

Abstract
Factor VIII and factor V share structural homology and bind to phospholipid membranes via tandem, lectin-like C domains. Their respective C2 domains bind via 2 pairs of hydrophobic amino acids and an amphipathic cluster. In contrast, the factor V-like, homologous subunit (Pt-FV) of a prothrombin activator from Pseudonaja textilis venom is reported to function without membrane binding. We hypothesized that the distinct membrane-interactive amino acids of these proteins contribute to the differing membrane-dependent properties. We prepared mutants in which the C2 domain hydrophobic amino acid pairs were changed to the homologous residues of the other protein and a factor V mutant with 5 amino acids changed to those from Pt-FV (FVMTTS/Y). Factor VIII mutants were active on additional membrane sites and had altered apparent affinities for factor X. Some factor V mutants, including FVMTTS/Y, had increased membrane interaction and apparent membrane-independent activity that was the result of phospholipid retained during purification. Phospholipid-free FVMTTS/Y showed increased activity, particularly a 10-fold increase in activity on membranes lacking phosphatidylserine. The reduced phosphatidylserine requirement correlated to increased activity on resting and stimulated platelets. We hypothesize that altered membrane binding contributes to toxicity of Pt-FV.
Introduction
Factor VIII (FVIII) is an essential cofactor for the intrinsic branch of the coagulation cascade. The clinical importance of FVIII is illustrated by hemophilia A, a disease in which deficient or defective FVIII leads to a severe clinical bleeding phenotype. FVIII has sequence homology with FV, an essential cofactor for the common final pathway of the coagulation cascade. Complete deficiency of FV is incompatible with life,1 although a partial deficiency causes a bleeding disorder termed “parahemophilia.”
Both FVIII and FV bind to membranes containing phosphatidylserine (PS) and serve, respectively, as cofactors for FXase and prothrombinase enzyme complexes.2-5 Excessive activity of the FXase6,7 and prothrombinase complexes8 is linked to an elevated risk of thrombosis. The importance of binding to PS-containing membranes is illustrated by nearly complete loss of activity when this interaction is blocked by an antibody9 or when membrane binding sites are blocked by a competing protein.10 Similarly, activity is diminished by point mutations that diminish the interaction with PS-containing membranes.11,12 There are no reported disorders linked to increased activity related to enhanced membrane binding. The functional consequences of altered membrane binding are the foci of this investigation.
To that end, we also investigated a form of FV that has less need for phospholipid membranes. The venom of the eastern brown snake (Pseudonaja textilis) contains a potent prothrombin activating protein complex (pseutarin C) that is homologous to FXa bound to FVa in the prothrombinase complex.13 Recent results have shown that the FVa-homologous subunit (Pt-FV) is constitutively active and does not require phospholipid membranes to function.14 We hypothesized that differences in the membrane-binding region of this subunit contribute to its unusual activity and the ability of pseutarin C to induce massive disseminated intravascular coagulation in the snake's prey.15
FVIII and FV both have domain structures of A1-A2-B-A3-C1-C2 in which the A domains share homology with ceruloplasmin, the B domains are unique to each protein, and the C domains are homologous with the discoidin class of lectins.16,17 The C2 domains contain determinants of membrane binding.18,19 High-resolution crystal structures have been published for the C2 domains of both FV20 and FVIII.21 The common feature is a β-barrel core with 3 relatively long loops protruding from one end. These loops have been dubbed “spikes” and both the FV C2 domain and the FVIII C2 domain have 2 water-exposed hydrophobic residues protruding from spike 1 and either 2 (FVIII) or one hydrophobic residue (FV) protruding from spike 3 (Figure 1). This feature led to a hypothesis that membrane binding is mediated by insertion of the hydrophobic residues into the membrane. A fourth loop (spike 4) in the C2 domain has also been identified as a possible membrane-interactive region because of its association with a small-molecule inhibitor of both FV and FVIII.22,23 Membrane-binding functionality of the hydrophobic residues on spike 1 is illustrated by the presence of mild hemophilia and defective phospholipid binding in patients with a single amino acid deletion near the tip of the loop.24 The membrane-binding functions of spikes 1, 3, and 4 have been confirmed by site-directed mutagenesis.11,12,25
Membrane binding of FVIII and FV is usually modeled as a simple, reversible equilibrium interaction.2,26 This interaction is mediated, in part, by stereoselective preference for phosphatidyl-L-serine over other membrane lipids,27,28 and both proteins bind with dissociation constants in the 1 to 10nM range.2 However, several lines of data indicate that the binding is more complex than the simple models imply. Both proteins have rapid and slow phases of membrane association and dissociation.29 FVIII requires more PS molecules per binding site than FV.2 Furthermore, neither protein competes fully for the binding sites the other recognizes on a phospholipid membrane.30 It is possible that these differing membrane-binding properties are partially explained by the membrane-interactive amino acids within their respective C2 domains. After stimulation by thrombin, platelets transiently expose small quantities of PS on the plasma membrane.31 Blood platelets therefore provide biologic membranes that support activity of FVIII and FV.
Prior investigations indicate that membrane binding of FVIII32 and FV33 lead to allosteric activation. The possibility that allosteric activation can be decoupled from membrane binding is suggested by partial activation of the FVa-FXa complex34 and the FVIIIa-FIXa complex35 by soluble PS. Furthermore, we have found that inhibitory antibodies directed against the C2 domain of FVIII and mutations of membrane-interactive residues of the C1 domain can decrease membrane-induced activation while binding is preserved.36 This work investigates the possibility that mutations may also enhance activation of the respective membrane-bound enzyme complexes.
The crystal structures of FVIII37,38 and inactivated FV (FVai)39 indicate that the membrane-binding C2 domains are positioned so that they might also make contact with the respective proteases, FIXa for FVIIIa, and FXa for FVa. They may also make contact with the respective substrates, FX for FVIIIa, and prothrombin for FVa.40 Indeed, the Gla domain of FIXa contacts FVIII, with the best candidate domains being either the C1 or C2 domain.41 Likewise, the Gla domain of prothrombin makes contact with FVa.40 Thus, mutations in the C2 domains could plausibly influence the interaction of either FVIIIa or FVa with the respective enzymes and/or substrates.
This study was motivated by questions about the tolerance for conservative mutations in the membrane-interactive amino acids of the C2 domains of both FV and FVIII. Thus, we prepared mutants in which the MF pair from the first membrane interactive loop of FVIII was swapped for the WW pair at the homologous position of the FV C2 domain (Figure 1). Likewise, the LL pair, protruding from the second membrane interactive loop of FVIII, was swapped for the LS pair at the same position in FV. Complementary swaps within FV were also prepared (Table 1). To investigate the activity of FV from P textilis venom, 5 amino acids in the membrane-binding region of FV were mutated to their corresponding amino acids from Pt-FV (see Figure 5). The results indicate that conservative mutations of these hydrophobic amino acids are consistent with maintained overall function. The impact of the mutations appears to influence the number and type of binding sites that support activity, the affinities for the respective substrates, and the degree of allosteric activation on membranes with little or no PS.
Methods
See supplemental Methods (available on the Blood Web site; see the Supplemental Materials link at the top of the online article) for materials, details of mutagenesis of full-length FVIII and FV, purification, and details of FXase activity and prothrombinase activity assays. Approval for the use of blood from normal donors (to purify platelets for study) was obtained from the Harvard Medical School and VA Boston Healthcare System Institutional Review Board. Subjects signed the Institutional Review Board–approved ICFs as part of the consent process in accordance with the Declaration of Helsinki.
VWF binding assay
The affinity of FVIII for VWF was measured in a competition ELISA.11 Wild-type or mutant FVIII, at 1 unit/mL, was incubated with various concentrations of VWF for 60 minutes at 23°C to enable equilibrium binding.42 Subsequently, the FVIII-VWF mixtures were placed in microtiter wells coated with mAb B02C11. VWF competes for the mAb B02C11 epitope9 so that only free FVIII was available to bind to immobilized mAb B02C11. After 60 minutes, the wells were washed and bound FVIII was detected with antibody ESH8, followed by HRP-conjugated goat anti–mouse antibody. The wells were developed with o-phenylenediamine dihydrochloride (Sigma-Aldrich) and read in kinetic mode. Wells without FVIII were used to obtain background values (which ranged from 5 mAU/min to 16 mAU/min over the experiments performed) that were subtracted from all data points (maximum signal from 77 mAU/min to 89 mAU/min) before further analysis.
Direct measurement of phospholipid binding
mAbs to FVIII (Green Mountain Antibodies, GMA-8021) and FV (CBC-MOR101; Abcam) were coupled to cyanogen bromide-activated cross-linked sepharose beads (Superose 12). These antibodies were selected for their high affinity and lack of interference with phospholipid-membrane binding. The antibody-Superose beads were incubated with the their respective mutants (≥ 0.5nM) overnight at 4°C. The beads were then mixed with various concentrations of fluorescein-labeled sonicated vesicles (1:4:20:75, phosphatidylethanolamine [PE]–CF:PS:PE:phosphatidylcholine [PC]) in 50mM Tris, 150mM NaCl, 0.2% BSA, 1.5mM Ca++, pH 7.85, to a final volume of 50 μL. Tubes were incubated at room temperature for 30 minutes with moderate speed shaker mixing. Samples were diluted with 250 μL buffer immediately before reading by flow cytometry. Background readings, taken using antibody-coupled beads that were not incubated with FV or FVIII, were subtracted from the raw data before analysis.
Data analysis
The competition binding data were interpreted assuming that mAb B02C11 bound all FVIII that was not bound to VWF. The ELISA data were normalized to the maximum signal in the absence of VWF for each dataset and then subtracted from 1 to generate upright binding curves. The data from 2 experiments were combined and analyzed according to the equation:
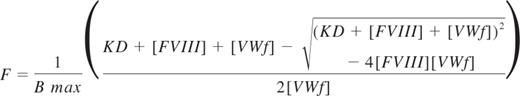
Where F is the fraction of FVIII bound to VWF, Bmax is the maximum achievable concentration of VWF-bound FVIII, KD is the equilibrium dissociation constant of FVIII with VWF, FVIII represents the total FVIII concentration, and VWF represents the total concentration of VWF subunits. Data were fitted using Prism Version 4.0 for the Macintosh.
Data from prothrombinase, FXase, and direct lipid-binding assays were fitted by nonlinear least squares analysis using the 1 site–binding function in the Prism Version 4.0 software package and normalized to the respective Bmax values. The normalized data from multiple FXase and prothrombinase experiments were combined and fitted to obtain the values shown in Figures 2C and 3C.
Results
FVIII mutants
We prepared 3 FVIII mutants, substituting the hydrophobic spike amino acids of FV for either and for both of the pairs of hydrophobic spike amino acids of FVIII (Figure 1; Table 1). The mutants, together with wild-type FVIII (FVIIIWT) were expressed from COS-7 cells and purified by immunoaffinity chromatography. The mutants were evaluated for activity in a commercial aPTT assay and for apparent affinities for phospholipid vesicles, FIXa, and FX in a defined FXase assay (Figure 2). For all assays, the activity of the mutants was referenced to FVIIIWT purified in the same manner for each batch of mutants. The specific activity of FVIIIWT averaged 9400 units/mg. Affinities for FIXa were not clearly different from wild-type FVIII and are not shown.

Crystal structures of the FVIII C2 domain (PDB entry 1D7P) and FV C2 domain (PDB entry 1CZT). The membrane-interactive hydrophobic amino acids for spikes 1 and 3 of FVIII (left) and FV (right) are shown with side chains in dark gray, whereas the rest of the structures show the backbone only in light gray. Residue numbers relate to the full-length proteins.
Crystal structures of the FVIII C2 domain (PDB entry 1D7P) and FV C2 domain (PDB entry 1CZT). The membrane-interactive hydrophobic amino acids for spikes 1 and 3 of FVIII (left) and FV (right) are shown with side chains in dark gray, whereas the rest of the structures show the backbone only in light gray. Residue numbers relate to the full-length proteins.

Relative activities, membrane interaction, and affinities for FVIII mutants. (A) Membrane interaction for FVIIIWT (■) or FVIIILS (▾) was evaluated by mixing FIXa, FX, and various concentrations of sonicated vesicles. The quantity of FXa formed was measured with chromogenic substrate S-2765. The membrane reactivity of FVIIILS was less than FVIIIWT. The vesicles had a composition of PS:PE:PC 4:20:76; concentration of FVIII or mutants was 0.2nM; FIXa, 4nM; and FX, 130nM. Results represent the mean ± SEM for at least 3 experiments, each performed in duplicate and fitted to a single-site binding model (smooth curves). (B) Apparent KM for the FVIIIa-FIXa complex was determined as in panel A, except that the phospholipid concentration was constant (100μM) and the FX concentration varied. (A-B) Fitted results were normalized to the Bmax value for each curve to optimize comparison of the relative affinities. (C) The specific activities, apparent phospholipid interactivity, and apparent FX affinities for each mutant are displayed with reference to control experiments performed with wild-type FVIII (as in panels A and B for FVIIILS). The displayed values represent the mean ± SEM. Each fit was performed on a minimum of 3 separate experiments, each performed in duplicate. (D) Apparent VWF affinity was determined by incubating FVIIIWT and mutants with various concentrations of VWF for 60 minutes before addition to a microtiter well coated with a monoclonal antibody that competes for the VWF-binding epitope. Bound FVIII was detected with a second antibody. The fraction of VWF-bound FVIII was calculated as the difference between the maximum signal in the absence of VWF and the signal at each concentration of VWF. Smooth curves represent best-fit curves corresponding to KD values of 0.7 ± 0.1nM, 2.2 ± 0.7nM, and 4.8 ± 0.8nM for FVIIIWT (■), FVIIIWW (▴), and FVIIILS (▾), respectively. Displayed results are the mean ± SEM of at least 2 experiments, each performed in duplicate.
Relative activities, membrane interaction, and affinities for FVIII mutants. (A) Membrane interaction for FVIIIWT (■) or FVIIILS (▾) was evaluated by mixing FIXa, FX, and various concentrations of sonicated vesicles. The quantity of FXa formed was measured with chromogenic substrate S-2765. The membrane reactivity of FVIIILS was less than FVIIIWT. The vesicles had a composition of PS:PE:PC 4:20:76; concentration of FVIII or mutants was 0.2nM; FIXa, 4nM; and FX, 130nM. Results represent the mean ± SEM for at least 3 experiments, each performed in duplicate and fitted to a single-site binding model (smooth curves). (B) Apparent KM for the FVIIIa-FIXa complex was determined as in panel A, except that the phospholipid concentration was constant (100μM) and the FX concentration varied. (A-B) Fitted results were normalized to the Bmax value for each curve to optimize comparison of the relative affinities. (C) The specific activities, apparent phospholipid interactivity, and apparent FX affinities for each mutant are displayed with reference to control experiments performed with wild-type FVIII (as in panels A and B for FVIIILS). The displayed values represent the mean ± SEM. Each fit was performed on a minimum of 3 separate experiments, each performed in duplicate. (D) Apparent VWF affinity was determined by incubating FVIIIWT and mutants with various concentrations of VWF for 60 minutes before addition to a microtiter well coated with a monoclonal antibody that competes for the VWF-binding epitope. Bound FVIII was detected with a second antibody. The fraction of VWF-bound FVIII was calculated as the difference between the maximum signal in the absence of VWF and the signal at each concentration of VWF. Smooth curves represent best-fit curves corresponding to KD values of 0.7 ± 0.1nM, 2.2 ± 0.7nM, and 4.8 ± 0.8nM for FVIIIWT (■), FVIIIWW (▴), and FVIIILS (▾), respectively. Displayed results are the mean ± SEM of at least 2 experiments, each performed in duplicate.
FVIIILS had approximately 10-fold lower reactivity with phospholipid vesicles (Figure 2A), suggesting that L2252 may make a substantial contribution to binding site interaction. We use the term “reactivity” to denote the product of binding site affinity and number of binding sites/phospholipid vesicle because the mutations may affect either or both parameters. The apparent affinity of FVIIILS for FX was decreased approximately 50% (Figure 2B). These results indicate that interaction with FX is altered by changes in membrane binding or that L2252 interacts directly with FIXa and/or FX. The specific activity, membrane reactivity, and apparent affinity of FVIIILS for FX are displayed as a ratio with FVIIIWT in Figure 2C. The unaltered specific activity of FVIIILS may be rationalized with the decreased phospholipid reactivity by noting that the commercial aPTT reagent has a very high phospholipid concentration of unknown composition that may influence results.
Mutant FVIIIWW had approximately 2-fold increased specific activity (Figure 2C). Correlating with the increased specific activity was an approximately 6-fold increase in phospholipid reactivity. The apparent affinity curves, analogous to those in Figure 2A and B, can be found in supplemental Figure 2. FVIIIWW also had increased apparent affinity for FX. These results indicate the hydrophobic WW sequence of FV supports these 3 components of FVIII activity with higher affinities than the native MF sequence.
Mutant FVIIIWW/LS, incorporating FV amino acids for both hydrophobic pairs, had normal specific activity (Figure 2C). Furthermore, the interaction with phospholipid and apparent affinity for FX were within 2-fold of FVIIIWT. The results indicate that these contacts result from constituent interactions of both hydrophobic spikes that are additive in effect.
Direct binding studies of phospholipid vesicles to immobilized FVIII (Table 2; supplemental Figure 1A) indicated that the WW substitution for residues of spike 1 conferred a 2-fold increase in affinity. This increase is consistent with the increased phospholipid vesicle reactivity (Figure 2C). In contrast, the preserved affinity of FVIIILS suggests that the decreased PL interaction observed in the FXase complex results from another change, such as functional activity on fewer membrane sites.
Because both hydrophobic spikes of FVIII affect the affinity for VWF,11 we evaluated the apparent VWF-binding affinities of our mutants (Figure 2D). In this assay, the interaction between FVIIIWT and VWF was modeled with a KD of 0.7 ± 0.1nM, similar to previously measured FVIII-VWF affinity using a similar assay.11 FVIIIWW had 3-fold decreased apparent affinity for VWF, indicating that the increased affinity for phospholipid does not correlate to increased VWF affinity. FVIIILS had a 7-fold decreased apparent affinity for VWF, indicating that L2252 in the second hydrophobic spike makes a significant contribution to VWF affinity that is not compensated by interaction with Ser. In the absence of VWF, FVIIIWT and FVIIIMF showed similar binding to the capture antibody B02C11, whereas FVIIILS showed approximately half the signal strength and FVIIIWW/LS had sufficiently decreased affinity for mAb B02C11 that measurement of apparent affinity for VWF was unreliable (data not shown). These results indicate that the native MF sequence in the first hydrophobic spike has interactions with VWF that are more selective than interactions with phospholipid.
FV mutants with FVIII residues
Wild-type FV and FV mutants (Table 1) were expressed from COS-7 cells and purified by ion exchange chromatography. The specific activity of purified FVWT averaged 560 units/mg. Apparent affinities of FV mutants for FXa were not clearly different from wild-type FV and are not shown.
FVLL supported measurable activity of the prothrombinase complex in the absence of phospholipid vesicles (Figure 3A). This activity averaged approximately 20% of the plateau activity when phospholipid vesicles were added. The basis for the phospholipid vesicle-independent activity was further investigated, as described in “Phospholipid vesicle-independent activity” (Figure 4). Furthermore, FVLL had increased phospholipid vesicle reactivity compared with FVWT. The results indicated that changing S → L in the second hydrophobic spike either increases phospholipid affinity or number of effective binding sites, or the activity of phospholipid-bound FV.

Relative activities, membrane interaction, and affinities for FV mutants. (A) Membrane reactivity for FVWT (■) or FVLL (▾) was determined by mixing with FXa, prothrombin, and various concentrations of sonicated vesicles. The quantity of thrombin formed was measured with chromogenic substrate S-2238. The apparent affinity of FVLL was higher than FVWT. The vesicles had a composition of PS:PE:PC 4:20:76; concentrations of FV and mutants were 0.2nM; FXa, 2.5pM; and prothrombin, 2μM. Displayed represent the mean ± SEM, fitted to a single-site binding model (smooth curves). (B) Apparent FXa affinity was determined as in panel A, except that the phospholipid concentration was held constant (50μM) and the FXa concentration was varied. (C) Apparent KM for the FVa-FXa complex was determined as in panel A, except that the phospholipid concentration was constant and the prothrombin concentration varied. (A-C) Fitted results were normalized to the Bmax value for each curve to optimize comparison of the relative affinities. (D) The specific activities, phospholipid vesicle reactivity, apparent FXa affinities, and apparent prothrombin affinities (KM) for each mutant are displayed with reference to control experiments performed with wild-type FV (as in panels A-C for FVLL). The displayed values represent the mean ± SEM. Each fit was performed on a minimum of 3 separate experiments, each performed in duplicate.
Relative activities, membrane interaction, and affinities for FV mutants. (A) Membrane reactivity for FVWT (■) or FVLL (▾) was determined by mixing with FXa, prothrombin, and various concentrations of sonicated vesicles. The quantity of thrombin formed was measured with chromogenic substrate S-2238. The apparent affinity of FVLL was higher than FVWT. The vesicles had a composition of PS:PE:PC 4:20:76; concentrations of FV and mutants were 0.2nM; FXa, 2.5pM; and prothrombin, 2μM. Displayed represent the mean ± SEM, fitted to a single-site binding model (smooth curves). (B) Apparent FXa affinity was determined as in panel A, except that the phospholipid concentration was held constant (50μM) and the FXa concentration was varied. (C) Apparent KM for the FVa-FXa complex was determined as in panel A, except that the phospholipid concentration was constant and the prothrombin concentration varied. (A-C) Fitted results were normalized to the Bmax value for each curve to optimize comparison of the relative affinities. (D) The specific activities, phospholipid vesicle reactivity, apparent FXa affinities, and apparent prothrombin affinities (KM) for each mutant are displayed with reference to control experiments performed with wild-type FV (as in panels A-C for FVLL). The displayed values represent the mean ± SEM. Each fit was performed on a minimum of 3 separate experiments, each performed in duplicate.

Apparent membrane-independent activity of FV mutants resulting from copurification of phospholipid. (A) The potential presence of phospholipid was evaluated by testing the capacity of the factor FVWT and mutants to support the FXase complex. FV preparations were mixed with FVIIIa, FIXa, and FX in the absence of phospholipid vesicles. FV mutants supported FXase activity, whereas FVWT supported little or no activity. Data represent the mean ± SEM for 2 experiments for FVLL and FVWT and one experiment for the other mutants each performed in duplicate. (B) Lactadherin inhibition of FVMF-supported FXase activity was evaluated using increasing concentrations of lactadherin. Activity was inhibited with half-maximal inhibition at approximately 4nM. Data represent the mean ± SEM for a single experiment performed in duplicate. (C) Prothrombinase activity supported by CHAPS-washed FVWT and FV mutants in the absence of phospholipid vesicles was evaluated in the presence of various concentrations of lactadherin. Lactadherin inhibited activity supported by FVMF (▵) with half-maximal inhibition at approximately 16nM lactadherin. Half-maximal inhibition of activity supported by FVLL (▿) occurred at approximately 1nM, whereas half-maximal inhibition of FVMF/LL (♢) occurred at an intermediate concentration. Data represent the mean ± SEM for 2 experiments. (D) Inhibition of prothrombinase activity by phospholipase A2 was evaluated for FVLL (▿) in the absence of phospholipid vesicles and for FVWT (□) in the presence of phospholipid vesicles (inset). Phospholipase A2 inhibited prothrombinase activity under both conditions. Data represent the mean ± SEM for 3 experiments performed in duplicate.
Apparent membrane-independent activity of FV mutants resulting from copurification of phospholipid. (A) The potential presence of phospholipid was evaluated by testing the capacity of the factor FVWT and mutants to support the FXase complex. FV preparations were mixed with FVIIIa, FIXa, and FX in the absence of phospholipid vesicles. FV mutants supported FXase activity, whereas FVWT supported little or no activity. Data represent the mean ± SEM for 2 experiments for FVLL and FVWT and one experiment for the other mutants each performed in duplicate. (B) Lactadherin inhibition of FVMF-supported FXase activity was evaluated using increasing concentrations of lactadherin. Activity was inhibited with half-maximal inhibition at approximately 4nM. Data represent the mean ± SEM for a single experiment performed in duplicate. (C) Prothrombinase activity supported by CHAPS-washed FVWT and FV mutants in the absence of phospholipid vesicles was evaluated in the presence of various concentrations of lactadherin. Lactadherin inhibited activity supported by FVMF (▵) with half-maximal inhibition at approximately 16nM lactadherin. Half-maximal inhibition of activity supported by FVLL (▿) occurred at approximately 1nM, whereas half-maximal inhibition of FVMF/LL (♢) occurred at an intermediate concentration. Data represent the mean ± SEM for 2 experiments. (D) Inhibition of prothrombinase activity by phospholipase A2 was evaluated for FVLL (▿) in the absence of phospholipid vesicles and for FVWT (□) in the presence of phospholipid vesicles (inset). Phospholipase A2 inhibited prothrombinase activity under both conditions. Data represent the mean ± SEM for 3 experiments performed in duplicate.
The apparent affinity of FVLL for prothrombin was increased approximately 2-fold (Figure 3B). The specific activity of FVLL was increased approximately 5-fold in a plasma-based FV activity assay (Figure 3C), suggesting that increased phospholipid vesicle reactivity can contribute to increased activity.
Experiments like those depicted in Figure 3A and B were used to evaluate the affinities of FVMF and FVMF/LL for phospholipid, FXa, and prothrombin (supplemental Figure 3). The results are summarized in Figure 3C. FVMF had phospholipid affinity and FXa affinity that was similar to FVWT. However, this mutant supported measurable activity of the prothrombinase complex in the absence of phospholipid vesicles, similar to FVLL. The basis for this is investigated in the next section. The apparent affinity for prothrombin was decreased approximately 10-fold. However, the specific activity in a commercial prothrombin time assay remained similar to FVWT.
The specific activity and phospholipid vesicle reactivity of FVMF/LL were increased, similar to FVLL. In addition, FVMF/LL supported prothrombinase activity in the absence of phospholipid vesicles. The affinity for FXa was decreased by approximately 50%, whereas the affinity for prothrombin remained similar to FVWT. Thus, the effects of the mutation in the second hydrophobic spike dominated the phospholipid interaction and specific activity, whereas the opposing tendencies of the separate mutations were offset with near-normal prothrombin affinity.
Phospholipid vesicle-independent activity
We asked whether the phospholipid vesicle-independent activity of FV mutants (Figure 3A) was the result of phospholipid that copurified with FV from the tissue culture source. Thus, we tested the capacity of these preparations to support activity of the FXase complex (Figure 4A). The 3 mutants supported activity of the FXase complex in the absence of phospholipid vesicles, whereas FVWT supported little or no activity. There was no measurable activity in the absence of FVIII indicating that the mutants were not substituting for FVIII in the FXase complex. To confirm that the mechanism of FXase support related to phospholipid, we added lactadherin to the FXase reactions supported by FVMF (Figure 4B). Lactadherin inhibited FXase activity confirming that the FV mutant preparation was contaminated by phospholipid.10
We next asked whether the contaminating phospholipid could be stripped from the FV mutants. Wild-type FV and mutants were washed with 1% CHAPS detergent (in addition to Tween 80 and PVP-40 already present) before elution from the ion exchange column. Mutants prepared in this way no longer supported the FXase complex, indicating that contaminating phospholipid had been reduced (not shown). However, the mutants prepared in this manner still exhibited a lesser degree of prothrombinase activity without phospholipid vesicles. Thus, we performed further experiments to determine whether the phospholipid vesicle-independent prothrombinase activity of mutants was entirely the result of contaminating phospholipid.
Lactadherin efficiently inhibited prothrombinase activity of FV mutants in the absence of phospholipid vesicles (Figure 4C). The extent of inhibition was complete, as judged by the prothrombinase assay. To confirm that phospholipid vesicle-independent prothrombinase activity could be attributed to contaminating phospholipid, we evaluated inhibition by phospholipase A2 (Figure 4D). Control experiments indicated that phospholipase greatly reduced the capacity of phospholipid vesicles to support prothrombinase activity with FVWT (Figure 4D inset). Addition of phospholipase A2 to FVLL reduced the capacity to support prothrombinase activity in the absence of phospholipid vesicles. Thus, the capacity of these FV mutants to support FXase activity in the absence of phospholipid vesicles can be attributed to the presence of contaminating phospholipid that copurifies with mutant FV.
The FV mutants were purified with a CHAPS wash for use in direct phospholipid binding assays (Table 2; supplemental Figure 1B). FVLL showed lower lipid affinity than FVWT, although there was no significant difference between FVMF/LL and FVWT. FVMF did not show measurable binding, probably because of insufficient binding of the mutant to the beads. These results indicate that quasi-equilibrium affinity of immobilized FV mutants for vesicles is comparable with wild-type FV. Thus, the most likely explanation for retained phospholipid is increased importance of the slow phase of dissociation.29
FV mutants related to P textilis.
Because the FV-like molecule from P textilis is reported to have membrane-binding properties that are distinct from human FV, we hypothesized that these differences might result from altered membrane-interactive residues of the C2 domain. Sequence alignment of the C2 domains of human FV, bovine FV, human FVIII, and the FV-homologous subunit from P textilis venom (Pt-FV) revealed sequence differences in spike 3 and the amphipathic cluster on spike 4 (Figure 5). Spike 3 contains an MTTS sequence that is similar to FVIII. The sequence around spike 4 has an S → Y substitution at residue 2183. To test whether these changes in the putative membrane-binding region account for the unusual behavior of Pt-FV, we prepared a mutant with the following 5 changes: L2116M, S2117T, S2118T, E2119S, and S2183Y (FVMTTS/Y).

P textilis–motivated FV mutant. (Left) Sequence alignment of the C2 domains from human FVIII (H-FVIII-C2), human FV (H-FV-C2), bovine FV (B-FV-C2), and the FV-homologous subunit of pseutarin C from P textilis venom (Pt-FV-C2). Black bars represent the 4 lipid binding spikes with the hydrophobic residues marked with triangles. Amino acids that were mutated to make FVMTTS/Y are starred. (Right) Locations of the mutated amino acids on the C2 domain of human FV are shown with original side chains in dark gray with the rest of the backbone in light gray. Residue numbers relate to the full-length human FV. Numbering references follow HGVS standard, using Met of respective propeptides as 1.
P textilis–motivated FV mutant. (Left) Sequence alignment of the C2 domains from human FVIII (H-FVIII-C2), human FV (H-FV-C2), bovine FV (B-FV-C2), and the FV-homologous subunit of pseutarin C from P textilis venom (Pt-FV-C2). Black bars represent the 4 lipid binding spikes with the hydrophobic residues marked with triangles. Amino acids that were mutated to make FVMTTS/Y are starred. (Right) Locations of the mutated amino acids on the C2 domain of human FV are shown with original side chains in dark gray with the rest of the backbone in light gray. Residue numbers relate to the full-length human FV. Numbering references follow HGVS standard, using Met of respective propeptides as 1.
When purified without a CHAPS wash, FVMTTS/Y showed phospholipid-independent activity at least 5-fold greater than FV (Figure 6A). This activity was abolished by addition of phospholipase A2 (data not shown) or by washing with CHAPS during purification. This confirmed that the activity was the result of copurified phospholipid. CHAPS-washed FVMTTS/Y exhibited approximately twice the specific activity of FVWT in a commercial prothrombin time assay (data not shown). In the presence of saturating phospholipid, there was no difference in apparent KM (Figure 6B). In a direct phospholipid-binding assay, FVMTTS/Y showed similar phospholipid affinity as FVWT (Figure 6C). Thus, the increased activity of FVMTTS/Y may be the result of increased affinity for FXa and/or reactivity with a greater number of phospholipid binding sites.

Membrane-independent activity, apparent prothrombin and FXa affinities, and phospholipid binding affinity of FVMTTS/Y. (A) Various concentrations of FVWT or FVMTTS/Y with or without a 1% CHAPS wash during purification were mixed with FXa (0.4nM) and prothrombin (1μM) in the absence of phospholipid vesicles. Washing with 1% CHAPS during purification abolished all apparent lipid-independent activity. (B) Apparent KM for the prothrombinase complex with mutant or WT CHAPS-washed FV was determined with saturating concentrations of vesicles of composition 4:20:76 (PS:PE:PC). FVMTTS/Y showed the same apparent affinity for prothrombin as FVWT. Values represent mean ± SEM for at least 2 experiments, each performed in duplicate. (C) Direct affinity for phospholipid vesicles was evaluated for FVWT or FVMTTS/Y by immobilizing FV to mAb CBC-MOR101 covalently linked to Superose beads. Beads were incubated overnight with either FVWT or FVMTTS/Y and then mixed with various concentrations of fluorescein-labeled vesicles of composition 4:5:20:71 (PS:PE-CF:PE:PC). After 30 minutes, the quantity of vesicles bound to FVWT or FVMTTS/Y was measured by flow cytometry. The vesicle dissociation constants were 4.8 ± 1.1μM for FVMTTS/Y and 1.7 ± 0.4μM for FVWT. Data represent the mean ± SEM for at least 4 experiments.
Membrane-independent activity, apparent prothrombin and FXa affinities, and phospholipid binding affinity of FVMTTS/Y. (A) Various concentrations of FVWT or FVMTTS/Y with or without a 1% CHAPS wash during purification were mixed with FXa (0.4nM) and prothrombin (1μM) in the absence of phospholipid vesicles. Washing with 1% CHAPS during purification abolished all apparent lipid-independent activity. (B) Apparent KM for the prothrombinase complex with mutant or WT CHAPS-washed FV was determined with saturating concentrations of vesicles of composition 4:20:76 (PS:PE:PC). FVMTTS/Y showed the same apparent affinity for prothrombin as FVWT. Values represent mean ± SEM for at least 2 experiments, each performed in duplicate. (C) Direct affinity for phospholipid vesicles was evaluated for FVWT or FVMTTS/Y by immobilizing FV to mAb CBC-MOR101 covalently linked to Superose beads. Beads were incubated overnight with either FVWT or FVMTTS/Y and then mixed with various concentrations of fluorescein-labeled vesicles of composition 4:5:20:71 (PS:PE-CF:PE:PC). After 30 minutes, the quantity of vesicles bound to FVWT or FVMTTS/Y was measured by flow cytometry. The vesicle dissociation constants were 4.8 ± 1.1μM for FVMTTS/Y and 1.7 ± 0.4μM for FVWT. Data represent the mean ± SEM for at least 4 experiments.
In the presence of vesicles containing excess PS, FVMTTS/Y had a 3-fold increase in vesicle reactivity compared with FVWT (Figure 7A). This suggested that FVMTTS/Y resembles FVLL in interacting with a greater number of phospholipid binding sites while retaining a similar membrane affinity. With vesicles containing 2% PS, FVMTTS/Y had a 2-fold higher membrane reactivity than FVWT (Figure 7B) but at least 3-fold higher Vmax. When PS was eliminated from the vesicles, the Vmax of FVMTTS/Y was at least 10-fold higher than FVWT (Figure 7A-C). Under these conditions, the phospholipid vesicle reactivity of FVWT appeared lower, but the prothrombinase activity was too low to allow a quantitative comparison. Thus, these results support an interpretation in which FVMTTS/Y is able to support activity on membranes with PS content too low to support activity of the normal prothrombinase complex.

Decreased PS dependence of FVMTTS/Y activity on vesicles and platelets. (A) Activity on vesicles with high PS content was determined with 5pM CHAPS-washed FVWT or FVMTTS/Y and various concentrations of vesicle of composition 15:20:65 (PS:PE:PC). (B) Activity on vesicles with limiting PS was evaluated with vesicles of composition 2:20:78 (PS:PE:PC) using 10pM FV. (C) Activity on vesicles lacking PS was evaluated on vesicles of composition 20:80 (PE:PC) using 50pM FV. FVMTTS/Y showed increasingly higher relative prothrombinase activity with lower vesicle PS content. Concentrations of FXa and prothrombin were 1nM and 1μM, respectively. Data represent the mean ± SEM for 3 experiments, each performed in duplicate. (D) Prothrombinase activity of FVMTTS/Y on unstimulated platelets or platelets incubated for 10 minutes with 10μM thrombin receptor agonist protein was measured in the presence of FXa and prothrombin. Platelets were suspended at 4 × 107/mL, and concentrations of FV and mutant were 20pM; FXa, 25pM; and prothrombin 1μM. Data represent the mean ± SD for 2 experiments, each performed in duplicate. Probabilities were calculated using the Student t test.
Decreased PS dependence of FVMTTS/Y activity on vesicles and platelets. (A) Activity on vesicles with high PS content was determined with 5pM CHAPS-washed FVWT or FVMTTS/Y and various concentrations of vesicle of composition 15:20:65 (PS:PE:PC). (B) Activity on vesicles with limiting PS was evaluated with vesicles of composition 2:20:78 (PS:PE:PC) using 10pM FV. (C) Activity on vesicles lacking PS was evaluated on vesicles of composition 20:80 (PE:PC) using 50pM FV. FVMTTS/Y showed increasingly higher relative prothrombinase activity with lower vesicle PS content. Concentrations of FXa and prothrombin were 1nM and 1μM, respectively. Data represent the mean ± SEM for 3 experiments, each performed in duplicate. (D) Prothrombinase activity of FVMTTS/Y on unstimulated platelets or platelets incubated for 10 minutes with 10μM thrombin receptor agonist protein was measured in the presence of FXa and prothrombin. Platelets were suspended at 4 × 107/mL, and concentrations of FV and mutant were 20pM; FXa, 25pM; and prothrombin 1μM. Data represent the mean ± SD for 2 experiments, each performed in duplicate. Probabilities were calculated using the Student t test.
We asked whether FVMTTS/Y might have greater activity on platelets that have little PS exposed on the outer membrane leaflet. After activation by thrombin, platelets transiently expose a small quantity of PS31 that supports prothrombinase activity. FVMTTS/Y supported 2-fold more prothrombinase activity on unstimulated platelets than FVWT (Figure 7D). FVMTTS/Y also supported at least 2-fold more activity on platelets stimulated by thrombin receptor agonist peptide. Indeed, the activity of FVMTTS/Y on unstimulated platelets was similar to FVWT on platelets stimulated via thrombin receptor agonist peptide. These results indicate that the MTTS/Y residues of Pt-FV confer the capacity to support increased prothrombinase activity both on vesicles lacking PS and on platelets.
Discussion
We have found that conservative mutations of the membrane-interactive amino acids of the C2 domains (Figure 1) can enhance as well as diminish function of FVIII and FV. The effects of these mutations relate to number of binding sites recognized, membrane affinity, and requirement for PS. In addition to the changes in membrane interaction for FVIII and FV, there were effects on affinity for the respective substrates of the FXase and prothrombinase complexes. Further, the mutations altered the linkage between membrane binding and activation of the respective cofactor-enzyme complexes. Thus, the results indicate that the specific membrane-interactive amino acids influence cofactor functionality and reversibility of membrane interaction as well as membrane-binding affinity.
The results in this report are in agreement with prior reports evaluating the membrane-interactive roles of amino acids on spike 1 and spike 3.11,12 The apparent affinity of immobilized FVIII for binding sites on phospholipid vesicles (Table 2) can be estimated by dividing the phospholipid concentration by the number of molecules per binding site on sonicated vesicles, approximately 500.2,43 The resulting dissociation constant, approximately 1nM, is in general agreement with solution measurements. Prior results did not clearly identify a membrane interactive role for the LS motif on spike 3 of FV,12 although a role was clear for the corresponding LL motif in FVIII. Results in this report indicate that the LL motif can have a major membrane-interactive role when substituted into FV. This implies that the native LS sequence either participates in membrane binding or is positioned in intimate contact with the membrane. Likewise, our results are consistent with the reported participation of the amphipathic cluster on loop 2313 to 2315 (spike 4) of FVIII in membrane binding.22
Our results are also in agreement with prior reports indicating that the 2 hydrophobic spikes of the FVIII C2 domain contribute to VWF binding.11 These spikes also contribute to affinity for mAb B02C11, with the MF motif making a relatively higher contribution than LL.44 However, we saw little difference between bound FVIIIWT and FVIIIWW in the absence of VWF, suggesting that the MF to WW mutation has relatively little effect on B02C11 binding except synergistically in FVIIIWW/LS. In addition, because B02C11 was the capture antibody for our assay, any lower affinity of B02C11 for FVIIILS would cause an overestimation of the affinity of VWF for that mutant. The results indicate that the VWF interaction has greater specificity for the amino acids of the hydrophobic spikes than the phospholipid membrane sites.
The current results underscore our recent observation that cooperative membrane binding of the FVIII C1 and C2 domains influences activity of FVIII as well as membrane binding affinity.36,45 The prior report indicated that membrane binding of FVIII via the C1 and C2 domains can be decoupled from full activation of the FXase complex. A similar finding for FV was evident in the current study where FVMTTS/Y had little change in affinity for membranes but 10-fold greater activity in the prothrombinase complex when PS was absent. These results imply that a change in orientation or conformation is linked to membrane binding and confers full cofactor activity.
Our results imply that conservative mutations in the C2 domain can alter membrane-dependent activity of FVIII or FV in 3 ways. First, they can affect membrane affinity or slow membrane dissociation. This is evident with mutants FVIIIWW and FVIIIWW/LS that have 2-fold increase in membrane affinity versus the wild-type control. It also appears evident in the FV mutants that copurify with phospholipid. Second, the mutations appear to affect binding site specificity. This is evident for mutants FVLL and FVMF/LL. These have preserved affinity for phospholipid vesicles and normal activity in a clotting activity with excess phospholipid. However, they have increased phospholipid interaction, probably representing capacity to bind and gain full activity on phospholipid binding sites that would not support binding of the respective wild-type cofactor. Third, and possibly most interesting, mutant FVMTTS/Y is activated by vesicles that bind wild-type FV but confer a much lower degree of FV activity. The same property is evident on unstimulated and stimulated platelets, where there is little exposed PS. Further studies will be required to determine whether these properties translate to increased prothrombotic activity in a more complex biologic system.
We note an apparent discrepancy between the normal membrane affinities for FVLL, FVMF/LL, and FVMTTS/Y and the capacity of these mutants to retain more phospholipid during purification than FVWT. We rationalize the apparent discrepancy by noting the slow second phase of membrane dissociation for FV.29 Our quasi-equilibrium membrane binding experiments had incubation times long enough to interpret the membrane reactivity of mutant FV for prothrombinase assays and binding of vesicles to immobilized FV (Figures 3 and 6; supplemental Figures 1B and 3A). However, the incubation times were not long enough to enable the slow phase of dissociation to have its full impact on the measured membrane affinity. Thus, we hypothesize that these mutants have a slower second component of membrane dissociation that explains the retained phospholipid.
Prior studies indicate that both activated FV and activated FVIII, in complex with FXa and FIXa, respectively, are partially activated by water-soluble PS.34,35 These data indicated partial decoupling of membrane binding and cofactor activation. Our current report is a counterpart, indicating enhanced coupling of membrane binding and cofactor activation in the absence of PS. Progress in testing hypotheses about this coupling will be enhanced by information about the conformation and dynamics of membrane bound FVIII or FV versus the respective phospholipid-free structures.46,47
The FV mutants exhibited phospholipid binding that was not reversible over the time course of purification. The implication is that the dissociation of FV from COS-7 membrane fragments is slowed to the point where it is not completed during purification. We postulate that this mechanism may have led to retained phospholipid and may have been a contributing factor to the apparent phospholipid independence of recombinant, purified Pt-FV.14 The persistent phospholipid binding cannot be readily explained by change in the hydrophobic surface area because FVMF had diminished hydrophobic surface area compared with FVWT. Our current hypothesis is that dissociation of FV from a phospholipid membrane is accompanied by the conformational change previously noted in the crystal structures of the FV C2 domain.20 The mutations may prevent completion of the conformational change. Regardless of the mechanism, these results indicate that high affinity, reversible membrane binding function of FV is delicately balanced so that modest changes can interfere with full functionality.
The C2 domains of both FVIII and FV undergo conformational changes that are related to membrane binding. The conformational flexibility of the FV C2 domain is illustrated by the different conformation of the largest membrane-interactive loop in the 3 crystal structures.20 Some conformational flexibility of the FVIII C2 domain is illustrated by comparison of the crystal structures with the cocrystal of the FVIII C2 domain with monoclonal antibody B02C11 and with the low molecular weight inhibitor of membrane binding.22 The hairpin turns displaying the membrane interactive spikes 1 and 3 exhibit lateral flexibility, and the 2 strands of the third loop on the membrane-interactive surface rotate approximately 70 degrees.48 The C2 domain of FVIII also participates in a conformational change in response to ESH8, a monoclonal antibody with an epitope that does not overlap with B02C11.49,50 The functional consequences of ESH8-induced conformational change include increased affinity for von Willebrand factor and decreased affinity for phospholipid membranes.49,51 The structural nature of this conformational change remains unknown. Thus, it is possible that the conservative substitutions constrained the normal conformational changes as a mechanism that influenced membrane binding.
Conservative mutations in both the first and third hydrophobic spikes influenced apparent affinities of both the FXase complex and the prothrombinase complex for the respective substrates. In both cases, the WW sequence in the first spike led to increased affinity and LL in the second spike led to increased affinities for FX, and prothrombin, respectively. The mechanism for these changes could relate to altered geometry of the enzyme complex related to the interaction of each spike with the membrane, or they could be explained by a direct interaction between the respective hydrophobic spike(s) and the substrates as recently proposed.37 Further experiments will be necessary to distinguish between these possible explanations.
Our experiments with FVMTTS/Y highlight the potential importance of properties these residues may confer to the membrane binding of Pt-FV. Our results show that these conservative mutations lead to tenacious phospholipid binding, leading to retained phospholipid during purification. Retained phospholipid is unlikely to have in vivo significance because P textilis venom proteins include phospholipases, which seem likely to degrade any retained phospholipid and therefore inhibit corresponding activity.52 More interesting is the activity of FVMTTS/Y on platelets and membranes lacking PS, a lipid that is normally critical for full FV function. These results suggest that Pt-FV may be responsible for the ability of pseutarin C to activate prothrombin on cell membranes that would normally not support significant coagulation activity, such as unstimulated platelets.
Our results suggest the possibility that the phospholipid-independent activity attributed to recombinant Pt-FV14 may actually be the result of retained phospholipid. This possibility will probably soon be resolved by experiments, such as those in this report. However, our results may also be interpreted while assuming that that the report of Pt-FV phospholipid-independent activity is accurate. Our results suggest that the mutations we identified are not sufficient to allow phospholipid-independent activity, even though they allow for prothrombinase activity on membranes that would not normally support it. If the phospholipid-independent activity of Pt-FV is subsequently confirmed, our results indicate that other areas of the protein are probably responsible for this phenomenon. In that case, the membrane-binding properties of Pt-FV might still function to localize the fully active prothrombinase-like complex to membrane sites that would not support binding and assembly of the native prothrombinase complex.
Presented in abstract form at the 49th Annual Meeting of the American Society of Hematology, Atlanta, GA, December 2007 and at the 53rd Annual Meeting of the American Society of Hematology, San Diego, CA, December 2011.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Jialan Shi MD, PhD, VA Boston Healthcare System, for assistance with assay development; Patricia Price MS, VA Boston Healthcare System, for excellent technical assistance; and Colin Kretz PhD, University of Michigan, for purification of the FV MTTS/Y mutant.
This work was supported in part by the Department of Veterans Affairs (G.E.G.), the National Hemophilia Foundation (G.E.G.), and the National Institutes of Health (grants PO1 HL057346 and RO1 HL52173, R.J.K.).
National Institutes of Health
Authorship
Contribution: G.E.G. designed research, analyzed results, and wrote the manuscript; V.A.N. performed experiments, analyzed results, and edited figures and the manuscript; H.M. produced and purified mutants and performed experiments; and S.W.P. and R.J.K. designed research and revised the manuscript.
Conflict-of-interest disclosure: G.E.G. owns intellectual property related to the use of lactadherin. No revenues have accrued to the inventors from these patents. The remaining authors declare no competing financial interests.
The current affiliation for R.J.K. is Sanford Burnham Medical Research Institute, La Jolla, CA.
Correspondence: Gary E. Gilbert, Veterans Administration Boston Healthcare System, 1400 VFW Parkway, West Roxbury, MA 02132; e-mail: gary_gilbert@hms.harvard.edu.

