

Key Points
Acquired selective mutations in Bcl2 and BAX conferred resistance to ABT-199 in experimental models of lymphoma.
Monitoring the potential development of such mutations in patients treated with ABT-199 is advised.
Abstract
Acquired resistance to targeted drugs is emerging as an obstacle to successful cancer treatment. Recently, a BCL2-selective BH3 mimetic termed ABT-199 showed promising therapeutic results in BCL2-dependent tumors. Based on its high affinity for BCL2, we studied potential mechanisms conferring resistance upon ABT-199 therapy, aiming to anticipate its occurrence in the clinic. Two models of resistant lymphomas were established by continuous ABT-199 exposure. In resistant Bcl2-expressing mouse lymphoma cells, 2 missense mutations within the Bcl2 BH3 domain were identified. Both F101C and F101L mutations impeded ABT-199 binding to the BH3 domain, therefore suppressing mitochondrial apoptosis. In resistant human lymphoma cells, a missense mutation in the C-terminal transmembrane domain of proapoptotic BAX (G179E) was found, which abrogated BAX anchoring to mitochondria and blocked ABT-199–induced apoptosis both in vitro and in vivo. Importantly, G179E BAX mutation also induced partial cross-resistance to other antineoplastic drugs. Our study reveals the acquisition of mutations in BCL2 family proteins as a novel mechanism of apoptosis resistance in cancer. These results anticipate the potential development of such mutations in patients treated with ABT-199, providing a basis to preventing their occurrence and to designing drugs able to circumvent the acquired resistance.
Introduction
The BCL2 family of proteins, which comprise prosurvival members such as BCL2, BCL-XL, BCL-W, MCL1, and BFL1, proapoptotic BH3-only proteins such as BIM and BAD, and the proapoptotic final effectors BAK and BAX, are critical regulators of the mitochondrial apoptotic pathway.1-3 The development of drugs that inhibit the interaction between antiapoptotic BCL2 family members and BH3-only proteins is having a major impact on cancer therapy.4-7 Among them, the BH3 mimetic ABT-737 was highly effective by inducing apoptosis through BCL2, BCL-XL, and BCL-W targeting in solid tumors and hematologic malignancies overexpressing BCL2.8 Its efficacy, however, was largely diminished in tumors expressing high levels of MCL1, BFL1 and BCL-XL, or in cancer cells lacking BAK and BAX.9-11
Despite the promising preclinical therapeutic efficacy of ABT-263, which is the orally available analog of ABT-737, phase II clinical trials showed a rapid, dose-dependent thrombocytopenia due to the inhibition of BCL-XL, limiting the ability to achieve drug concentrations at an efficacious range in cancer patients.12-14 By reengineering of ABT-263, a potent and high-affinity BCL2-selective BH3 mimetic with low avidity for BCL-XL, termed ABT-199, has been developed.15 In vitro, ABT-199 inhibited the growth of BCL2-expressing leukemia, lymphoma, and myeloma cell lines more effectively than ABT-737.15 Additionally, a single dose of ABT-199 administered to patients with chronic lymphocytic leukemia (CLL) resulted in a rapid reduction of palpable lymphadenopathy within 24 hours and a >95% reduction in peripheral blood lymphocytosis.15 Importantly, ABT-199 inhibited tumor growth while sparing platelets not only in the patients with CLL but also in mice with Myc/Bcl2-driven B-cell lymphomas.15,16 More recently, ABT-199 was shown to induce apoptosis in acute myeloid leukemia, acute lymphoblastic leukemia, mantle cell lymphoma (MCL), multiple myeloma, and breast cancer cells.17-23 These results predict a promising role of ABT-199 in the treatment of BCL2-dependent cancers.
Acquired resistance to antitumor agents, and particularly to new targeting drugs, is a nearly universal clinical challenge for cancer patients. Examples include kinase domain mutations that ablate drug efficacy of imatinib in chronic myeloid leukemia and mutations in the extracellular domain of epidermal growth factor receptor (EGFR) causing cetuximab resistance in colorectal cancer.24-26 The development of similar mutations, however, has not been demonstrated for proapoptotic drugs targeting BCL2.9 Based on the high affinity of ABT-199 for BCL2, we hypothesized that this drug might induce selective target mutations conferring therapeutic resistance. To test this hypothesis, we established ABT-199–resistant cell clones derived from mouse and human lymphoma cell lines by continuous exposure to the drug. Here, we address the genetic mechanisms leading to acquired ABT-199 therapeutic resistance in lymphoma and provide a basis for preventing and solving such potential complication.
Methods
Cell lines
Two newly established murine human-like MCL cell lines (LyBcl2-6 and LyBcl2-9), generated as previously reported with modifications, were used.27 Briefly, CyD1-4 cells were established by cloning the human CCND1 gene into the Combit-TA vector, which was stably transfected into mouse interleukin (IL)-3–dependent BaF3 pro-B lymphocytes. Then, the murine Bcl2 complementary DNA (cDNA) cloned in the pBABE-puro vector (Addgene) was transduced by retroviral infection into CyD1-4 cells, which were also transduced with the SFG-nesTGL expression vector containing a fusion protein with GFP, luciferase, and HSV1-tk. To test the tumorigenecity of 2 of the CyD1-4-Bcl2 cell clones (LyBcl2-6 and LyBcl2-9 cells), these were inoculated in RAG2−/−γc−/−, giving rise to human-like MCL tumors that could be propagated into new mice. All retroviral infections were performed as previously reported.28 The human-derived MCL cell lines HBL2 and Mino were also included.
Establishment of resistant cells to ABT-199
Resistant cell line models were established by continuous exposure to the BH3 mimetics, beginning with 5 nM of ABT-737. When cells were able to grow at a rate similar to the parental cell lines in the presence of ABT-737, the drug dose was doubled until reaching 200 nM or 1 µM of ABT-737 for HBL2 or murine cells, respectively. After that, partially resistant cells were grown in the presence of ABT-199 (1 µM) for several months and until cells became totally resistant. Parental cells were cultured in parallel using the same culture medium without ABT-737 or ABT-199 during the same period of time.
Reagents
Drugs used in the study were the BH3 mimetics (ABT-737 and ABT-199), purchased from Chemie Tek; Suberoylanilide hydroxamic acid (Vorinostat, Merck); Roscovitine (A.G. Scientific); Bortezomib (Millennium Pharmaceuticals); Doxorubicin (Pfizer); Verapamil, Etoposide, and Cisplatin (Sigma); Paclitaxel (Hospira); Temsirolimus (Wyeth); and tumor necrosis factor–related apoptosis-inducing ligand (TRAIL, Chemicon). The fluorescent Bad-peptide (5-FAM)-AAAAAQRYGRELRRMSDEFVDSFKK and the unlabeled 25-residue peptide from human Bad (NLWAAQRYGRELRRMSDEFVDSFKK) were purchased from Peptides & Elephants GmbH.29
Western-blot analysis
Western blots were performed as previously described.28 Antibodies used were: Bax (2772), Citocrome C (4272), Caspase 3 (9662), and Caspase 9 (C9) from Cell Signaling Technology; Cyclin D1 (sc-718) and Bcl2 (sc-783) from Santa Cruz Biotechnology; Actin (ab-1) from Calbiochem; BclXL (ab2568), CoxIV (ab16056), and Mcl1 (ab32087) from Abcam; and Bak (AAP-030) and Bim (AAP-330) from Stressgen. Protein quantification was performed by densitometry using the Quantity One 4.5 software (Biorad). Data, presented as the protein:actin ratio, are means of 3 independent experiments.
Quantitative real-time PCR and gene sequencing
Total messenger RNA was isolated using TRIzol Reagent (Invitrogen, Carlsbad, CA). cDNA was synthesized with the M-MLV RT enzyme (Invitrogen). Quantitative real-time polymerase chain reaction (PCR) was performed in triplicate by SYBR Green Master Mix (Applied Biosystems) and analyzed on an ABI Prism 7500 System (Applied Biosystems). Results were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and 2-∆Ct values were used for expression analysis. For direct sequencing of BCL2/Bcl2 and BAX/Bax genes, genomic DNA was purified using a QIAamp DNA Mini Kit (Qiagen), PCR amplified using TAQ polymerase (Invitrogen), and sequenced in the ABI PRISM 3730 sequencer (Applied Biosystems). Primers used in Quantitative real-time PCR and gene sequencing analysis are listed in supplemental Table 1 on the Blood Web site.
Cell viability and apoptosis assays
Lymphoma cells were seeded in triplicate in 96-well plates in the presence of the different drugs. After 48 hours, cell growth was quantified using the Cell Proliferation Assay kit (MTS) (Promega). Apoptosis was measured using the Annexin V-FITC Apoptosis Detection Kit (BD Biosciences) or the Cell Death Detection ELISA KIT (Roche), as indicated in the corresponding figure. Fluorescence was detected on a FACSCalibur (Becton Dickinson) using the CellQuest Pro software (version 6.0), as reported.28 In addition, activation of caspase 9 and caspase 3 proteins was evaluated by western-blot analysis.28
Immunoprecipitation and subcellular fractionation
For immunoprecipitation studies, murine cells were incubated overnight with 1 µM ABT-199, collected, and washed with lysis buffer (25 mM 4-2-hydroxyethyl-1-piperazine ethanesulfonic acid [HEPES], pH 7.4, 150 mM NaCl, 5 mM EDTA, 0.5 mM CaCl2, 0.2% NP40, 0.2% Tween-20, 10% glycerol) supplemented with complete protease inhibitor tablets (Roche). Cells were lysed using a Dounce Tissue Grinder. Then, lysates were cleared by centrifugation at 16 000 × g for 10 minutes and incubated with anti-Bcl2 antibody bound to protein-A beads (Pierce) for 4 hours at 4°C with agitation. Immunoprecipitates were washed 4 times with lysis buffer and boiled in sample buffer. Proteins were separated electrophoretically on 15% sodium dodecyl sulfate polyacrylamide gel electrophoresis and analyzed by western blot. For subcellular fractionation, cytosol and mitochondrial proteins were purified using the Mitochondria Isolation Kit (Thermo Scientific).
In vivo treatment with ABT-199
All mouse experiments were performed in the Animal Core Facilities of the Center for Applied Medical Research (University of Navarra) after approval by the local Animal Ethics Committee. For evaluation of the lymphoma resistance to ABT-199 in vivo, 4 groups of seven 6- to 8-week-old Rag2−/−γc−/− mice were inoculated intravenously via the tail vein with 5 × 105 cells from HBL2 or HBL2 resistant (HBL2R) cell lines, which then received either ABT-199 or vehicle. Cell inoculation and monitoring of tumor development were performed as previously reported.30 ABT-199 was prepared for oral dosing with 60% phosal 50 propylene glycol, 30% polyethylene glycol, and 10% ethanol for oral administration and was administrated weekly (20 mg/kg) starting day 3 for 4 weeks. For survival analyses, time of death was considered to be that occurring either spontaneously or by elective killing because of pain or suffering, in accordance with ethical criteria.
Functional studies of G179E BAX mutant
BAX cDNA was obtained from parental HBL2 total RNA using random primers and SuperScripIII (Invitrogen), amplified by PCR using specific primers, digested with HindIII and XhoI, and ligated to the pCMV6 vector (OriGene) at HindIII and XhoI sites. G179E BAX mutant was generated using the QuikChange Site-Directed Mutagenesis Kit from Stratagene. For protein expression, plasmids were transfected into HBL2R cells using the Amaxa Nucleofector device (solution V, program X-001). After that, cells were incubated with 200 nM ABT-199 for 6 hours, and cell viability and apoptosis were evaluated as described above. A list with the description of the primers used for BAX cloning is shown in supplemental Table 1.
Expression and purification of murine Bcl2
The fragment coding amino acids 1 to 204 of mBcl2 (lacking transmembrane domain) was obtained by standard PCR using the pBABE-puro-mBCL2 plasmid as template27 and then was subcloned into the expression vector pETM-14 using the GIBSON Assembly Cloning Kit (New England BioLabs) according to the manufacturer’s instruction. F101L and F101C mutants were generated using site-directed mutagenesis as describe above. Primers used for PCR amplification and the GIBSON Assembly Cloning reaction are listed in supplemental Table 1. The pETM plasmids carrying the desired constructions were used to transform the Escherichia coli Rosetta BL21 strain (Novagen).
Microscale thermophoresis assay
A microscale thermophoresis assay was performed for murine Bcl2 (wild-type [WT], F101L, and F101C mutants) on a Monolith NT.115 instrument (NanoTemper Technologies) as previously described.31 To evaluate the impact of the Bcl2 mutations in the interaction with ABT-199, a competitive approach with the FAM-Bad peptide was used. For evaluation of the FAM-Bad binding to murine Bcl2 protein, 10 µL of 140 nM stock solution of FAM-Bad peptide was mixed with 10 µL of a serial dilution of murine Bcl2 protein (WT or mutants). The samples were incubated in assay buffer for 5 minutes at room temperature and measured at an infrared laser power of 15% and a light-emitting diode power of 50% with a laser on time of 30 seconds and a laser off time of 5 seconds. For competition experiments, a stock solution of murine Bcl2 proteins (WT or mutants) and FAM-Bad peptide at saturating concentrations was prepared. Ten µL of the stock solution of FAM-Bad peptide-Bcl2 protein complex was mixed with a serial dilution of the unlabeled Bad peptide or ABT-199. These samples were measured at the same conditions as before. Dissociation constants were determined from each titration curves by using the NT Analysis software (NanoTemper technologies). The affinity of the nonlabeled small molecules (unlabeled Bad peptide and ABT-199 to Bcl2 proteins were determined precisely by using the equation
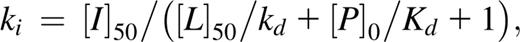
as previously described.32 Kd is the dissociation coefficient of the FAM-Bad peptide, [P]0 is the concentration of the protein at 0% binding, [I]50 is the concentration of free ligand at 50% binding, L50 the concentration of the free tracer at 50% binding, and Ki is the affinity of the nonlabeled small molecule for the unlabeled protein.
Immunofluorescence studies
Immunofluorescence (IF) was performed as previously described.33 Briefly, cells grown in culture flasks were labeled with 50 nM of Mitotracker Red CMXRos (Molecular Probes) for 30 minutes prior to drug treatment. After 1 hour of ABT-199 treatment, cells were washed twice in phosphate buffered saline (PBS) and fixed for 15 minutes at 37°C with prewarmed growth medium containing 3.7% formaldehyde. After fixation, cells were washed several times in PBS and permeabilized for 5 minutes with 0.2% Triton X-100 in PBS. Nonspecific binding was blocked with 1% bovine serum albumin (BSA) in PBS for 1 hour. Cells were then incubated with Anti-BAX diluted 1:50 in 1% BSA/PBS overnight at 4°C in a humidified chamber. Cells were then incubated for 1 hour protected from light with AlexaFluor 488-labeled (Invitrogen, Molecular probes) goat anti-mouse secondary antibody diluted 1:200 in 1% BSA/PBS. After washing 3 times with PBS, slides were mounted with Prolong Gold Antifade mounting reagent (Molecular Probes). Slides were analyzed with a LSM 510 confocal microscope and the Fiji image analysis software.34 For IF quantification analysis of BAX/Mitotracker colocalization in HBL2 and HBL2R in both ABT-199–untreated and –treated conditions, a 3-dimensional segmentation of every cell nuclei in each image was done using the Dapi channel. This segmentation was used as a mask input for the colocalization analysis performed cell by cell with Coloc 2 software. Coloc 2 is Fiji's plugin for colocalization analysis. It implements and performs the pixel intensity correlation over space methods of Pearson, Manders, Costes, Li, and more for scatterplots, analysis, automatic thresholding, and statistical significance testing.34 For comparison of IF images, the Pearsons for image above thresholds (Rcoloc) were analyzed.
Statistical analyses
Statistical analyses were performed using GraphPad Prism 5.0 software (GraphPad). Residual values of gene expression were used to test for normal distribution using D’Agostino-Pearson omnibus normality test. Two-tailed Student t test (for normally distributed data) or Mann-Whitney U test (for nonnormally distributed data) was used for evaluating significance of differences of gene expression values. Additionally, Mann-Whitney U test was used for comparison of IF quantification image values (Rcoloc). Overall survival was estimated by the Kaplan-Meier method.
Results
Establishment and characterization of mouse lymphoma models of resistance to ABT-199
To ascertain whether ABT-199 might induce mutations in target proteins and thus confer therapeutic resistance, we chose MCL as a model disease, because this aggressive cyclin-D1–expressing B-cell lymphoma frequently shows constitutive expression of BCL2 and responds to ABT-737 and ABT-199.23,27 We used human-like MCL cell lines developed by stably coexpressing cyclin-D1 and Bcl2 in mouse B lymphocytes (supplemental Figure 1).27 Two mouse cell lines, LyBcl2-6 and LyBcl2-9, were cultured with increasing concentrations of ABT-737, followed by sustained exposure to ABT-199 for 5 months (Figure 1A). After this period, the initially sensitive lymphoma cells maintained viability and minimized apoptosis upon exposure to ABT-199, as shown by Annexin V-FITC detection and the lack of caspase 3 and caspase 9 cleavage (Figure 1B-C). The acquired resistance was selective to the BH3 mimetics, because treatment with different antitumor agents did not yield differences in cell viability between parental and Bcl2-resistant cells (supplemental Figure 2). Assessment of changes in the expression of Bcl2 family proteins revealed increased expression of Bcl2 and Mcl1 and decreased expression of Bim in the resistant clones with respect to the parental cell lines (Figure 1D). Quantitative real-time PCR showed higher Bcl2 messenger RNA levels in the resistant cells, but gene expression changes in Mcl1 and Bcl2l11 (encoding Bim) were not detected (Figure 1E). These data illustrate that cells with acquired resistance to the BH3 mimetics display changes in the expression levels of selected Bcl2 family proteins.

Acquired mutations in the BH3 domain of Bcl2 conferring resistance to ABT-199. (A) Scheme showing the generation of the resistant murine model. A detailed description is provided in “Methods.” (B) Cell lines were incubated with the BH3 mimetics for 48 hours; cell proliferation was measured using the MTS assay. Ly2Bcl2-6 and Ly2Bcl2-9 resistant clones did not show activation of the mitochondrial apoptotic pathway upon exposure to the BH3 mimetics, as shown by the annexin V-FITC measurement and the lack of caspases 3 and 9 cleavage. A representative example from 3 independent experiments is shown. (C) Treatment with 1 µM ABT-199 induced activation of the mitochondrial apoptotic pathway in sensitive but not in resistant cells. Expression analysis of Bcl2 family members by western blot (D) or quantitative real-time PCR (E). (F) Resistant cell lines cultured without ABT-199 (labeled as SA) remained resistant to the BH3 mimetics. (G-H) The protein expression profile in the SA cells was similar to those of the sensitive cell lines. (C-G) Representative data from 1 of 3 independent experiments performed in triplicate (mean ± SD, where indicated). (I) Sequence of Bcl2 identified 2 de novo missense mutations at the same codon (F101) located within the BH3 domain. These mutations were found in the ectopic Bcl2 mouse gene but not in the endogenous mouse Bcl2. Arrows indicate the nucleotide change.
Acquired mutations in the BH3 domain of Bcl2 conferring resistance to ABT-199. (A) Scheme showing the generation of the resistant murine model. A detailed description is provided in “Methods.” (B) Cell lines were incubated with the BH3 mimetics for 48 hours; cell proliferation was measured using the MTS assay. Ly2Bcl2-6 and Ly2Bcl2-9 resistant clones did not show activation of the mitochondrial apoptotic pathway upon exposure to the BH3 mimetics, as shown by the annexin V-FITC measurement and the lack of caspases 3 and 9 cleavage. A representative example from 3 independent experiments is shown. (C) Treatment with 1 µM ABT-199 induced activation of the mitochondrial apoptotic pathway in sensitive but not in resistant cells. Expression analysis of Bcl2 family members by western blot (D) or quantitative real-time PCR (E). (F) Resistant cell lines cultured without ABT-199 (labeled as SA) remained resistant to the BH3 mimetics. (G-H) The protein expression profile in the SA cells was similar to those of the sensitive cell lines. (C-G) Representative data from 1 of 3 independent experiments performed in triplicate (mean ± SD, where indicated). (I) Sequence of Bcl2 identified 2 de novo missense mutations at the same codon (F101) located within the BH3 domain. These mutations were found in the ectopic Bcl2 mouse gene but not in the endogenous mouse Bcl2. Arrows indicate the nucleotide change.
To determine whether the acquired resistance was reversible, Bcl2-resistant cells were cultured without ABT-199 for 4 weeks. Then, the previously detected Bcl2, Mcl1, and Bim expression changes were not found, being the expression pattern of LyBcl2-resistant cells identical to that of the parental cell lines (Figure 1F-G). However, reexposure to the BH3 mimetics did not affect cell viability or apoptosis, suggesting that mechanisms other than protein level variations sustained resistance (Figure 1H). To further investigate this hypothesis, sequencing of Bcl2 family genes was performed in LyBcl2 parental and resistant cells. Notably, 2 missense mutation in the same codon within the BH3 domain of Bcl2 (F101C and F101L) were detected in LyBcl2-6 and LyBcl2-9 resistant cells, respectively (Figure 1I). These changes were not present in the parental cells and were retained upon ABT-199 treatment discontinuation. These results show that the continuous exposure to the BH3 mimetics induced selective mutations in the target BH3 domain of Bcl2, possibly conferring apoptotic resistance.
Characterization of human MCL cell lines resistant to ABT-199
To evaluate whether a similar strategy could induce resistance in human lymphoma cells, the ABT-737–sensitive MCL cell line HBL227 was cultured with increasing concentrations of ABT-737, followed by ABT-199 for 7 months (Figure 2A). After this period, treatment of HBL2 cells with ABT-737 or ABT-199 did not affect proliferation, viability, or apoptosis, as shown by annexin V-FITC staining and the lack of caspase 3 and caspase 9 cleavage (Figure 2B-C). To determine whether the HBL2R cells were also resistant in vivo, Rag2−/−γc−/− mice were inoculated intravenously with 5 × 105 cells from HBL2 or HBL2R cell lines, which then orally received either ABT-199 or vehicle. HBL2R cells did not respond to ABT-199 in comparison with HBL2 parental cells xenografted in Rag2−/−γc−/− immunodeficient mice (Figure 2D-E). HBL2R cells showed an increase in MCL1 protein expression, which was accompanied by a moderate decrease of BIM at the RNA and protein levels (Figure 2F-G). Discontinuation of ABT-199 exposure for 4 weeks was associated with the reversion of MCL1 and BIM expression changes in HBL2R cells, but these remained resistant to the drug (Figures 2B,F-G). However, and differently from the murine LyBcl2 lymphoma cells, incubation of HBL2R with 9 anticancer drugs revealed partial resistance to 6 of them (taxol, doxorubicine, bortezomib, cisplatin, temsirolimus, and TRAIL) in comparison to parental cells, indicating that the ABT-199-induced resistance was not exclusive to this drug (supplemental Figure 3). Sequencing analysis did not identify changes in BCL2 gene but instead, a missense mutation in the gene encoding the proapoptotic final effector protein BAX (G179E) was detected in HBL2R cells (Figure 2H). This change was located in the hydrophobic segment at the C-terminal transmembrane domain, which is essential for anchoring BAX to the mitochondrial membrane, suggesting that the G179E mutation may impede apoptosis through blocking BAX translocation to mitochondria.35-37

Mutation in the transmembrane domain of BAX abolished cell death induced by ABT-199. (A) Scheme showing the generation of HBL2R cells. A detailed description is provided in “Methods.” (B) Both HBL2 and HBL2R cells cultured without ABT-199 for 4 weeks (labeled as HBL2SA) were resistant to the BH3 mimetics. (C) HBL2R cells did not show activation of the mitochondrial apoptotic pathway induced by the BH3 mimetics, as shown by the annexin V-FITC FACS and the lack of caspases 3 and 9 cleavage. Experiments were performed in duplicate. (D) Kaplan-Meier overall survival curves for Rag2−/−IL2γc−/− mice transplanted with HBL2 or HBL2R cells. (E) A representative image showing differences in the tumor volume in mice inoculated with HBL2 and HBL2R cells and treated with ABT-199 (marked with arrows). (F-G) Expression profiling of BCL2 family proteins did not show changes between HBL2 and HBL2SA cells. Shown are representative data from 1 of 3 independent experiments. (H) Cells with acquired resistance to ABT-199 showed a missense mutation in BAX transmembrane domain (G179E). Arrows indicate the base change. cHBL2R, cDNA; gHBL2R, genomic DNA.
Mutation in the transmembrane domain of BAX abolished cell death induced by ABT-199. (A) Scheme showing the generation of HBL2R cells. A detailed description is provided in “Methods.” (B) Both HBL2 and HBL2R cells cultured without ABT-199 for 4 weeks (labeled as HBL2SA) were resistant to the BH3 mimetics. (C) HBL2R cells did not show activation of the mitochondrial apoptotic pathway induced by the BH3 mimetics, as shown by the annexin V-FITC FACS and the lack of caspases 3 and 9 cleavage. Experiments were performed in duplicate. (D) Kaplan-Meier overall survival curves for Rag2−/−IL2γc−/− mice transplanted with HBL2 or HBL2R cells. (E) A representative image showing differences in the tumor volume in mice inoculated with HBL2 and HBL2R cells and treated with ABT-199 (marked with arrows). (F-G) Expression profiling of BCL2 family proteins did not show changes between HBL2 and HBL2SA cells. Shown are representative data from 1 of 3 independent experiments. (H) Cells with acquired resistance to ABT-199 showed a missense mutation in BAX transmembrane domain (G179E). Arrows indicate the base change. cHBL2R, cDNA; gHBL2R, genomic DNA.
Mechanisms of therapeutic resistance to ABT-199 in lymphoma cells
The functional consequence of Bcl2 and BAX mutations in therapeutic resistance was explored. Mutations in the BCL2 gene are common in certain subtypes of B-cell lymphoma such as diffuse large cell lymphoma (DLBCL) and follicular lymphoma, but these are rarely detected in MCL, CLL, and primary mediastinal B-cell lymphoma.38,39 Interestingly, most of the 77 nonsynonymous BCL2 mutations found in 2 series comprising 353 patients with DLBCL occurred in the flexible loop domain containing the P53-binding site, but only one affected the BH3 domain.38,39 The fact that only 1 of 353 patients with untreated lymphomas (0.0028%) showed mutations in this BCL2 domain strongly suggests that the F101C and F101L mutations found in the murine lymphoma cells are related to the continuous ABT-199 exposure. To functionally characterize such Bcl2 mutations, competitive binding experiments were performed using a fluorescent BH3-Bad peptide.8,15 The Bad peptide was able to bind WT Bcl2 and both Bcl2 mutants (Ki values: WT, 35.31 ± 2.1; F101L, 5.64 ± 0.1; F101C, 182 ± 17 nM). Conversely, whereas ABT-199 bound to WT Bcl2 with high affinity (WT Ki: 0.1 ± 0.01 nM), both mutations inhibited its binding (Figure 3A). Further, immunoprecipitation experiments detected binding of Bcl2 to Bim in both LyBcl2-sensitive and -resistant cells; this physical interaction was disrupted by ABT-199 only in LyBcl2-sensitive cells, but not in the resistant clones (Figure 3B). These results functionally demonstrate that the acquired Bcl2 mutations impede ABT-199 binding to the Bcl2 hydrophobic groove, thus suppressing apoptosis and conferring therapeutic resistance without interfering with downstream Bcl2 functions.

Functional evaluation of Bcl2 and BAX mutations. (A) ABT-199 was unable to bind F101C and F101L Bcl2 mutant proteins. (B) Bcl2 mutations had no impact on Bcl2 binding to Bim in both ABT-199–sensitive and –resistant lymphomas. Immunoprecipitation experiments were performed in duplicate. (C) Ectopic expression of WT BAX (but not G179E) in HBL2R cells restored ABT-199 sensibility. Impact of ABT-199 on cell viability (left). Induction of apoptosis with and without ABT-199 treatment (right). Experiments were performed in triplicate. (D) IF analysis showing that ABT-199 induced BAX translocation from cytoplasm to mitochondria in HBL2 but not in HBL2R cells, as shown by colocalization of BAX and mitotraker (Mito). Experiments were performed in duplicate: a representative example is shown. Quantification values of colocalization BAX and mitotraker IF images are provided in the main text. DAPI, 4,6 diamidino-2-phenylindole. (E) Subcellular localization of BAX performed by cell fractionation experiments showed that mutant BAX remained cytoplasmic after ABT-199 treatment. In addition, BAX G179E mutation prevented release of cytochrome C to cytoplasm. Experiments were performed in duplicate.
Functional evaluation of Bcl2 and BAX mutations. (A) ABT-199 was unable to bind F101C and F101L Bcl2 mutant proteins. (B) Bcl2 mutations had no impact on Bcl2 binding to Bim in both ABT-199–sensitive and –resistant lymphomas. Immunoprecipitation experiments were performed in duplicate. (C) Ectopic expression of WT BAX (but not G179E) in HBL2R cells restored ABT-199 sensibility. Impact of ABT-199 on cell viability (left). Induction of apoptosis with and without ABT-199 treatment (right). Experiments were performed in triplicate. (D) IF analysis showing that ABT-199 induced BAX translocation from cytoplasm to mitochondria in HBL2 but not in HBL2R cells, as shown by colocalization of BAX and mitotraker (Mito). Experiments were performed in duplicate: a representative example is shown. Quantification values of colocalization BAX and mitotraker IF images are provided in the main text. DAPI, 4,6 diamidino-2-phenylindole. (E) Subcellular localization of BAX performed by cell fractionation experiments showed that mutant BAX remained cytoplasmic after ABT-199 treatment. In addition, BAX G179E mutation prevented release of cytochrome C to cytoplasm. Experiments were performed in duplicate.
Despite the fact that loss-of-function mutations within BH3 or BH1 domains of BAX are common in solid tumors and hematologic malignancies,40-42 mutations in the C-terminal transmembrane domain have not been reported.43 To evaluate the relevance of such acquired mutation (G179E), WT and mutant G179E BAX proteins were expressed in HBL2R cells, which were then incubated with ABT-199. As shown in Figure 3C, the acquired resistance could be reversed only in cells expressing WT BAX, but not the G179E mutant. Additionally, IF studies showed BAX translocation to mitochondria after exposure to ABT-199 in HBL2-sensitive but not in -resistant clones. Indeed, quantification of BAX/Mitotracker colocalization images showed an increase from basal 0.05 to 0.17 after ABT-199 incubation in HBL2 cells (P = .0016) but not in HBL2R cells (0.071 vs 0.076; P = .84) (Figure 3D). Accordingly, ABT-199 induced cytochrome C release from mitochondria to cytoplasm in HBL2-sensitive but not in HBL2R cells (Figure 3E). To determine whether ABT-199 can also translocate BAX to mitochondria and induce apoptosis in the absence of BIM, the BIM-deficient MCL cell line Mino was incubated with ABT-199. ABT-199 induced apoptosis in Mino by translocating BAX to mitochondria, similarly to the BIM-expressing HBL2 cell line, indicating that BIM is not essential in ABT-199-induced apoptosis (supplemental Figure 4). Overall, these data demonstrate that the acquired G179E mutation abrogates BAX translocation to mitochondria upon ABT-199 therapy, thus blocking apoptosis. These results also explain the partial refractoriness observed in HBL2R cells to other cytotoxic drugs including TRAIL.
Discussion
Resistance to targeted therapies is a major obstacle to successful cancer treatment.24 The most representative examples are mutations in the extracellular domain of EGFR causing cetuximab resistance in colorectal cancer,25 kinase domain mutations that block the effect of imatinib in chronic myeloid leukemia,26 and the dimerization of aberrantly spliced BRAF (V600E) conferring RAF inhibitor resistance in melanoma.44 More recently, an epigenetic mechanism of resistance to targeted therapy using γ-secretase inhibitors in T-cell acute lymphoblastic leukemia has been reported.45 Using a strategy similar to ours, Yecies et al9 generated models of acquired resistance to ABT-737 in DLBCL cell lines cells, which exhibited increased expression of antiapoptotic MCL1 and BFL-1 conferring the induced resistance. However, this report did not identify molecular changes underlying the abnormal expression. Konopleva et al and Mazumder et al46,47 reported that phosphorylation of BCL2 or increased levels of MCL1 impeded ABT-737–induced apoptosis in cancer cells, which could be restored upon inhibition of BCL2 phosphorylation and reduction of MCL1 expression. Again, these manuscripts did not report mutations in BCL2-family proteins associated with ABT-737 resistance. We noticed similar changes in expression of several proteins, including BCL2, MCL1, and BIM (and of BFL1, data not shown) upon continuous exposure to ABT-199, but these abnormal expression levels were reversible and thus returned to normal values after drug withdrawal. Importantly, and differently from previous reports, we found genetic mutations in Bcl2 and BAX that conferred resistance to ABT-199. Overall, the development of similar mutations has not been demonstrated for proapoptotic drugs targeting BCL2.1,6,9,13,48-50
Here, we have attempted to elucidate the molecular basis of the acquired resistance to the BH3 mimetic ABT-199, which may be expected based on its high affinity for BCL2. Our study shows the acquisition of Bcl2 and BAX mutations as mechanisms of resistance to ABT-199 in experimental models of lymphoma (Figure 4). We think that the rapid identification and validation of the acquired resistance mechanisms arising in the setting of “targetable” tumor dependencies will conduct the development of novel reengineered drugs that can bypass the acquired resistance, together with a close molecular monitoring of patients receiving these drugs. Currently, at least 14 phase I-II clinical trials using ABT-199, alone or in combination with other compounds, are being conducted in patients with hematologic malignancies and solid tumors (www.clinicaltrials.gov).51 Therefore, our results may be clinically relevant in the near term, as the development of mutations conferring ABT-199 resistance could follow high-dose therapy with this BH3 mimetic. Certainly, development of the ABT-199–induced BCL2 target mutations could be expected, because similar changes have occurred following various high-affinity targeted therapies.24 The BAX mutation was, however, unpredictable and, most importantly, led to cross-resistance to other antineoplastic agents, highlighting a dangerous mechanism of apoptotic resistance.

Proposed model of acquired resistance to ABT-199 in experimental models of human and mouse lymphoma. Mutations in the BH3 domain of BCL2 abolish ABT-199 binding and thus block apoptosis. Mutation in BAX transmembrane domain prevents mitochondrial translocation of BAX and thus impedes apoptosis.
Proposed model of acquired resistance to ABT-199 in experimental models of human and mouse lymphoma. Mutations in the BH3 domain of BCL2 abolish ABT-199 binding and thus block apoptosis. Mutation in BAX transmembrane domain prevents mitochondrial translocation of BAX and thus impedes apoptosis.
Although our results have to be interpreted with caution, these data predict the potential acquisition of such mutations in patients treated with ABT-199. We think that our data provide the framework to prevent their development, possibly by changing the schedule of administration to avoid long-term continuous exposure to the drug. In addition, patients receiving ABT-199 could be monitored during follow-up for the development of such mutations, particularly if signs of resistance appear. Finally, it will also be of interest to determine whether ABT-199 or other BCL2-targeting molecules can induce similar mutations conferring resistance in diverse tumor types.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
We thank Miguel Galarraga and Carlos Ortiz de Solorzano for IF quantification images.
This work was supported by grants from the Instituto de Salud Carlos III, Spanish Ministry of Economy and Competitiveness: FIS-PI12/00202 and RTICC-RD12/0036/0063-FEDER (to J.A.M.-C.), and FIS-PS09/02437 (to V.F.). The Biomolecular Screening and Protein Technologies Unit at the Centre for Genomic Regulation (CRG) is cofunded by the European Regional Development Fund (ERDF) in the framework of the ERDF operational Programme of Catalonia 2007-2013.
Authorship
Contributions: M.R. and M.J.G.-B. performed research and analyzed data; C.C. designed and performed research; and V.F. and J.A.M.-C. designed, coordinated, and supervised the study, performed research, analyzed data, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Vicente Fresquet, Center for Applied Medical Research, University of Navarra, Avda Pio XII, 55, Pamplona 31008, Spain; e-mail: vfresquetarnau@gmail.com; and Jose A. Martinez-Climent, Center for Applied Medical Research, University of Navarra, Avda Pio XII, 55, Pamplona 31008, Spain; e-mail: jamcliment@unav.es.

