections of 4 HSCT-treated patients with refractory AID were sorted. These cell samples were used for TCR TCRβ sequencing. Per patient, each time point of blood sampling is shown. Per time point, 2 pie charts show the number and abundance of TCR sequences found for the Tregs and non-Tregs sorted from that cell sample. Color overlap between different pie charts does not represent the same TCR sequence. N represents the number of different TCR sequences found per sample, Di indicates the sample’s diversity (0 = no diversity, 1 = maximal diversity). (B) For patients 1 to 4, the changes in Di prior to HSCT and during follow up is shown in graphs. (C) TCRβ sequencing results of Tregs derived from 4 healthy controls are shown in a similar fashion as in panel A. aSCT, autologous stem cell transplantation; HC, healthy controls.
During BMT, injected Foxp3GFP+ Tregs are retraceable and remain functional over time in mice
Based on our findings in patients indicating the importance of renewal/diversification of the Treg compartment for disease remission after HSCT, we questioned if HSCT results could be improved by Treg infusion together with the graft for the suppression of proinflammatory residual effector T cells. To explore clinical improvement with the immediate presence of Tregs after BMT, Foxp3GFP+ Tregs were added in different numbers (250,000 or 500,000 cells) to the graft.
Longitudinal blood punctures were performed to examine if the infused Foxp3GFP+ Tregs were retraceable during the observation period. Three weeks post-BMT, Foxp3GFP+ Tregs comprised the majority of the total CD4+ T cell compartment. However, this prominence disappeared during the following weeks and did not differ between the 2 Foxp3GFP+ Treg treatment groups (Figure 5A left panel). The decrease in the percentage of Foxp3GFP+ Tregs coincided with an increase in the donor-derived CD4+ T-cell compartment (Figure 5A middle and right panels).

Extra Foxp3GFP+ Tregs infused at the time of BMT are retraceable during follow-up and remain functional. Arthritic mice were treated with 2 × 106 congenic BM cells (BMT). In addition, 2 treatment groups received 250,000 or 500,000, GFP+ Tregs in the BM graft. Blood was analyzed 1, 3, 5, and 7 weeks after BMT. (A) Percentages of additional infused Foxp3GFP+ Tregs (left), host TCRβ+CD4+ T cells (middle), donor TCRβ+CD4+ T cells (right). PGIA+BMT, N = 4; PGIA+BMT+250,000 Tregs, N = 5; PGIA+BMT+500,000 Tregs, N = 5. (B) Suppression assay of Tregs from PGIA+BMT-treated mice (white bar), non-GFP Tregs from mice treated with PGIA+BMT+250,000 (light gray), and Foxp3GFP+ Tregs from mice treated with BMT+250,000 Tregs (dark gray). Tregs were added in 1:2 and 1:10 ratios to healthy effector T cells (Teff). Tregs were pooled per treatment group before adding to the effector T cells. PGIA+BMT, N = 2 mice; PGIA+BMT+250,000 Tregs, N = 3.
Extra Foxp3GFP+ Tregs infused at the time of BMT are retraceable during follow-up and remain functional. Arthritic mice were treated with 2 × 106 congenic BM cells (BMT). In addition, 2 treatment groups received 250,000 or 500,000, GFP+ Tregs in the BM graft. Blood was analyzed 1, 3, 5, and 7 weeks after BMT. (A) Percentages of additional infused Foxp3GFP+ Tregs (left), host TCRβ+CD4+ T cells (middle), donor TCRβ+CD4+ T cells (right). PGIA+BMT, N = 4; PGIA+BMT+250,000 Tregs, N = 5; PGIA+BMT+500,000 Tregs, N = 5. (B) Suppression assay of Tregs from PGIA+BMT-treated mice (white bar), non-GFP Tregs from mice treated with PGIA+BMT+250,000 (light gray), and Foxp3GFP+ Tregs from mice treated with BMT+250,000 Tregs (dark gray). Tregs were added in 1:2 and 1:10 ratios to healthy effector T cells (Teff). Tregs were pooled per treatment group before adding to the effector T cells. PGIA+BMT, N = 2 mice; PGIA+BMT+250,000 Tregs, N = 3.
The Foxp3GFP+ Tregs present at the end of the observation period remained functional as shown in the suppression assay (Figure 5B). The suppressive capacity of Foxp3GFP+ Tregs was similar compared with total non-GFP+ Tregs of the same mice and of regular transplanted animals.
These results demonstrate that additional injected Foxp3GFP+ Tregs survive, expand, and remain functional.
The addition of Foxp3GFP+ Tregs to the graft does not improve clinical outcome but decreases proinflammatory cytokine production
Next, we questioned whether the addition of these Tregs led to an improvement of the clinical outcome. The arthritis scores of mice treated with Foxp3GFP+ Tregs were similar to those of mice treated with regular BMT (Figure 6A). To evaluate the clinical effect during the entire observation period, the area under the curve was calculated (Figure 6B). In both the PGIA+BMT and PGIA+BMT+250,000 Foxp3GFP+ Treg-treated groups, significant improvement in suppressing arthritis severity was seen compared with the untreated PGIA group. In the PGIA+BMT+500,000 Foxp3GFP+ Treg-dosing group, a similar but nonsignificant improvement was seen.

Additional Tregs in the graft reduces T-cell–produced proinflammatory cytokines but does not lead to a better clinical outcome. (A) Arthritis scores after transplantation. Arthritis scores were set to 100% on the day of transplantation, and the subsequent clinical effect was expressed as a percentage of the score at the time of transplantation. Mean arthritis scores are shown (±SEM error bars). Data are representative of 2 individually performed experiments. (B) Area under the arthritis score curve during the 7-week follow-up period. Mean area under the curve ± SEM error bars are shown. PGIA (black bar), N = 4; PGIA+BMT (white bar), N = 4; PGIA+BMT+250,000 Tregs (gray bar), N = 5; and PGIA+BMT+500,000 Tregs (dark gray bar), N = 5. *P < .05 compared to PGIA control group (Mann-Whitney U test). (C) Spleen cells were isolated 7 weeks after BMT and cultured in culture medium with the addition of anti-CD3 (1 μg/mL) for 96 hours. Supernatants were collected and analyzed with Multiplex Immuno Assay for interferon-γ (IFNγ), IL-17, IL-10, and tumor necrosis factor-α (TNFα) production. PGIA, N = 4; PGIA+BMT, N = 4; PGIA+BMT+250,000 Tregs, N = 5; and PGIA+BMT+500,000 Tregs, N = 5. *P < .05 compared with PGIA+BMT (Mann-Whitney U test).
Additional Tregs in the graft reduces T-cell–produced proinflammatory cytokines but does not lead to a better clinical outcome. (A) Arthritis scores after transplantation. Arthritis scores were set to 100% on the day of transplantation, and the subsequent clinical effect was expressed as a percentage of the score at the time of transplantation. Mean arthritis scores are shown (±SEM error bars). Data are representative of 2 individually performed experiments. (B) Area under the arthritis score curve during the 7-week follow-up period. Mean area under the curve ± SEM error bars are shown. PGIA (black bar), N = 4; PGIA+BMT (white bar), N = 4; PGIA+BMT+250,000 Tregs (gray bar), N = 5; and PGIA+BMT+500,000 Tregs (dark gray bar), N = 5. *P < .05 compared to PGIA control group (Mann-Whitney U test). (C) Spleen cells were isolated 7 weeks after BMT and cultured in culture medium with the addition of anti-CD3 (1 μg/mL) for 96 hours. Supernatants were collected and analyzed with Multiplex Immuno Assay for interferon-γ (IFNγ), IL-17, IL-10, and tumor necrosis factor-α (TNFα) production. PGIA, N = 4; PGIA+BMT, N = 4; PGIA+BMT+250,000 Tregs, N = 5; and PGIA+BMT+500,000 Tregs, N = 5. *P < .05 compared with PGIA+BMT (Mann-Whitney U test).
Cytokine production of each treatment group was assessed by ex vivo stimulation of splenocytes. All BMT-treated groups showed lower proinflammatory cytokine production compared with the PGIA group, which is in line with the clinical amelioration. In addition, both Foxp3GFP+ Treg-treated groups demonstrated a tendency toward decreased interferon-γ and significantly decreased IL-17 compared with regular transplanted animals (Figure 6C). For IL-10 and tumor necrosis factor-α, no difference was observed among the 3 BMT treatment groups.
These data demonstrate that the addition of Foxp3GFP+ Tregs to the graft did not lead to a better clinical outcome, despite reduced proinflammatory cytokine production in these groups.
Infusion of extra Foxp3GFP+ Treg with the BMT graft causes a delay in donor-derived T-cell and Treg reconstitution
Tregs are known to suppress T-cell proliferation. Therefore, we hypothesized that the additional Tregs present in the graft could also influence T-cell reconstitution following BMT. Seven weeks post-BMT, the total number of CD4+ T cells in blood and spleen of the PGIA+BMT+Foxp3GFP+ Treg-treated groups was smaller compared with PGIA+BMT-treated animals (Figure 7A). Although the total number of Tregs remained similar in the 3 different treatment groups (Figure 7B), donor-derived Treg numbers were smaller in the Treg-treated groups, with a significant reduction in the 500,000–treated group (Figure 7B). The delay in donor Treg reconstitution was already evident at 5 weeks post-BMT (supplemental Figure 3).

Addition of extra Foxp3GFP+ Treg results in a reconstitution delay of donor-BM–derived Tregs. (A-B) Seven weeks post-BMT, blood and spleen cells were isolated and stained with congenic markers to distinguish extra injected Foxp3GFP+ Treg (gray bar), host (black bar), and donor (white bar) CD4+ T cells (TCRβ+CD4+) and Tregs (TCRβ+CD4+CD25+Foxp3+). (A) CD4+ T-cell reconstitution in blood (left) and spleen (right). (B) Treg reconstitution in blood (left) and spleen (right). All figures show absolute numbers ± SEM error bars. PGIA, N = 3; PGIA+BMT, N = 4; PGIA+BMT+250,000 Tregs, N = 5; and PGIA+BMT+500,000 Tregs, N = 5. *P < .05 for donor cell compartment.
Addition of extra Foxp3GFP+ Treg results in a reconstitution delay of donor-BM–derived Tregs. (A-B) Seven weeks post-BMT, blood and spleen cells were isolated and stained with congenic markers to distinguish extra injected Foxp3GFP+ Treg (gray bar), host (black bar), and donor (white bar) CD4+ T cells (TCRβ+CD4+) and Tregs (TCRβ+CD4+CD25+Foxp3+). (A) CD4+ T-cell reconstitution in blood (left) and spleen (right). (B) Treg reconstitution in blood (left) and spleen (right). All figures show absolute numbers ± SEM error bars. PGIA, N = 3; PGIA+BMT, N = 4; PGIA+BMT+250,000 Tregs, N = 5; and PGIA+BMT+500,000 Tregs, N = 5. *P < .05 for donor cell compartment.
Together, these data indicate that the addition of Foxp3GFP+ Tregs to the graft leads to a delay in donor-derived CD4+ T-cell and Treg reconstitution.
Discussion
Autologous stem cell transplantation is the only treatment that can induce long-term drug-free disease remission in refractory AID. Tregs are thought to be important players for remission after HSCT. In the clinical setting, exploring immunologic mechanisms that underlie disease remission is difficult as no distinction can be made between conditioning survivor cells and graft-derived cells. With the use of a congenic T-cell marker and the PGIA model, we investigated host and donor Treg dynamics following congenic BMT. Here, the results indicate that BMT, in addition to renewal of the effector T-cell compartment,8 also renews the Treg compartment with donor-derived Tregs that were superior in function to the remaining host Tregs.
Most donor-derived Tregs were initially found in the thymus, followed by the peripheral organs. In the first weeks after BMT, host Treg proliferation occurred with a decrease in naive Tregs followed by an increase in naive donor-derived Tregs. These observations suggest that in the current model, donor-derived Tregs originate from precursor cells that develop in the thymus and not by peripheral proliferation of donor-derived CD4+ T cells in the graft. To further explore this in humans, TCRβ sequencing was performed in 2 JIA and 2 juvenile dermatomyositis autologous-HSCT–treated patient samples. We found a remarkable Treg oligoclonality in these 4 patients before HSCT, which was not present in the non-Treg CD4+ T cell compartment or in Tregs from healthy children. Limited data are available on clonal diversity of the TCR repertoire of Tregs, but in healthy individuals, the CD4+FOXP3+ Treg repertoire seems as diverse as the CD4+ non-Treg repertoire.27-29 Our data are the first to show that chronic inflammation and Treg oligoclonality exist simultaneously and to a much larger extent than the CD4+ non-Treg compartment. Whether this contributes to autoimmune inflammation or is caused by autoimmune inflammation or medical treatment remains to be investigated. The development of an improved suppressive Treg compartment after autologous HSCT has been shown in humans30 and is shown here in mice. As this is accompanied with the increased diversity of the Treg repertoire, one can speculate that Treg diversity is linked to suppressive function. We found no TCR sequence overlap between preexisting Tregs and non-Tregs vs post-treatment Treg and non-Treg samples. In addition, there was a clear broadening of the Treg TCR repertoire following successful HSCT and induction of disease remission. Together, this suggests that post-HSCT, there is a renewal and diversification of the naturally occurring thymic-derived Treg compartment. Muraro and colleagues have recently demonstrated renewal of the general CD4 TCR repertoire in multiple sclerosis patients undergoing HSCT. This was in contrast to CD8+ T cells, which showed expansion of existing TCRs.10 The TCR diversity of the non-Treg compartment also expanded after HSCT, although not as striking as the Treg compartment, indicating that the Treg compartment is more affected. In conclusion, thymic reconstitution of both CD4+ T-cell and Treg compartments seems to play a pivotal role after HSCT. In JIA patients, 90% of disease relapses occur during the first year post-HSCT, and this indeed correlates with the absence of thymus involvement in T-cell reconstitution.15,31 Whether our data can be extrapolated to other (non)–juvenile autoimmune patients undergoing HSCT remains to be established. Recently, immune monitoring and biobanking of HSCT-treated AID patient sample guidelines have been published32 that may make it possible to answer specific research questions that require unique patient samples.
It is known that lymphopenic conditions are able to reduce Foxp3 expression of Tregs and thereby their suppressive function.33-36 In addition, host-derived Tregs survived a strong conditioning regimen when present post-BMT. These events may together lead to activated Tregs that (temporarily) lose their suppressive function after transplantation. This could explain the functional difference between host and donor Tregs.
Given the importance of the Treg compartment in preventing disease relapse following BMT,17 we initiated a therapeutic approach by infusing Tregs at the time of BMT to control early effector T-cell activation and potentially improve clinical outcome. We show that despite decreased proinflammatory T-cell cytokine production, no additional clinical improvement was detected. Importantly, infusion of extra Tregs resulted in a delayed donor-derived T-cell and Treg reconstitution. This delayed donor naive CD4+ T-cell/Treg reconstitution may abolish the potential beneficial effect of the additional Foxp3 Tregs. Even though the total number of Tregs was equal in all BMT-treated groups after transplantation, it is likely that the extra infused Tregs will disappear after a certain period.37 Remaining Tregs will then be essential for retaining disease remission, and their numbers may be insufficient due to delayed reconstitution induced by the infused Tregs.38 Peripheral Tregs have indeed recently been shown to inhibit their precursors in the thymus resulting in less Treg output by the thymus,39 suggesting that extra infused Tregs may delay donor Treg reconstitution via inhibition of the thymic output. Together, our data suggest that there is a delicate balance between the addition of Tregs to the graft and optimizing reconstitution of the donor-derived T-cell compartment. Murine and human studies have investigated Treg infusions to prevent graft-versus-host disease in the allogenic HSCT setting. These studies observed no T-cell reconstitution delay and even showed enhanced T-cell reconstitution after treatment.40,41 This may underline a difference between autologous and allogeneic HSCT.
In summary, we here showed that BMT induces a functional and stable donor-derived Treg compartment in an experimental arthritis model. In addition, total Treg TCR repertoire renewal was observed after successful autologous HSCT in humans. With the renewed interest in HSCT due to the use of nonmyeloablative conditioning,7 it is important to realize that replacing the immune system may be crucial for inducing and maintaining long-term immune tolerance. More knowledge about the immunologic mechanisms that lead to renewed immune balance will help us adjust current treatment regimens and develop new approaches with outcomes similar to HSCT but with less severe side effects.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Sarah Roord for her advice, Joost Swart for his help with the synovial fluid punctures, Jef Yildiz for his work with TCRβ sequencing analysis, and the Luminex Core, University Medical Center Utrecht for their work with the multiplex assay.
This work was supported by an AGIKO grant (E.M.D.) and a Veni grant (F.v.W.) from The Netherlands Organization for Scientific Research (NWO, ZonMw). T.v.d.B. was supported by Center for Translational Molecular Medicine (CTMM, TRACER).
Authorship
Contribution: E.M.D., T.v.d.B., M.B., B.J.P., and F.v.W. designed the study; E.M.D., T.v.d.B., and F.v.W. wrote the manuscript; E.M.D., T.v.d.B., G.M., J.M., E.J.W., and S.O. performed experiments; M.J.C.v.H. and F.B. provided several mouse strains; A.v.R. and N.M.W. contributed clinical samples; E.M.D., T.v.d.B., and E.S. analyzed data; and E.M.D. and T.v.d.B. conducted statistical analyses.
The current affiliation for E.J.W. is Division of Biological Sciences, University of California San Diego, La Jolla, San Diego, CA.
Conflict-of-interest disclosure: The authors declare no competing financials interests.
Correspondence: Femke van Wijk, Laboratory of Translational Immunology (LTI), Department of Paediatric Immunology, Wilhelmina Children’s Hospital, University Medical Center Utrecht, Lundlaan 6, 3508 AB Utrecht, The Netherlands; e-mail: f.vanwijk@umcutrecht.nl.

