
Abstract
Current immunosuppressive treatment (IST) induces remissions in 50%-70% of patients with aplastic anemia (AA) and result in excellent long-term survival. In recent years, the survival of refractory patients has also improved. Apart from relapse and refractoriness to IST, evolution of clonal diseases, including paroxysmal nocturnal hemoglobinuria and myelodysplastic syndrome (MDS), are the most serious long-term complications and constitute a strong argument for definitive therapy with BM transplantation if possible. Consequently, the detection of diagnostic chromosomal abnormalities (mostly monosomy 7) is of great clinical importance. Newer whole-genome scanning technologies such as single nucleotide polymorphism (SNP) array–based karyotyping may be a helpful diagnostic test for the detection of chromosomal defects in AA due to its precision/resolution and lack of reliance on cell division.
Introduction
In aplastic anemia (AA), immunosuppressive therapy (IST) with antithymocyte globulin (ATG) and cyclosporine A (CsA) produces excellent results. With single or repeated courses of IST, the majority of patients respond and have excellent survival.1 Refractory patients have a less favorable prognosis, but due to recent progress in supportive care, their survival has improved.2 However, for younger and selected older patients with a suitable donor, hematopoietic stem cell transplantation appears to be the treatment of choice that, unlike conservative therapies, can provide a durable cure and alleviate the risk of long-term complications such as clonal evolution or relapse.
Progress in further improving the results of IST has been slow. Restoration of blood counts so that they do not fulfill “severity” criteria and achievement of transfusion independence leads to long-term survival. However, many patients who respond to IST continue to display decreased counts, indicating that only a partial remission can be achieved in most instances. Many patients continue to require IST on a chronic basis. However, the most serious complication of severe AA treated with IST is the development of clonal disease, including the acquisition of clonal chromosomal abnormalities and the development of myelodysplastic syndrome (MDS) and expansion of paroxysmal nocturnal hemoglobinuria (PNH) clones. This review focuses on the pathogenesis, diagnosis, and therapy of this complication.
Diagnosis of AA versus hypocellular MDS
The diagnosis of AA requires documentation of depressed counts of at least 2 blood cell lineages and a hypocellular BM. Whereas macrocytosis is often present, a dysplastic myeloid series and megakaryocytes (severely depressed in AA) should be absent. However, hypocellularity of the BM may preclude assessment of dysplastic changes and thereby the diagnosis of hypocellular MDS (hMDS). Karyotyping, if positive for typical MDS abnormalities, is diagnostic for MDS, but often traditional metaphase cytogenetics is not informative in hypocellular cases due to a lack of evaluable metaphases. Whereas in some cases the presence of a cytogenetic abnormality, in particular when present in only a portion of metaphases, is compatible with AA, such abnormalities usually disappear after therapy.3
Newer cytogenetic detection methods such as single nucleotide polymorphism (SNP) array–based karyotyping allow for the detection of unbalanced chromosomal abnormalities in interphase DNA and therefore provide the advantage of not being dependent on cell culture.4–6 This technique also permits the detection of small clonal lesions such as microdeletions and gains, as well as copy number neutral loss of heterozygosity.7,8 These additional features of SNP-array karyotyping method may complement metaphase cytogenetics in the detection of chromosomal defects in AA and other hematologic diseases. Systematic application of this technique to cases of hMDS and AA led to the identification of previously cryptic defects in 3 of 24 patients with a clinical diagnosis of hMDS and in 10 of 33 cases of AA at presentation.9 Similar to metaphase cytogenetics, some SNP-array–detected lesions (in particular, microdeletions) disappear upon further treatment, proving their somatic nature and consistent with the oligoclonal nature of hematopoiesis at presentation.10
Current results of conservative therapy of AA
Recently reported results of large multicenter or single-center trials or cumulative analysis demonstrate responses to classical IST in the range of 58%-77% and survival of 58% at 11 years (Table 1).13–23 It is possible that the long-term complications such as clonal evolution are a by-product of sufficiently long survival after IST.24 Whereas early reports linked G-CSF therapy with monosomy 7 clonal evolution in patients with BM failure, large multi-institutional studies did not reveal excessive rates of clonal progression.21,25 Nonetheless, given its limited utility in this setting, judicious use of this agent is important.
Results of IST in major clinical trials
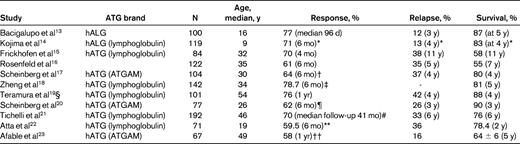
hALG indicates horse anti-lymphocyte globulin; hATG, horse ATG.
*Patients randomized to cohort with largest patients (ATG/CsA/DAN/rhG-CSF, N = 50).
†hATG/CsA/mycophenolate mofetil.
‡hATG/CsA arm.
§hATG/CsA arm.
¶hATG/CsA arm.
#hATG/CsA ± G-CSF.
**hATG/CsA arm.
††hATG/CsA arm (median follow-up of 43.5 mo).
Clinical aspects of clonal evolution
MDS
Evolution of MDS occurs either early or late in the course of the disease (Figures 1 and 2). Because of the limited number of cells in patients with hypoplastic BM, there is no robust and reliable way to consistently distinguish all patients with AA from those with hMDS. In some patients with hypocellular BM who are initially suspected to have AA, a rapid progression to AML can be observed and may be consistent with what has been historically described as aleukemic leukemia.26 More typical is the late evolution of the disease seen in most of the refractory cases or in those who did not achieve a robust response.16,27–29 The reported rates vary from 1.7%-57% in the observation period of 5-11 years (Table 2).15,16,21,28,30,31–33 Most common is the evolution to MDS with monosomy 7 as the sole abnormality.24,34 Other karyotypes are by far less common, including trisomy 8, del 13, del 13q, and del Y.24

Clonality in AA. True clonal evolution and progression to MDS or AML has to be distinguished from pseudoclonality, which can be a result of a contracted stem cell pool and is characterized by a transient nature, lower proportion of metaphases with clonal markers, and often unusual breaks not typical of MDS.
Clonality in AA. True clonal evolution and progression to MDS or AML has to be distinguished from pseudoclonality, which can be a result of a contracted stem cell pool and is characterized by a transient nature, lower proportion of metaphases with clonal markers, and often unusual breaks not typical of MDS.

Pathogenesis of clonal evolution. Top, clonal evolution represents an escape mechanism from immune pressure exerted by the immune system. In this process, antigens on normal stem cells or tumor-associated antigens can trigger the autoimmune attack or unselective tumor immune surveillance response, respectively. Bottom, clonal initiation due to recruitment of a defective stem cells in the context of a contracted stem cell pool, clonal promotion in a pro-oncogenic milieu due to increased growth factor drive and increased cycling to keep up with the demand, and increased rate of chromosomal defects leading to clonal expansion in the context of excessive telomere shortening or endogenous oncogenesis during inflammatory reactions.
Pathogenesis of clonal evolution. Top, clonal evolution represents an escape mechanism from immune pressure exerted by the immune system. In this process, antigens on normal stem cells or tumor-associated antigens can trigger the autoimmune attack or unselective tumor immune surveillance response, respectively. Bottom, clonal initiation due to recruitment of a defective stem cells in the context of a contracted stem cell pool, clonal promotion in a pro-oncogenic milieu due to increased growth factor drive and increased cycling to keep up with the demand, and increased rate of chromosomal defects leading to clonal expansion in the context of excessive telomere shortening or endogenous oncogenesis during inflammatory reactions.
Evolution rate to clonal disease including PNH and MDS
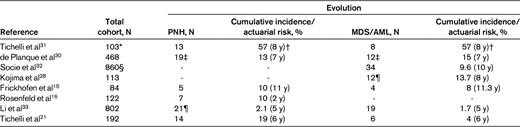
*Patients who were treated only with ALG.
†Probability of developing all hematological complications (PNH, MDS/AML).
‡Frequency based on 209 patients who are long-term survivors; MDS, n = 11 (5 of which subsequently evolved to acute leukemia); acute leukemia, n = 1.
§Patients treated with IST; MDS, n = 19; AML, n = 15.
¶Two of these patients had concurrent MDS.
As explained above, SNP-array karyotyping allowed for the detection of previously cryptic clonal abnormalities in patients with normal cytogenetics both at diagnosis of AA and at follow-up.9 In several cases, serial testing using SNP-array karyotyping showed that monosomy 7 was detected earlier than by metaphase cytogenetics or led to the identification of aberration when metaphase cytogenetics was not helpful (Figure 3).9 In 3 of 93 patients examined by SNP-array, we have identified loss of heterozygosity (2 deletions and 1 copy number neutral loss of heterozygosity) involving the HLA locus. One of these lesions disappeared upon remission induced by IST. We have theorized that deletion of the HLA-A locus could be a result of selection pressure and lead to loss of the antigenic peptide–presenting HLA allele. Once the immune response has been therapeutically suppressed, the aberrant clone is outgrown by normal hematopoiesis. A similar phenomenon has been observed after partially mismatched allogeneic BM transplantation.35

Detection of monosomy 7 in a patient with otherwise typical AA and failed cytogenetic examination due to lack of evaluable metaphases. SNP-A karyotyping was performed at 2 time points during the course of disease: at presentation (normal male karyogram) and at 12 months (noninformative cytogenetic examination). In general, the total copy number is around 2N (left). The second SNP-A karyotyping (right) demonstrates the deletion of one arm of chromosome 7, shown here as reduction of copy number (2N to 1N).
Detection of monosomy 7 in a patient with otherwise typical AA and failed cytogenetic examination due to lack of evaluable metaphases. SNP-A karyotyping was performed at 2 time points during the course of disease: at presentation (normal male karyogram) and at 12 months (noninformative cytogenetic examination). In general, the total copy number is around 2N (left). The second SNP-A karyotyping (right) demonstrates the deletion of one arm of chromosome 7, shown here as reduction of copy number (2N to 1N).
No biomarkers that predict MDS evolution are currently known in AA. However, those AA patients with complete and durable responses to IST are at lower risk of progressing to MDS.16,27–29 Consequently, in cases of decreasing counts, a BM examination is needed to rule out the possibility of evolution to MDS by cytogenetics and to avoid futile therapy with ATG, which is usually ineffective. In various studies, the evolution of chromosomal abnormalities including monosomy 7 has been associated with a poor prognosis.24,34 Consequently, BM transplantation should be a therapy of choice for such patients whenever possible. A few anecdotal cases of post-AA monosomy 7 treated at our institution did respond to hypomethylating agents.
PNH
Sensitive flow cytometric methods allow for the detection of even tiny glycosylphosphatidylinositol (GPI) anchor-deficient clones.36,37 Using these methods, PNH cells can be identified in a large number of patients with AA.38 In the course of their disease, the number of clonal cells will steadily increase in some of these patients, who will develop manifest hemolytic PNH with evolution rates ranging from 2.1%-19% in 2-11 years (Table 2).15,16,21,28,30–33 In contrast to PNH progressors, a significant proportion of AA patients have stable PNH clone size. What factors govern the progression remains unclear. Whereas most of these patients have received IST, not all of them progress, and occasional patients with initially moderate AA develop manifest PNH without IST. In various studies, the presence of PNH has constituted a favorable prognostic factor for response to IST.23,39 However, the immune privilege theory of PNH evolution would imply that in patients who were successfully treated (responders), immune pressure leading to the escape of PNH has been alleviated. In contrast, many patients who respond to ATG or CsA show progression of PNH to manifest hemolytic disease with seeming uninhibited proliferation of the PNH clone. This observation is consistent with the possibility (explained below) that the PNH phenotype may be a trigger for the immune reaction that is not selective and that, upon inhibition, PNH clones proliferate more freely.
In many aspects, PNH evolution is similar to the evolution of clones with chromosomal abnormalities. In fact, cases of PNH due to microdeletion of the PNH locus (true PNH-MDS) have been described based on SNP-array–based study of patients with PNH.40 Therefore, the pathogenesis of PNH and secondary MDS may be similar and is inherently but not exclusively linked to depletion of stem cell compartments.
The therapy of PNH in the setting of AA has not been well studied. True hemolytic PNH does not benefit from ATG,40 but patients in whom AA and PNH coexist (AA/PNH syndrome) may respond to CsA. With the advent of eculizumab, patients who have hemolytic PNH will respond to this drug, although it does not seem to improve platelet and neutrophil counts. In cases with reticulocytopenia, anemia is likely a result of deficient red cell production rather than hemolysis, but BM failure and hemolysis can coexist.
Pathogenesis of clonal evolution
Irrespective of the pathogenetic mechanisms leading to AA, the main pathologic process is contraction of the stem cell compartment leading to decreased numbers of available stem cells. It is likely that at the peak of disease, hematopoiesis in AA is oligoclonal if not clonal. Unlike in MDS, this is due to depletion rather than clonal outgrowth.42 The presence of clonal hematopoiesis is indicated by a large number of patients within whom PNH clones can be detected even at the onset of the disease (Figure 2). By analogy, it is possible that defective stem cells with clonal chromosomal abnormalities exist early in the course of the disease. In fact, the occasional detection of chromosomal abnormalities has been often reported at the initial diagnosis of AA, and some experts believe that these abnormalities are compatible with the diagnosis of AA. In some of these patients, chromosomal abnormalities are of a transient nature and may disappear, in particular after successful IST. Therefore, it is important that when using chromosomal abnormalities as clonal markers, the true clonal, malignant process is distinguished from the possibility that the appearance of a seemingly clonal cytogenetic marker is a result of stem cell pseudoclonality (Figure 1). Depletion of the stem cell pool may predispose to recruitment of a genetically defective hematopoietic clone. For example, certain mutations or chromosomal breaks may randomly affect individual stem cells, but the chance of their recruitment may be higher in the severely contracted stem cell pool, particularly when the genetic defect involves survival pathways. Conversely, one could rationally hypothesize that the appearance of such clones may trigger an autoimmune tumor surveillance response that could successfully eliminate abnormal cells but also lead to decimation of normal stem cells (Figure 2).
In addition to stem cell depletion and the recruitment of a defective stem cell clone that initiates clonal evolution, other pathogenetic mechanisms may exist (Figure 2). For example, excessive telomere shortening may constitute a risk factor for the acquisition of chromosomal damage.43 Both telomerase machinery defects and decreased telomere length have been identified in some patients with AA, and some patients with MDS and AA with clonal evolution did show decreased telomere length (Figure 2).44 Further clonal events such as facilitating mutations may lead to more rapid evolution of cryptic clones and progression to more advanced disease. For example, we have identified several mutations in patients with post-AA monosomy 7, including mutations in TET2, CBL, and DNMT3A (accepted abstract). In particular, CBL mutations were found in 2 of 17 cases of monosomy 7 that developed in AA.
Finally, cytogenetically abnormal cells may trigger an immune response that at first eliminates abnormal cells at the cost of collateral damage to normal stem and progenitor cells. With time, the selection pressure would lead to the generation and escape of mutant clones that outgrow normal hematopoiesis. Before the advent of effective IST, severe AA patients had high mortality rates, precluding the recognition of these late clonal complications. Therefore, one can theoretically argue that effective IST may have significantly improved the survival of AA patients, but in such prolonged survival there might be an increased risk of clonal evolution. Irrespective of the initiating events, the expansion of aberrant clones is likely a result of immune selection, and clonal progression may be regarded as a form of immunologic escape (Figure 1).
Chromosomal lesions constitute markers of clonal evolution. Similarly, PIG-A mutation may also serve as a clonal marker due to the inherent deficiency of GPI-anchored proteins that are easily recognizable by flow cytometry and therefore amenable to serial testing. It has been hypothesized that PNH cells enjoy a growth advantage in the context of an autoimmune attack on stem cells and therefore expand in immune-mediated AA. The nature of this growth advantage is not clear, but is likely due to a deficiency of one of the GPI-linked proteins. Alternatively, it is also possible that GPI-anchor deficiency is sensed by immune surveillance, which attempts to eliminate the intrinsically abnormal PNH cells but lacks selectivity.
In summary, differentiating between AA and hMDS may be difficult, particularly when clonal cytogenetic markers are absent. Transient chromosomal abnormalities may be present in AA and my reflect oligoclonality of the stem cell compartment, whereas true clonal expansions are of prognostic significance (Figure 1). Despite improving survival, evolution to MDS constitutes a significant clinical problem in the management of AA. Monosomy 7 is the most frequent cytogenetic lesion observed during such evolution and is associated with a poor prognosis. This evolution is most often associated with persistence of low counts or further worsening of cytopenia. Whereas surveillance may not be needed, better diagnostic resolution of cytogenetic status with, for example, SNP- or CGH-array–based cytogenetics may be helpful in distinguishing AA from hMDS and/or in the early detection of clonal progression.
Disclosures
Conflict-of-interest disclosures: M.G.A. has been affiliated with the speakers' bureau for Alexion; R.V.T. has consulted for, received honoraria and research funding from, and been affiliated with the speakers' bureaus for Alexion, Novartis, INCYTE, the AA MDS International Foundation, the MDS Foundation, and Allos Therapeutics. J.P.M. declares no competing financial interests. Off-label drug use: thymoglobulin, CsA, and prednisone.
Correspondence
Jaroslaw P. Maciejewski, MD, PhD, Department of Translational Hematology and Oncology Research, Taussig Cancer Institute R-40, Cleveland Clinic, 9500 Euclid Ave, Cleveland, OH 44195; Phone: (216) 445-5962; Fax: (216) 636-2498; e-mail: maciejj@ccf.org.
