Abstract
A major obstacle to using bone marrow cell-based therapies for ischemic cardiovascular disease is that transplanted cells must survive in an ischemic microenvironment characterized by low oxygen, glucose, and pH. We demonstrate that treatment of bone marrow-derived angiogenic cells (BMDACs) with dimethyloxalylglycine, an α-ketoglutarate antagonist that induces hypoxia-inducible factor 1 (HIF-1) activity, results in metabolic reprogramming of these cells, with increased glucose uptake, decreased O2 consumption, increased lactate production, decreased reactive oxygen species, and increased intracellular pH. These effects are dependent on HIF-1, which transactivates target genes encoding metabolic enzymes and membrane transporters. Dimethyloxalylglycine-treated BMDACs have a significant survival advantage under conditions of low O2 and low pH ex vivo and in ischemic tissue. Combined HIF-1α-based gene and cell therapy reduced tissue necrosis even when BMDAC donors and ischemic recipient mice were 17 months old, suggesting that this approach may have therapeutic utility in elderly patients with critical limb ischemia.
Introduction
Ischemia induces the production of angiogenic factors, which trigger sprouting of new capillaries (angiogenesis) and remodeling of preexisting vessels (arteriogenesis). In mouse models, limb ischemia induced by femoral artery ligation leads to the mobilization of a heterogeneous population of bone marrow-derived angiogenic cells (BMDACs), which promote the rapid recovery of perfusion that is required to maintain tissue viability. Included among BMDACs are endothelial progenitor cells, which are incorporated into new or remodeling vessels,1 and myeloid, mesenchymal, and hematopoietic stem/progenitor cells, which may secrete angiogenic factors/cytokines that activate resident vascular cells.2-4 Recent data suggest that the majority of BMDACs identified in many experimental models are proangiogenic myeloid cells.5
The recruitment of BMDACs to ischemic tissue is mediated by angiogenic factors that are produced in response to tissue hypoxia through the action of hypoxia-inducible factor 1 (HIF-1). HIF-1 is a heterodimer consisting of O2-regulated HIF-1α and constitutively expressed HIF-1β subunits.6 HIF-2α is also O2-regulated, dimerizes with HIF-1β, and has been implicated in vascular responses to ischemia.7,8 HIF-1α and HIF-2α are subjected to prolyl hydroxylation in a reaction that requires O2 and α-ketoglutarate and leads to their ubiquitination and proteasomal degradation.9 Hydroxylation is inhibited under hypoxic conditions, leading to the accumulation of HIF-1α and HIF-2α. The hydroxylases are also inhibited by the administration of a competitive antagonist of α-ketoglutarate, such as dimethyloxalylglycine (DMOG), which leads to HIF-1 activation under nonhypoxic conditions.10 HIF-1 directly activates transcription of the genes encoding vascular endothelial growth factor, stromal-derived factor 1, stem cell factor, and angiopoietin 2, which are angiogenic cytokines that both activate resident vascular cells as well as serve as homing signals for the recruitment of BMDACs.11-14
BMDACs hold considerable promise as a therapeutic tool for ischemic diseases, provided that major obstacles to their use are overcome. One therapeutic approach has involved the local injection of BMDACs directly into ischemic tissue. However, the introduction of millions of additional cells at a site where oxygen and energy substrates are already insufficient to maintain tissue viability is problematic. The alternative strategy of systemic injection relies on the hypothesis that those BMDACs capable of responding to signals arising in the ischemic tissue will home to (and be retained at) the desired site, thereby allowing in vivo selection of proangiogenic cells.
Recent studies have demonstrated that the ischemia-induced activation of HIF-1 and subsequent recruitment of BMDACs are impaired by aging11,15,16 and diabetes,17,18 which was overcome by local intramuscular injection of AdCA5, a recombinant adenovirus encoding a constitutively active form of the HIF-1α subunit, resulting in improved recovery of blood flow and prevention of tissue damage after femoral artery ligation of young and middle-aged mice.11,19 Local AdCA5 injection was not sufficient to mediate recovery of blood flow in old mice, reflecting a deficiency of responding BMDACs. However, the combination of IM AdCA5 injection followed by intravenous BMDAC injection resulted in limb salvage.20 This approach was only successful if the BMDACs were cultured for 4 days in the presence of the HIF-1 inducer DMOG before injection. Whereas intramuscular AdCA5 injection was sufficient to induce homing of injected BMDACs to ischemic tissue, DMOG treatment of the cells improved their retention in the ischemic limb because of the HIF-1-dependent expression of β2-integrins, which facilitated binding of BMDACs to hypoxic vascular endothelial cells.20
Although these results provided molecular mechanisms for the homing and retention of BMDACs in ischemic tissue, they did not address whether HIF-1 activation also increased the survival of BMDACs once recruited. In tissue culture cell lines, HIF-1 has been shown to mediate a series of metabolic responses to hypoxia that maintain energy, pH, and redox homeostasis. First, HIF-1 coordinates a subunit switch in the composition of cytochrome c oxidase (COX4I2 on, COX4I1 off) that increases its efficiency under hypoxic conditions.21 Second, HIF-1 activates transcription of the PDK1 gene encoding pyruvate dehydrogenase kinase, thereby inactivating pyruvate dehydrogenase and shunting pyruvate away from mitochondria.22,23 Third, HIF-1 activates BNIP3 expression, which triggers selective mitochondrial autophagy.24 These 3 adaptations serve to reduce the generation of reactive oxygen species (ROS) under hypoxic conditions. Fourth, HIF-1 activates transcription of genes encoding glucose transporters (GLUT1 and GLUT3) and glycolytic enzymes, including lactate dehydrogenase A (LDHA), to increase flux from glucose to lactate.25-27 Fifth, HIF-1 maintains intracellular pH homeostasis through the expression of carbonic anhydrase 9 (CAR9) and sodium-hydrogen exchanger 1 (NHE1), which export H+, as well as the monocarboxylate transporter 4 (MCT4), which exports lactate.28-30
In this study, we investigated whether DMOG-induced metabolic reprogramming increases BMDAC survival ex vivo and in vivo. Moreover, because advanced peripheral arterial disease has highest incidence in the elderly, we investigated whether combined AdCA5 and DMOG-treated BMDAC therapy is effective when both donor and recipient mice are of advanced age.
Methods
Mouse strains
The Hif1atm1jhu mouse strain was generated by homologous recombination to replace exon 2 of the Hif1a gene with a neo expression cassette in the J1 line of embryonic stem cells, originally derived from 129/Sv mice, followed by injection of the genetically modified cells into C57BL/6J blastocysts.25 The strain has been maintained by Hif1a+/− × Hif1a+/+ intercrosses for the last 12 years, and mice therefore segregate the Hif1atm1jhu allele on an outbred 129 × BL/6 genetic background. The study was approved by the Johns Hopkins University Animal Care and Use Committee.
Bone marrow cell isolation, culture, and DMOG treatment
Bone marrow was isolated from 3- or 17-month-old male mice. BMDACs were cultured for 4 days in microvascular endothelial growth media 2 (EGM2-MV; Lonza) in the presence of vehicle (0.01% dimethyl sulfoxide [DMSO]) or DMOG (100μM in DMSO).20
Hypoxic cell culture
BMDACs were cultured in EGM2-MV and subjected to hypoxia in a modular chamber (Billups-Rothenberg) flushed for 3 minutes at 2 psi with a hypoxic gas mixture (1% O2/5% CO2/94% N2). Afterward, the chamber was sealed and incubated at 37°C for 96 hours. Nonhypoxic cultures (20% O2) were kept in a standard tissue culture incubator (95% air and 5% CO2).
Ex vivo cell viability and apoptosis measurements
Cell viability was determined using the Trypan blue exclusion method. In addition, for the hypoxia/acidosis challenge experiments, we used the LIVE/DEAD Violet Fixable Dead Cell Stain Kit (Invitrogen) to quantify cell death by flow cytometry. At least 5000 events per sample were acquired. Apoptosis was measured by flow cytometry using an annexin V-7-amino-actinomycin D apoptosis detection kit (BD Biosciences) according to the manufacturer's instructions.
Cell cycle analysis
Real-time RT-PCR
Total RNA was extracted from BMDACs using TRIzol (Invitrogen), precipitated, and treated with DNase I (Ambion). A total of 1 μg of total RNA was used for cDNA synthesis using the iScript kit (Bio-Rad). Real-time quantitative reverse-transcription polymerase chain reaction (RT-PCR) was performed using Maxima SYBR Green Master Mix (Fermentas) and the iCycler Real-time PCR Detection System (Bio-Rad). We tested for the best combination of reference genes for BMDACs using a model-based variance estimation approach33 and calculated a normalization factor for each biologic replicate as the geometric mean of the 3 most stable reference genes under our experimental conditions (B2m, Rpl13a, and Ppia). The expression of target mRNAs was corrected for efficiency, calculated from each fluorescence amplification curve.34 Relative expression changes were obtained as previously described.35 Validated primer pairs were obtained from the Harvard PrimerBank database (Table 1).36
Immunoblot assay
A total of 20 to 30 μg of protein extracted from cells with RIPA buffer was subjected to immunoblot assays with anti-LDHA (1:500, Novus), anti-PDK1 (1:1000, Novus), or anti-BNIP3 (1:1000, Abcam) antibodies.
Extracellular lactate measurements
Conditioned media was obtained from BMDAC cultures and immediately frozen in liquid N2 until assayed using a lactate assay kit (K607–100; Biovision) according to the manufacturer's instructions.
Measurement of mitochondrial mass, ROS, and glucose uptake
Mitochondrial mass, intracellular ROS levels, or glucose uptake was measured by staining cells with 10nM nonyl acridine orange, 1μM dihydrodichlorofluorescein diacetate, or 150μM 2-[N-(7-nitrobenz-2oxa-1,3-diazol-4-yl)amino]-2-deoxy-D-glucose (Invitrogen), respectively, at 37°C for 15 to 30 minutes in phosphate-buffered saline (PBS). Stained cells were analyzed immediately in a LSR II flow cytometer (BD Biosciences) with a minimum of 30 000 events acquired per sample.
Mitochondrial DNA quantification
Total DNA was isolated from BMDAC lysates. The amount of mitochondrial DNA relative to nuclear genomic DNA was determined by quantitative PCR using primers (listed in Table 2) for adenosine triphosphate synthase 6 (mt-ATP6; mitochondrial) and Nme1 (nuclear). Normalized mitochondrial (Mt) DNA levels were corrected for differences in PCR efficiency using the following equation35 :
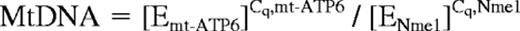
where E is the efficiency for each primer pair and Cq is the threshold cycle obtained as described previously.34 The ratio of mitochondrial to nuclear DNA after DMOG or vehicle treatment was calculated as MtDNADMOG/MtDNAvehicle.
Oxygen consumption measurements
BMDACs were resuspended in serum-free EGM2-MV at 2 × 106 cells/mL, and oxygen consumption (JO2) was measured with an Oxygraph device (Hansatech Instruments), which uses a Clark-type electrode to measure JO2. Partial pressure of dissolved O2 in water was calculated as described previously.37
HIF-1α flow cytometry
BMDACs were detached with PBS/1mM ethylenediaminetetraacetic acid and resuspended in PBS, fixed in 4% paraformaldehyde, permeabilized in PBS/0.5% Triton X-100, and incubated with a rabbit polyclonal anti-HIF-1α antibody (Novus Biologicals) at a 1:100 dilution for 30 minutes at room temperature. After washing, cells were incubated with an Alexa 594-conjugated goat antirabbit secondary antibody (1:500; Invitrogen) for 15 minutes. BMDACs incubated only with the secondary antibody were used as negative controls. The percentage of HIF-1α+ cells was quantified in a LSR-II flow cytometer (BD Biosciences). A minimum of 100 000 events per biologic replicate were acquired. Specificity of the antibody was confirmed by analyzing HIF-1α wild-type and null mouse embryonic fibroblasts38 using the same protocol.
Intracellular pH and NHE activity measurements
Intracellular pH was measured as described previously.28 BMDACs were incubated with the membrane-permeant pH-sensitive fluorescent dye BCECF-AM for 1 hour at 37°C under 20% O2/5% CO2, washed with N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid-buffered salt solution (130mM NaCl, 5mM KCl, 1.2mM MgCl2, 1.5mM CaCl2, 10mM glucose, 10mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid [pH 7.4]) for 15 minutes at 37°C to remove extracellular dye and allow complete deesterification of cytosolic dye. A 2-point calibration was created from fluorescence measurements as intracellular pH was adjusted with KOH from 6.5 to 7.5. Baseline intracellular pH was measured in BMDACs for 3 minutes, and values were averaged to obtain a mean value for each cell. A standard ammonia pulse technique was used to measure NHE activity by exposure to NH4Cl at pH 7.4 for 3 minutes, causing alkalinization because of NH3 influx and buffering of intracellular H+. Washout of NH4Cl in the absence of extracellular Na+ using a Na+- and NH4+-free solution for 10 minutes resulted in acidification because of rapid diffusion and washout of NH3, leaving behind H+ ions. Restoration of extracellular Na+ activates Na+/H+ exchange and recovery to basal pH. The rate of Na+-dependent recovery from intracellular acidification corresponds to Na+/H+ exchange activity.
CAR activity assay
Total CAR activity was measured by the Tris-imidazole method.39 Briefly, the change in absorbance at 407 nm of BMDACs protein homogenates was recorded in a Biomate 3 UV-Vis spectrophotometer (Thermo Scientific). Mouse erythrocyte lysates were used as positive controls. CAR activity was corrected for total protein content as measured by the Bradford method. An equal amount of homogenate from each biologic replicate was boiled for 30 minutes and used as a measure for the uncatalyzed CAR reaction, to calculate specific CAR activities.
Detection of BMDAC homing to ischemic muscle
Gastrocnemius muscles were excised bilaterally and incubated in tissue lysis buffer (5mM ethylenediaminetetraacetic acid, 0.2% SDS, 200mM NaCl, 100mM Tris-HCl [pH 7.5], and 100 μg/mL Proteinase K) for 18 hours at 55°C. Genomic DNA was isolated, adjusted to 25 ng/μL and frozen at −70°C until assayed. Quantitative PCR for Sry gene sequences was performed using DNA isolated from the ischemic and nonischemic limb of each mouse as previously described.20 Uniform template loading was confirmed with primers against the autosomal gene Nme1. Nontemplate controls were included in each quantitative PCR run and were devoid of amplification. Primer sequences are listed in Table 2.
Femoral artery ligation
Ischemia was induced by complete ligation and excision of the left femoral artery and its branches, as previously described.11,20 The femoral artery of 3- or 17-month-old female mice was dissected and excised between the origin of the superficial epigastric and saphenous arteries. Preoperative and postoperative limb perfusion was measured by laser Doppler perfusion imaging (Moor Instruments) using a near-infrared laser to measure subcutaneous blood flow as a function of light scattering by Doppler shift. After confirmation of successful limb ischemia, perfusion on days 3, 7, 14, 21, and 28 was measured and expressed as the limb perfusion ratio, which was calculated as the flow signal in the ischemic foot divided by the signal in the nonischemic foot. Ischemic tissue damage and motor impairment were assessed using a semiquantitative clinical score.40 The tissue damage scoring system was modified to take into account the severity of necrosis in very old mice.
Adenoviral treatment
Before closure of the surgical site after femoral artery ligation, mice were injected with either saline or adenovirus AdCA5, which encodes a constitutively active form of HIF-1α that is expressed in an O2-independent manner. A total of 2 × 108 plaque-forming units (pfu) were injected, divided among 3 sites in the adductor muscle and one site in the gastrocnemius muscle of the ischemic limb.
Data analysis
All data were expressed as mean plus or minus SEM. Differences between 2 experimental groups were assessed using Student t test. Three or more groups were analyzed with one- or 2-way analysis of variance (ANOVA) followed by post hoc comparisons with the Bonferroni test. Multivariate analysis was used to analyze the effect of DMOG, hypoxia, low pH, and glucose on ex vivo BMDAC survival. Cell cycle data were analyzed with the χ2 test. Ratios and percentages were normalized using a log10 conversion before analyses with parametric tests. Results were considered statistically significant if P < .05.
To estimate the survival half-life of Sry signal in ischemic gastrocnemius muscles, we modeled the data using a one phase exponential decay equation:
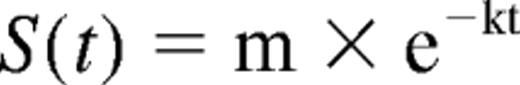
where S(t) is the Sry signal, expressed as copy numbers per 100 ng of total genomic DNA at time t; m represents S at t = 0 and k is the decay constant. Survival half-life was calculated as ln(2)/k, or 0.69/k (measured in hours). The effect of DMOG on survival half-life was analyzed with an F test. Correlation coefficients were consistently high (r > 0.95; P < .01) in all experiments.
Results
DMOG increases BMDAC proliferation and survival under nonstress conditions
To study the effect of DMOG on BMDACs ex vivo, we isolated bone marrow from 3-month-old female mice, cultured equal numbers of cells in endothelial growth medium for 72 to 96 hours in the presence of 100μM DMOG (dissolved in DMSO) or vehicle (DMSO) under standard tissue culture conditions of 95% air/5% CO2 (i.e., 20% O2), and counted the number of live cells using the Trypan blue exclusion method. The majority of bone marrow cells, which are hematopoietic cells and their progenitors, die under these conditions, whereas (by definition) BMDACs survive and proliferate. After 4 days, more than 90% of cells take up acetylated low-density lipoprotein and bind Bandeira simplificifolia agglutinin-1, which are commonly used criteria for identifying BMDACs.11,41,42 DMOG treatment significantly increased the percentage of viable cells by 1.9-fold at 72 hours and 4.1-fold at 96 hours (P < .001; Figure 1A). The increased cell numbers in the DMOG-treated cultures were the result of a 2.3-fold decrease in apoptosis (17.1% [vehicle] vs 7.6% [DMOG]; P < .001; Figure 1B) and a 4.0-fold increase in the percentage of cells in the S/G2/M phase of the cell cycle (6.4% [vehicle] vs 25.5% [DMOG]; P < .001; Figure 1C). Thus, culturing bone marrow cells in the presence of DMOG greatly increases the yield of BMDACs.

DMOG increases BMDAC viability by decreasing apoptosis and increasing proliferation. (A) Equal numbers of BMDACs were cultured in endothelial growth media in the presence of vehicle or DMOG for 4 days, and viable cells were counted using the Trypan blue exclusion method. *P < .001, DMOG vs vehicle. #P < .001 vs time 0; Bonferroni post hoc test after 2-way ANOVA. The overall effects of time and DMOG treatment on BMDAC survival were significant (P < .001). (B) Apoptosis was detected at day 4 as phycoerythrin-conjugated annexin V (annexin V-PE) positive/7-amino-actinomycin D (7-AAD) negative cells (green rectangle) using flow cytometry. (C) Cell cycle distribution of BMDACs stained with propidium iodide and analyzed by flow cytometry. Data are mean ± SEM (n = 3 or 4).
DMOG increases BMDAC viability by decreasing apoptosis and increasing proliferation. (A) Equal numbers of BMDACs were cultured in endothelial growth media in the presence of vehicle or DMOG for 4 days, and viable cells were counted using the Trypan blue exclusion method. *P < .001, DMOG vs vehicle. #P < .001 vs time 0; Bonferroni post hoc test after 2-way ANOVA. The overall effects of time and DMOG treatment on BMDAC survival were significant (P < .001). (B) Apoptosis was detected at day 4 as phycoerythrin-conjugated annexin V (annexin V-PE) positive/7-amino-actinomycin D (7-AAD) negative cells (green rectangle) using flow cytometry. (C) Cell cycle distribution of BMDACs stained with propidium iodide and analyzed by flow cytometry. Data are mean ± SEM (n = 3 or 4).
Altered glycolytic and mitochondrial metabolism in DMOG-treated BMDACs
Next, we investigated the metabolic consequences of DMOG treatment. Staining of BMDACs with nonyl acridine orange, which binds to cardiolipin in mitochondrial membranes independently of mitochondrial membrane potential, revealed a 30% decrease in mitochondrial mass in DMOG-treated cells (Figure 2A top panels). As an alternative means of estimating mitochondrial mass, we found a 42% decrease in the mitochondrial/nuclear DNA ratio (Figure 2B) in DMOG-treated compared with vehicle-treated BMDACs (hereafter referred to as BMDAC-D and BMDAC-V, respectively). Reduction in mitochondrial mass was accompanied by a 61% reduction in O2 consumption (Figure 2C) and 58% decrease in ROS levels as determined by dihydrodichlorofluorescein diacetate staining (Figure 2A middle panels). Underlying this metabolic reprogramming was a significant increase in the levels of mRNAs encoding BNIP3, COX4I2, and PDK1 as determined by quantitative RT-PCR (Figure 2D).
![Figure 2. DMOG elicits transcriptional and metabolic responses in BMDACs. (A) BMDACs were cultured in the presence of vehicle (V) or DMOG (D). Mitochondrial mass (top panels), ROS (middle panels), and glucose uptake (bottom panels) were measured in BMDACs by flow cytometry after staining with the fluorescent dyes nonyl acridine orange (NAO), 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA), and 2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino]-2-deoxy-D-glucose (2-NBDG), respectively (n = 4-6). (B) Mitochondrial DNA content relative to nuclear genomic DNA was determined by quantitative PCR (n = 6). (C) Oxygen consumption (JO2) was analyzed in BMDACs suspended in serum-free EGM2-MV media (n = 6). (D) Quantitative RT-PCR analysis of HIF-1 target genes encoding metabolic regulators in BMDACs cultured in vehicle or DMOG for 4 days (n = 4-6). ETFA and TBP are not HIF-1 targets and were used as negative controls. Gray shaded area indicates ± 2-fold of control. (Inset) Immunoblot assay showing BNIP3, LDHA, and PDK1 protein levels. (E) Extracellular lactate content of BMDAC cultures (n = 9). (F) HIF-1α protein expression in BMDACs was determined by immunofluorescence using flow cytometric analysis (left panel). Gray histogram corresponds to the negative control omitting the primary antibody. Data are mean ± SEM (n = 6; right panel). *P < .05, DMOG vs vehicle, by Student t test in all panels of this figure. Data are mean ± SEM.](/view-large/figure/7155764/zh89991170380002.jpeg)
DMOG elicits transcriptional and metabolic responses in BMDACs. (A) BMDACs were cultured in the presence of vehicle (V) or DMOG (D). Mitochondrial mass (top panels), ROS (middle panels), and glucose uptake (bottom panels) were measured in BMDACs by flow cytometry after staining with the fluorescent dyes nonyl acridine orange (NAO), 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA), and 2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino]-2-deoxy-D-glucose (2-NBDG), respectively (n = 4-6). (B) Mitochondrial DNA content relative to nuclear genomic DNA was determined by quantitative PCR (n = 6). (C) Oxygen consumption (JO2) was analyzed in BMDACs suspended in serum-free EGM2-MV media (n = 6). (D) Quantitative RT-PCR analysis of HIF-1 target genes encoding metabolic regulators in BMDACs cultured in vehicle or DMOG for 4 days (n = 4-6). ETFA and TBP are not HIF-1 targets and were used as negative controls. Gray shaded area indicates ± 2-fold of control. (Inset) Immunoblot assay showing BNIP3, LDHA, and PDK1 protein levels. (E) Extracellular lactate content of BMDAC cultures (n = 9). (F) HIF-1α protein expression in BMDACs was determined by immunofluorescence using flow cytometric analysis (left panel). Gray histogram corresponds to the negative control omitting the primary antibody. Data are mean ± SEM (n = 6; right panel). *P < .05, DMOG vs vehicle, by Student t test in all panels of this figure. Data are mean ± SEM.
DMOG elicits transcriptional and metabolic responses in BMDACs. (A) BMDACs were cultured in the presence of vehicle (V) or DMOG (D). Mitochondrial mass (top panels), ROS (middle panels), and glucose uptake (bottom panels) were measured in BMDACs by flow cytometry after staining with the fluorescent dyes nonyl acridine orange (NAO), 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA), and 2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino]-2-deoxy-D-glucose (2-NBDG), respectively (n = 4-6). (B) Mitochondrial DNA content relative to nuclear genomic DNA was determined by quantitative PCR (n = 6). (C) Oxygen consumption (JO2) was analyzed in BMDACs suspended in serum-free EGM2-MV media (n = 6). (D) Quantitative RT-PCR analysis of HIF-1 target genes encoding metabolic regulators in BMDACs cultured in vehicle or DMOG for 4 days (n = 4-6). ETFA and TBP are not HIF-1 targets and were used as negative controls. Gray shaded area indicates ± 2-fold of control. (Inset) Immunoblot assay showing BNIP3, LDHA, and PDK1 protein levels. (E) Extracellular lactate content of BMDAC cultures (n = 9). (F) HIF-1α protein expression in BMDACs was determined by immunofluorescence using flow cytometric analysis (left panel). Gray histogram corresponds to the negative control omitting the primary antibody. Data are mean ± SEM (n = 6; right panel). *P < .05, DMOG vs vehicle, by Student t test in all panels of this figure. Data are mean ± SEM.
To compensate for the observed reduction in oxidative metabolism, BMDAC-D manifested significantly increased glucose uptake, as determined by 2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino]-2-deoxy-D-glucose staining (Figure 2A bottom panels), as well as increased lactate production and secretion (Figure 2E). Underlying these metabolic alterations were significant increases in the levels of mRNAs encoding GLUT1, GLUT3, LDHA, and MCT4 as determined by quantitative RT-PCR (Figure 2D). We confirmed a sustained increase in HIF-1α protein levels in BMDAC-D at 96 hours using flow cytometry and indirect immunofluorescence (Figure 2F), thereby providing a putative molecular basis for the metabolic reprogramming of BMDAC-D because the BNIP3, COX412, GLUT1, GLUT3, PDK1, HK2, LDHA, and MCT4 mRNAs analyzed in Figure 2D are encoded by genes that are known to be transactivated by HIF-1 under hypoxic conditions.
Increased survival of DMOG-treated BMDACs under hypoxic and acidic conditions
When BMDACs home to ischemic tissue, they encounter an acidic, hypoxic, and nutrient-depleted environment. We simulated these conditions ex vivo by incubating BMDACs in the presence of N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid-buffered Tyrode solutions, at pH 7.4 or 6.4, under hypoxic (1% O2) or nonhypoxic (20% O2) conditions and at several different glucose concentrations. Survival time-course experiments revealed that DMOG treatment improved BMDAC survival under acidic conditions (pH 6.4), at both 20% and 1% O2 (Figure 3A). The beneficial effect of DMOG treatment under acidic conditions was observed over a wide range of glucose concentrations (Figure 3B). Multivariate analysis showed a significant interaction between hypoxia and acidosis (P < .001), such that DMOG treatment had an even greater effect on BMDAC survival under combined low O2/pH conditions. In contrast, there was no demonstrable effect of glucose concentration on BMDAC survival under these experimental conditions because the survival curves were superimposed over one another (Figure 3B).
![Figure 3. Increased survival of DMOG-treated BMDACs under hypoxic and acidic conditions ex vivo. (A) Effect of pH and hypoxia on BMDAC-D survival at 5.6mM glucose (▴). After culture for 4 days, equal numbers of BMDACs were exposed to low pH (6.4), nonhypoxic (20% O2), and hypoxic (1% O2) conditions. Cell viability was measured using the Trypan blue exclusion method. (B) Effect of glucose concentration on BMDAC-V and BMDAC-D survival under low pH (6.4) and hypoxia (1% O2). After culture for 4 days, BMDAC viability was measured with the LIVE/DEAD Violet fluorescent dye and flow cytometry. Survival curves under 3 glucose concentrations (0.56 [●], 2.8 [▴], and 5.6mM [■]) are shown. *P < .01, BMDAC-V vs BMDAC-D, by Bonferroni post hoc comparisons after 2-way ANOVA. Data are mean ± SEM (n = 3-5).](/view-large/figure/7155768/zh89991170380003.jpeg)
Increased survival of DMOG-treated BMDACs under hypoxic and acidic conditions ex vivo. (A) Effect of pH and hypoxia on BMDAC-D survival at 5.6mM glucose (▴). After culture for 4 days, equal numbers of BMDACs were exposed to low pH (6.4), nonhypoxic (20% O2), and hypoxic (1% O2) conditions. Cell viability was measured using the Trypan blue exclusion method. (B) Effect of glucose concentration on BMDAC-V and BMDAC-D survival under low pH (6.4) and hypoxia (1% O2). After culture for 4 days, BMDAC viability was measured with the LIVE/DEAD Violet fluorescent dye and flow cytometry. Survival curves under 3 glucose concentrations (0.56 [●], 2.8 [▴], and 5.6mM [■]) are shown. *P < .01, BMDAC-V vs BMDAC-D, by Bonferroni post hoc comparisons after 2-way ANOVA. Data are mean ± SEM (n = 3-5).
Increased survival of DMOG-treated BMDACs under hypoxic and acidic conditions ex vivo. (A) Effect of pH and hypoxia on BMDAC-D survival at 5.6mM glucose (▴). After culture for 4 days, equal numbers of BMDACs were exposed to low pH (6.4), nonhypoxic (20% O2), and hypoxic (1% O2) conditions. Cell viability was measured using the Trypan blue exclusion method. (B) Effect of glucose concentration on BMDAC-V and BMDAC-D survival under low pH (6.4) and hypoxia (1% O2). After culture for 4 days, BMDAC viability was measured with the LIVE/DEAD Violet fluorescent dye and flow cytometry. Survival curves under 3 glucose concentrations (0.56 [●], 2.8 [▴], and 5.6mM [■]) are shown. *P < .01, BMDAC-V vs BMDAC-D, by Bonferroni post hoc comparisons after 2-way ANOVA. Data are mean ± SEM (n = 3-5).
DMOG enhances intracellular pH homeostasis in BMDACs
The enhanced survival of BMDAC-D under acidic conditions suggested that DMOG treatment altered pH homeostasis. Measurement of intracellular pH in BMDAC-D revealed significant alkalinization compared with BMDAC-V (7.04 ± 0.09 vs 6.66 ± 0.09; P < .05; Figure 4A). No differences in extracellular pH were observed in conditioned media from BMDAC-V and BMDAC-D (7.60 ± 0.04 vs 7.46 ± 0.03, P > .05; Figure 4B). The levels of mRNA encoding NHE2 and NHE3, but not NHE1, were significantly increased in BMDAC-D (Figure 4C). Measurement of NHE activity showed differences between BMDAC-V and BMDAC-D that did not reach statistical significance (0.0676 ± 0.004 vs 0.1368 ± 0.072 minute−1; P > .05). Because changes in NHE activity did not provide a compelling mechanism for the intracellular alkalinization induced by DMOG in BMDACs, we analyzed the expression of mRNAs encoding all active CAR isoforms, which catalyze the reaction CO2 + H2O ↔ H2CO3 ↔ H+ + HCO3−.43-45 We detected mRNA for CAR 1, 2, 3, 5B, 6, 9, 13, 14, and 15 in BMDACs, whereas expression of CAR 4, 5A, 7, and 12 was not detected after 40 cycles of quantitative RT-PCR. DMOG treatment of BMDACs increased the expression of CAR 3, 6, 9, 14, and 15, which include 3 membrane-bound isoforms (Figure 4C). The mRNA levels for cytosolic (CAR13) and mitochondrial (CAR5B) isoforms were significantly decreased in BMDAC-D (Figure 4C). Total CAR enzymatic activity was increased 2.2-fold in BMDAC–D (P < .001; Figure 4D), providing a molecular basis for their increased intracellular pH (Figure 4B). To determine whether the increased carbonic anhydrase activity observed in BMDAC-D (Figure 4D) contributes to cell survival, we exposed cells to the carbonic anhydrase inhibitor acetazolamide at concentrations ranging from 1nM to 500μM (Figure 4E). Remarkably, acetazolamide potently reduced the viability of BMDAC-D in a dose-dependent manner (IC50 = 6.5nM), but only when the cells were subjected to both hypoxia (1% O2) and acidosis (pH 6.4). We conclude that induction of CAR activity by DMOG treatment contributes to cell survival under hypoxic and acidic conditions similar to those present in ischemic tissue.

DMOG induces intracellular alkalinization associated with increased CAR activity and altered expression of CAR isoforms. (A) Intracellular pH of BMDACs. (B) Extracellular pH of BMDACs. (C) Quantitative RT-PCR analysis of Na+/H+ exchangers and active CAR isoforms in BMDAC-D. Gray shaded area represents ± 2-fold difference in BMDAC-D versus BMDAC-V. CAR isoforms: (c) indicates cytosolic; (mt), mitochondrial; (mb), membrane-bound; and (s), secreted. (D) CAR activity of BMDAC homogenates was measured using a colorimetric assay and normalized to the total amount of protein. (E) Effect of CAR inhibition on BMDAC viability under low pH (6.4), glucose (0.56mM), and hypoxia (1% O2). BMDACs were incubated with the CAR inhibitor acetazolamide (1nM to 500μM) for 12 hours; viability was measured with the LIVE/DEAD Violet fluorescent dye and flow cytometry. (A-E) Cells were cultured for 4 days in the presence of vehicle (V) or DMOG (D) before analysis. *P < .05, BMDAC-V vs BMDAC-D, by Student's t test or Bonferroni post hoc comparisons after 1-way ANOVA. Data are mean ± SEM (n = 3).
DMOG induces intracellular alkalinization associated with increased CAR activity and altered expression of CAR isoforms. (A) Intracellular pH of BMDACs. (B) Extracellular pH of BMDACs. (C) Quantitative RT-PCR analysis of Na+/H+ exchangers and active CAR isoforms in BMDAC-D. Gray shaded area represents ± 2-fold difference in BMDAC-D versus BMDAC-V. CAR isoforms: (c) indicates cytosolic; (mt), mitochondrial; (mb), membrane-bound; and (s), secreted. (D) CAR activity of BMDAC homogenates was measured using a colorimetric assay and normalized to the total amount of protein. (E) Effect of CAR inhibition on BMDAC viability under low pH (6.4), glucose (0.56mM), and hypoxia (1% O2). BMDACs were incubated with the CAR inhibitor acetazolamide (1nM to 500μM) for 12 hours; viability was measured with the LIVE/DEAD Violet fluorescent dye and flow cytometry. (A-E) Cells were cultured for 4 days in the presence of vehicle (V) or DMOG (D) before analysis. *P < .05, BMDAC-V vs BMDAC-D, by Student's t test or Bonferroni post hoc comparisons after 1-way ANOVA. Data are mean ± SEM (n = 3).
HIF-1α haploinsufficiency impairs the metabolic response to hypoxia
Taken together, the data presented in Figures 1 to 4 demonstrate a profound reprogramming of metabolism associated with HIF-1 activation by DMOG treatment of BMDACs. To complement the analysis of pharmacologic gain-of-function for HIF-1, we analyzed the effect of genetic loss-of-function of HIF-1, by analyzing BMDACs derived either from Hif1a+/− (HET) mice, which are heterozygous for a null (knockout) allele at the locus encoding HIF-1α, or from their wild-type (WT) littermates. Consistent with the effects of DMOG treatment (Figure 1A), hypoxic BMDAC cultures from WT mice contained a higher percentage (Figure 5A) and greater total number (Figure 5B) of viable cells compared with nonhypoxic cultures. In contrast, hypoxia did not significantly increase viable BMDACs cultured from HET mice (Figure 5A-B). Quantitative RT-PCR analysis revealed a failure to activate transcription of HIF-1 target genes encoding metabolic enzymes and transporters in hypoxic BMDACs from HET mice (Figure 5C). As an additional loss-of-function strategy, BMDACs isolated from WT mice were incubated with digoxin, which inhibits HIF-1α (and HIF-2α) synthesis at nanomolar concentrations.46 Digoxin blocked DMOG-induced increases in GLUT1, PDK1, and LDHA mRNA (Figure 5D). In addition, the prosurvival advantage of BMDAC-D from WT mice was blocked by digoxin under nonstress conditions (Figure 5E). Taken together, the loss-of-function studies demonstrate that metabolic reprogramming induced by exposure of BMDACs to DMOG or hypoxia is mediated by HIF-1, as even partial HIF-1α deficiency in BMDACs from HET mice is associated with a complete loss of target gene activation.

Effect of HIF-1α loss of function on BMDAC survival and gene expression. (A-C) Bone marrow cells were isolated from Hif1a+/− (HET) mice, which are heterozygous for a null (knockout) allele at the locus encoding HIF-1α, or from their wild-type (WT) littermates and cultured for 4 days under nonhypoxic (N) or hypoxic (H) conditions in endothelial growth medium. The percentage of viable cells (A), number of viable cells (B), and expression of HIF-1 target genes (C) were determined. *P < .05 compared with nonhypoxic WT cells. #P < .01 compared with hypoxic WT cells in panels B and C, Bonferroni post hoc comparisons after 1-way ANOVA. (D) Quantitative RT-PCR analysis of HIF-1 target gene expression and cell viability (E) in BMDAC-V and BMDAC-D cultured for 4 days in the presence (+) or absence (−) of 100nM digoxin (Dig). *P < .05 compared with BMDAC-V without digoxin. #P < .01 compared with BMDAC-D without digoxin, Bonferroni post hoc comparisons after 1-way ANOVA. Data are mean ± SEM (n = 3 or 4).
Effect of HIF-1α loss of function on BMDAC survival and gene expression. (A-C) Bone marrow cells were isolated from Hif1a+/− (HET) mice, which are heterozygous for a null (knockout) allele at the locus encoding HIF-1α, or from their wild-type (WT) littermates and cultured for 4 days under nonhypoxic (N) or hypoxic (H) conditions in endothelial growth medium. The percentage of viable cells (A), number of viable cells (B), and expression of HIF-1 target genes (C) were determined. *P < .05 compared with nonhypoxic WT cells. #P < .01 compared with hypoxic WT cells in panels B and C, Bonferroni post hoc comparisons after 1-way ANOVA. (D) Quantitative RT-PCR analysis of HIF-1 target gene expression and cell viability (E) in BMDAC-V and BMDAC-D cultured for 4 days in the presence (+) or absence (−) of 100nM digoxin (Dig). *P < .05 compared with BMDAC-V without digoxin. #P < .01 compared with BMDAC-D without digoxin, Bonferroni post hoc comparisons after 1-way ANOVA. Data are mean ± SEM (n = 3 or 4).
DMOG treatment improves BMDAC survival in ischemic tissue
We isolated bone marrow cells from male mice and cultured them for 4 days in endothelial growth medium supplemented with DMOG or vehicle. Female mice were subjected to unilateral femoral artery ligation followed by intramuscular injection of 2 × 108 pfu of AdCA5, an adenovirus encoding a constitutively active form of HIF-1α, in the adductor and gastrocnemius muscles of the ischemic limb. Twenty-four hours later, the female mice were administered BMDACs from donor male mice by tail vein injection. Cohorts of mice were euthanized at 4 time points after intravenous injection of BMDACs and the ischemic and contralateral-nonischemic gastrocnemius muscles were harvested for isolation of genomic DNA. A DNA sequence at the Sry locus on the Y-chromosome was quantified by real-time PCR as a measure of the number of BMDACs remaining in the ischemic tissue. Nonlinear fitting to an exponential decay model revealed that the half-life of BMDAC-V was 7.2 hours, whereas the half-life of BMDAC-D was 13.4 hours (Figure 6), an increase of 1.86-fold (P < .05 by F test and 2-way ANOVA; n = 3 or 4). These data suggest that the effect of DMOG on BMDAC survival was preserved in the subset of cells that home to the ischemic tissue, although it remains a formal possibility that differences in retention, rather than survival, in the ischemic tissue account for the observed differences in half-life of the cells.

DMOG increases BMDAC survival in ischemic limbs. Genomic DNA was isolated from ischemic and nonischemic gastrocnemius muscles of female mice subjected to unilateral femoral artery ligation. All mice were injected with 2 × 108 pfu of AdCA5 distributed equally at 4 intramuscular injection sites in the ischemic limb immediately after surgery. A total of 5 × 105 BMDACs (isolated from male mice and cultured in vehicle or DMOG) were injected intravenously 24 hours after surgery. Muscle was harvested at 8, 16, 34, and 58 hours after injection. DNA was isolated and analyzed using quantitative PCR for a Y-chromosome-specific Sry gene sequence. No signal was detected in gastrocnemius muscles from nonischemic limbs. Decay curves were constructed using a 1-phase exponential decay model with nonlinear regression. BMDAC half-life was calculated from the data, and the difference between vehicle and DMOG was statistically significant (P < .05). Two-way ANOVA showed a statistically significant overall effect of DMOG treatment on BMDAC survival (P < .01 n = 3 or 4 animals per time point). Data are mean ± SEM.
DMOG increases BMDAC survival in ischemic limbs. Genomic DNA was isolated from ischemic and nonischemic gastrocnemius muscles of female mice subjected to unilateral femoral artery ligation. All mice were injected with 2 × 108 pfu of AdCA5 distributed equally at 4 intramuscular injection sites in the ischemic limb immediately after surgery. A total of 5 × 105 BMDACs (isolated from male mice and cultured in vehicle or DMOG) were injected intravenously 24 hours after surgery. Muscle was harvested at 8, 16, 34, and 58 hours after injection. DNA was isolated and analyzed using quantitative PCR for a Y-chromosome-specific Sry gene sequence. No signal was detected in gastrocnemius muscles from nonischemic limbs. Decay curves were constructed using a 1-phase exponential decay model with nonlinear regression. BMDAC half-life was calculated from the data, and the difference between vehicle and DMOG was statistically significant (P < .05). Two-way ANOVA showed a statistically significant overall effect of DMOG treatment on BMDAC survival (P < .01 n = 3 or 4 animals per time point). Data are mean ± SEM.
DMOG-treated BMDACs are effective when obtained from very old donors
We previously demonstrated a therapeutic effect when young (3-month-old) or old (13-month-old) mice were subjected to femoral artery ligation and treated with intramuscular AdCA5 followed 24 hours later by intravenous administration of BMDAC-D from young (3-month-old) donors.20 However, the anticipated clinical translation of this approach would involve the injection of autologous BMDACs in aged patients. Because ischemia-induced HIF-1α expression is impaired by aging,11,15,16 it was not clear whether DMOG-treated BMDACs from aged donors would promote recovery of perfusion and limb salvage. Very old (17-month-old) mice were used as both BMDAC donors and recipients. Blood flow was monitored serially by laser Doppler perfusion imaging (Figure 7A) and quantified as the ratio of perfusion in the ischemic/nonischemic limb (Figure 7B). Treatment with AdCA5 or saline resulted in virtually no recovery of blood flow over 28 days after femoral artery ligation. In contrast, mice treated with intramuscular injection of AdCA5 followed by intravenous injection of BMDAC-D showed a robust recovery of perfusion in the ischemic limb (Figure 7B). These mice also had a significant reduction in tissue damage and motor impairment in the ischemic limb (Figure 7C). In mice treated with saline or AdCA5 alone, femoral artery ligation resulted in complete limb amputation as a result of the failure to recover sufficient perfusion to maintain tissue viability (Figure 7A bottom panel). In contrast, treatment of mice with the combination of intramuscular AdCA5 plus intravenous BMDAC-D resulted in limb salvage with no permanent tissue damage. Thus, even in very old mice, DMOG pretreatment results in activated BMDACs which, in concert with local HIF-1α gene therapy, are capable of restoring sufficient perfusion to maintain viability in severely ischemic tissue.
![Figure 7. Combined therapy with DMOG-treated BMDACs and AdCA5 improves perfusion and clinical outcome after hindlimb ischemia in 17-month-old mice. (A) Laser Doppler perfusion imaging (top and middle panels) and photography (bottom panel) were performed at the indicated time after femoral artery ligation. The top panel shows typical scans of female 17-month-old mice immediately before (Pre) and after (Post) femoral artery ligation in the left hindlimb. Blood flow is quantified using a pseudocolor scale as shown at right (perfusion units [PU]). The middle panel shows representative follow-up images of mice treated with saline, AdCA5, or combined therapy (AdCA5 + BMDAC-D). Because saline- and AdCA5-treated mice had severe tissue necrosis, the last time point (day 14 [D14]) before autoamputation ensued is shown. Bottom panel: Representative photographs of mice at day 28 after surgery (D28). (B) Mean ischemic/nonischemic limb perfusion ratio, as determined by laser Doppler perfusion imaging, was calculated at the indicated time (in days) after hindlimb ischemia. Mice were treated with saline (black), intramuscular AdCA5 (blue), or intramuscular AdCA5 + intravenous BMDAC-D (green). Intramuscular AdCA5 was injected immediately after surgery and intravenous BMDAC-D were delivered 24 hours later. *P < .05 vs saline control by Bonferroni post hoc test after ANOVA (n = 3 mice per group). (C) Tissue damage (left panel) was scored as follows: 0 indicates no tissue damage; 1, cyanosis/discoloration indicative of soft tissue necrosis; 2, loss of 1 or 2 toes; 3, loss of 3 to 5 toes; 4, amputation of entire foot; 5, amputation of entire lower leg; and 6, amputation of entire leg. Motor impairment (right panel) was scored as follows: 0 indicates normal response (plantar/toe flexion in response to tail traction); 1, plantar but not toe flexion; 2, no plantar or toe flexion; and 3, dragging of foot. Motor impairment and ischemic tissue damage were measured 28 days after surgery. *P < .05 vs saline control by Bonferroni post hoc test after ANOVA (n = 3 mice per group). Data are mean ± SEM.](/view-large/figure/7155780/zh89991170380007.jpeg)
Combined therapy with DMOG-treated BMDACs and AdCA5 improves perfusion and clinical outcome after hindlimb ischemia in 17-month-old mice. (A) Laser Doppler perfusion imaging (top and middle panels) and photography (bottom panel) were performed at the indicated time after femoral artery ligation. The top panel shows typical scans of female 17-month-old mice immediately before (Pre) and after (Post) femoral artery ligation in the left hindlimb. Blood flow is quantified using a pseudocolor scale as shown at right (perfusion units [PU]). The middle panel shows representative follow-up images of mice treated with saline, AdCA5, or combined therapy (AdCA5 + BMDAC-D). Because saline- and AdCA5-treated mice had severe tissue necrosis, the last time point (day 14 [D14]) before autoamputation ensued is shown. Bottom panel: Representative photographs of mice at day 28 after surgery (D28). (B) Mean ischemic/nonischemic limb perfusion ratio, as determined by laser Doppler perfusion imaging, was calculated at the indicated time (in days) after hindlimb ischemia. Mice were treated with saline (black), intramuscular AdCA5 (blue), or intramuscular AdCA5 + intravenous BMDAC-D (green). Intramuscular AdCA5 was injected immediately after surgery and intravenous BMDAC-D were delivered 24 hours later. *P < .05 vs saline control by Bonferroni post hoc test after ANOVA (n = 3 mice per group). (C) Tissue damage (left panel) was scored as follows: 0 indicates no tissue damage; 1, cyanosis/discoloration indicative of soft tissue necrosis; 2, loss of 1 or 2 toes; 3, loss of 3 to 5 toes; 4, amputation of entire foot; 5, amputation of entire lower leg; and 6, amputation of entire leg. Motor impairment (right panel) was scored as follows: 0 indicates normal response (plantar/toe flexion in response to tail traction); 1, plantar but not toe flexion; 2, no plantar or toe flexion; and 3, dragging of foot. Motor impairment and ischemic tissue damage were measured 28 days after surgery. *P < .05 vs saline control by Bonferroni post hoc test after ANOVA (n = 3 mice per group). Data are mean ± SEM.
Combined therapy with DMOG-treated BMDACs and AdCA5 improves perfusion and clinical outcome after hindlimb ischemia in 17-month-old mice. (A) Laser Doppler perfusion imaging (top and middle panels) and photography (bottom panel) were performed at the indicated time after femoral artery ligation. The top panel shows typical scans of female 17-month-old mice immediately before (Pre) and after (Post) femoral artery ligation in the left hindlimb. Blood flow is quantified using a pseudocolor scale as shown at right (perfusion units [PU]). The middle panel shows representative follow-up images of mice treated with saline, AdCA5, or combined therapy (AdCA5 + BMDAC-D). Because saline- and AdCA5-treated mice had severe tissue necrosis, the last time point (day 14 [D14]) before autoamputation ensued is shown. Bottom panel: Representative photographs of mice at day 28 after surgery (D28). (B) Mean ischemic/nonischemic limb perfusion ratio, as determined by laser Doppler perfusion imaging, was calculated at the indicated time (in days) after hindlimb ischemia. Mice were treated with saline (black), intramuscular AdCA5 (blue), or intramuscular AdCA5 + intravenous BMDAC-D (green). Intramuscular AdCA5 was injected immediately after surgery and intravenous BMDAC-D were delivered 24 hours later. *P < .05 vs saline control by Bonferroni post hoc test after ANOVA (n = 3 mice per group). (C) Tissue damage (left panel) was scored as follows: 0 indicates no tissue damage; 1, cyanosis/discoloration indicative of soft tissue necrosis; 2, loss of 1 or 2 toes; 3, loss of 3 to 5 toes; 4, amputation of entire foot; 5, amputation of entire lower leg; and 6, amputation of entire leg. Motor impairment (right panel) was scored as follows: 0 indicates normal response (plantar/toe flexion in response to tail traction); 1, plantar but not toe flexion; 2, no plantar or toe flexion; and 3, dragging of foot. Motor impairment and ischemic tissue damage were measured 28 days after surgery. *P < .05 vs saline control by Bonferroni post hoc test after ANOVA (n = 3 mice per group). Data are mean ± SEM.
Discussion
In this study, we demonstrate that DMOG treatment of BMDACs cultured from whole bone marrow over 4 days results in metabolic reprogramming with decreased mitochondrial mass, O2 consumption, and ROS levels; increased glucose uptake, lactate production, and secretion; increased intracellular pH; increased survival under acidic and hypoxic conditions ex vivo; and increased survival after homing to ischemic tissue in vivo. This reprogramming of cellular metabolism reflected the increased expression of multiple HIF-1 target genes encoding membrane transporters (GLUT1, GLUT3, MCT4), metabolic enzymes (COX4I2, LDHA, PDK1), pH regulators (CAR2, CAR3, CAR6, CAR9, CAR14, CAR15), and an autophagy regulator (BNIP3), which play important roles in the metabolic alterations that are described in this study. Our data indicate that induction of HIF-1 is required for the increased expression of these gene products in BMDACs exposed to DMOG or hypoxia. Although the data in Figure 5 demonstrate that partial HIF-1α deficiency in BMDACs is sufficient to result in a significant decrease in cell survival and it is well established that many of the relevant genes (Bnip3, Car9, Hk2, Ldha, Pdk1) are regulated exclusively in a HIF-1α–dependent manner, it is possible that the survival benefit associated with DMOG treatment of BMDACs may in part the result of the induction of HIF-2α as well as HIF-1α.
Although both hypoxia47 and HIF-1α gene transduction48 have been reported to increase the proangiogenic properties of BMDACs in rodent ischemia models, the effect of these treatments was attributed to increased proliferation, differentiation, or homing of cells to the ischemic tissue. However, we previously demonstrated that DMOG treatment of BMDACs does not affect homing.20 Instead, the present study has revealed alterations in metabolism that allow cells to survive under the adverse environmental conditions that are present in ischemic tissue. Because they are pretreated, BMDAC-D are adapted to the ischemic microenvironment even before they reach it, whereas naive cells that home to ischemic tissue will take at least 24 hours to induce HIF-1 activity and express downstream target gene products that are required for metabolic adaptation and cell survival.
Taken together, the results of this study provide an important mechanistic basis for the recovery of limb perfusion when AdCA5 local gene therapy is combined in series with BMDAC-D cell therapy, resulting in prevention of limb necrosis and autoamputation in very old recipient mice treated with BMDACs from very old donors, which serves as a preclinical model for autologous cell therapy in elderly patients with critical limb ischemia in which perfusion is inadequate to maintain tissue viability. There are an increasing number of no-option patients for whom the only currently available treatment is surgical amputation, highlighting the need for novel therapeutic strategies.49
Our data show that, even with DMOG treatment, most of the BMDACs that home to the ischemic limb are no longer present by 3 days after injection, indicating that these cells play a role in initiating the vascular response to ischemia but are not required for the progressive increase in perfusion that occurs over the subsequent weeks. Given the short time span over which the proangiogenic role of recruited BMDACs occurs, the observed doubling of BMDAC half-life in ischemic tissue that results from DMOG treatment probably enhances the therapeutic efficacy of these cells when they are combined with local AdCA5 gene therapy, which creates the homing signal required for BMDAC recruitment to ischemic tissue.20 The demonstrated requirement for combined gene and cell therapy to restore perfusion to ischemic tissue in mice of advanced age provides a potential explanation for the failure of a clinical trial involving intramuscular administration of an adenoviral vector encoding a chimeric protein consisting of the amino-terminal half of HIF-1α fused to the herpesvirus VP16 protein,50 although it is also likely that the chimeric protein did not regulate the complete battery of HIF-1 target genes. Our preclinical data suggest that trials of combined HIF-1α gene therapy and DMOG-activated BMDACs are warranted in patients for whom surgical amputation is the only remaining therapeutic option.
In conclusion, although the poor survival of transplanted cells is of greatest concern in the context of ischemic cardiovascular disease, it remains an issue for all areas of regenerative medicine in which the introduction of cells into a tissue will inevitably result in reduced local availability of O2 and nutrients as well as increased levels of H+, ROS, and other metabolites that threaten cell survival. Thus, the metabolic reprogramming strategy that is induced by DMOG treatment of BMDACs may have general applicability as a strategy to promote effective tissue regeneration.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Luana Schito for useful suggestions to improve the carbonic anhydrase assay; Andrea Gambotto and Susan Schoonover (National Heart, Lung, and Blood Institute Vector Core Lab at the University of Pittsburgh) for large-scale preparation of AdCA5; Karen Padgett (Novus Biologicals) for antibodies against HIF-1α, LDHA, and PDK1; and Chi V. Dang for assistance with Oxygraph measurements.
This work was supported by the National Heart, Lung, and Blood Institute (Public Health Service grant R01-HL55338) and the Johns Hopkins Institute for Cell Engineering. G.L.S. is the C. Michael Armstrong Professor at the Johns Hopkins University School of Medicine.
National Institutes of Health
Authorship
Contribution: S.R. and G.L.S. designed the research and wrote the paper; S.R., W.L., and L.A.S. performed experiments; and S.R., W.L., L.A.S., and G.L.S. analyzed results.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Gregg L. Semenza, Broadway Research Bldg, Suite 671, 733 N Broadway, Baltimore, MD 21205; e-mail: gsemenza@jhmi.edu.