

TO THE EDITOR:
In a previous study by our group, we reported that pediatric early T-cell precursor acute lymphoblastic leukemia (ETP-ALL) is BCL-2 dependent, whereas pediatric T-cell acute lymphoblastic leukemia (T-ALL) is BCL-XL dependent.1 However, the therapeutic success of adult ALL is different from that of pediatric ALL and is less understood. Thus, this study aimed to identify antiapoptotic BCL-2 family protein dependencies and sensitivities to BH3 mimetics in adult ETP-ALL and T-ALL lymphoblasts. By performing direct mitochondrial permeabilization and cell death assays, we validated BH3 profiling as a predictor of BCL-2 dependence in adult ETP-ALL and BCL-2/BCL-XL codependence in adults with T-ALL. Furthermore, we revealed the substantial on-target cytotoxicity of venetoclax and navitoclax, suggesting that this combination of BH3 mimetics is potentially efficacious in adults with T-ALL.
T-ALL results from malignant transformation of immature T-cells, accounting for 10% to 15% of childhood and 20% to 25% of adult ALL cases.2 Long-term survival rates for standard risk childhood T-ALL have shown striking improvements to 80% to 90%, yet response outcomes in adults remain much lower (40%), possibly due to high induction therapy–related toxicities.3 In general, the causes for poor response in adult T-ALL are incompletely understood.
Among ALL, the high-risk ETP-ALL subtype originating from clonal expansion of recently immigrated thymocytes has a significantly worse outcome in adults.4 ETP-ALL retains multipotent differentiation capacities and resemblance to hematopoietic stem cells or myeloid progenitor cells. Targeted next-generation sequencing5 showed that adult ETP-ALL presents similar mutation profiles as its pediatric equivalent.6 Furthermore, similar to pediatric ETP-ALL, adult ETP-ALL has inferior outcomes to chemotherapy than non-ETP-ALL.6 However, the implementation of allogenic transplant after achieving the first complete response with standard chemotherapy regimens improved the overall prognosis of patients with ETP-ALL, similar to the rest of the high-risk patients in the T-ALL cohort.6 Nevertheless, the 5-year overall survival rate of adults with ETP-ALL is only 49%.6 These outcomes highlight the unmet need for new therapeutic approaches for adult ETP-ALL.
BH3 mimetics, small molecule antagonists of BCL-2 family proteins, have recently shown clinical success in various hematologic malignancies, including chronic lymphocytic leukemia (CLL),7,8 acute myeloid leukemia (AML),9 and ALL.10,11 Studies from our group1,12-15 and several others16-18 have shown that BH3 profiling can predict the functional dependency of tumor cells on BCL-2. In turn, BCL-2 dependence identified using BH3 profiling directed clinical success of the U.S. Food and Drug Administration-approved venetoclax (BCL-2 inhibitor) in CLL19 and AML.13,14 In practice, BH3 profiling involves exposing the mitochondria to synthetic BH3 peptides and subsequently measuring the ensuing mitochondrial outer membrane permeabilization.20 When selective peptides are used, eg, BAD, heightened mitochondrial sensitivity indicates greater dependence on an individual anti-apoptotic protein, eg, BCL-2 and BCL-XL.21
To determine the anti-apoptotic dependencies of adult ETP-ALL, we performed BH3 profiling on 15 primary adult ETP-ALL samples obtained at diagnosis (Figure 1A; supplemental Table). We exploited the different binding affinities of BAD and HRK BH3 peptides to distinguish between BCL-2 dependence (high BAD, low/no HRK priming) and BCL-XL dependence (equal BAD and HRK priming) (Figure 1B). All samples showed robust mitochondrial depolarization in response to the BAD peptide compared with the HRK and DMSO control, suggesting a primary dependence on BCL-2 (Figure 1C). Despite the heterogeneity among samples, the mean BAD-induced cytochrome c release was significantly higher than that of the HRK peptide, further confirming BCL-2 dependence in ETP-ALL (P < .0001, Figure 1D). Notably, blasts from only patient A showed higher priming above the threshold by HRK (>20%), whereas the remaining 14 patients crossed the priming threshold only in response to the BAD peptide (Spearman r = 0.71, P < .01; Figure 1E). Furthermore, BAD and BAD-HRK showed a statistically significant association, suggesting that mitochondrial depolarization caused by the BAD peptide in ETP-ALL samples was driven by BCL-2 (Spearman r = 0.54, P < .05; Figure 1E) and not BCL-XL.

Adult ETP-ALL has increased dependency on BCL-2 rather than BCL-XL for survival. (A) Experimental schematic of the baseline BH3 profiling. Cytochrome c release was measured by gating on blasts. (B) Binding affinities of BH3 peptides (BAD and HRK) and BH3 mimetics (venetoclax, navitoclax, and A-1331852) for antiapoptotic proteins BCL-2 and BCL-XL. (C/D) FACS-based BH3 profiles for BAD (BCL-2 and BCL-XL dependence) and HRK (BCL-XL dependence). One-way analysis of variance (ANOVA) for % cytochrome c release between BAD vs DMSO and BAD vs HRK. (E) Spearman correlation between % cytochrome c release for BAD vs HRK and BAD vs BAD-HRK. Data is normalized to DMSO. (F-G) FACS-based BH3 profiles for venetoclax, navitoclax, and A-1331852. One-way ANOVA analysis for % cytochrome c release between venetoclax vs DMSO, navitoclax vs DMSO, and venetoclax vs navitoclax. (H) Cell death assays using annexin V in adult ETP-ALL samples treated with venetoclax, navitoclax, or A-1331852 for 8 hours. Data are plotted as the percentage of live cells compared with the DMSO controls. Note: gating of adult ETP-ALL primary blast samples. ∗ P < .05; ∗∗ P < .01; ∗∗∗ P < .001; ∗∗∗∗ P < .0001. UN, untreated; ns, no significance. (C,F) The dotted line represents the threshold for significant priming determined using DMSO ± 3xSD.
Adult ETP-ALL has increased dependency on BCL-2 rather than BCL-XL for survival. (A) Experimental schematic of the baseline BH3 profiling. Cytochrome c release was measured by gating on blasts. (B) Binding affinities of BH3 peptides (BAD and HRK) and BH3 mimetics (venetoclax, navitoclax, and A-1331852) for antiapoptotic proteins BCL-2 and BCL-XL. (C/D) FACS-based BH3 profiles for BAD (BCL-2 and BCL-XL dependence) and HRK (BCL-XL dependence). One-way analysis of variance (ANOVA) for % cytochrome c release between BAD vs DMSO and BAD vs HRK. (E) Spearman correlation between % cytochrome c release for BAD vs HRK and BAD vs BAD-HRK. Data is normalized to DMSO. (F-G) FACS-based BH3 profiles for venetoclax, navitoclax, and A-1331852. One-way ANOVA analysis for % cytochrome c release between venetoclax vs DMSO, navitoclax vs DMSO, and venetoclax vs navitoclax. (H) Cell death assays using annexin V in adult ETP-ALL samples treated with venetoclax, navitoclax, or A-1331852 for 8 hours. Data are plotted as the percentage of live cells compared with the DMSO controls. Note: gating of adult ETP-ALL primary blast samples. ∗ P < .05; ∗∗ P < .01; ∗∗∗ P < .001; ∗∗∗∗ P < .0001. UN, untreated; ns, no significance. (C,F) The dotted line represents the threshold for significant priming determined using DMSO ± 3xSD.
Having observed the primary dependence of adult ETP-ALL on BCL-2, we next hypothesized that ETP-ALL tumors are sensitive to BH3 mimetics, particularly BCL-2 inhibitors. We measured the direct mitochondrial sensitivity to BH3 mimetics venetoclax (selectively antagonizes BCL-2) and navitoclax (antagonizes BCL-2 and BCL-XL). Both drugs showed increased cytochrome c release compared with DMSO, and venetoclax was a better inducer of mitochondrial priming than navitoclax, which can be explained by the higher binding affinity of venetoclax (Ki < 0.010 nM)22 over navitoclax (Ki = 0.044 nM)22 for BCL-2 (P < .05; Figure 1F-G). In 4 samples, we also measured the mitochondrial sensitivity to a selective BCL-XL antagonist (A-1331852). None of the samples induced a mitochondrial priming response to A-1331852 (Figure 1F-G). We further found that BH3 profiling using the BAD peptide predicted on-target cellular sensitivity to BH3 mimetics, as shown by the significant correlation between cytochrome c release induced by the BH3 peptide and cytochrome c release caused by venetoclax and navitoclax (supplemental Figure 1A). To validate whether the mitochondrial sensitivity to venetoclax predicted by BH3 profiling results in the apoptosis of ETP-ALL cells, we performed cell death assays using venetoclax, navitoclax, and A-1331852. Although the responses were heterogeneous, all samples tested were sensitive to both venetoclax and navitoclax, emphasizing that BCL-2 dependence sensitizes adult ETP-ALL blasts to apoptosis (Figure 1H).
Having shown the selective apoptotic dependence of adult ETP-ALL on BCL-2, we next evaluated whether adult T-ALL shows selective survival dependency on specific members of BCL-2 family proteins. We performed BH3 profiling of 22 primary adult T-ALL samples collected at diagnosis (Figure 1A; supplemental Table). In contrast to adult ETP-ALL, 18 out of 22 samples showed mitochondrial depolarization in response to both BAD and HRK peptides compared with DMSO (P < .0001; Figure 2A-B). Notably, the mean of BAD- and HRK-induced cytochrome c release was significantly higher than that of DMSO, indicating codependence on both BCL-2 and BCL-XL (P < .0001; Figure 2A-B). The BAD-induced mitochondrial sensitivity of T-ALL tumors was relatively higher than that of HRK (P < .0001), indicating greater sensitivity to BCL-2 inhibition. We found a modest association between BAD and HRK (Spearman r = 0.47; P < .05) and a moderate association between BAD and BAD-HRK (Spearman r = 0.62; P < .01), further indicating a primary dependence on BCL-2 and co-dependence on BCL-XL (Figure 2C). We verified our findings by measuring the direct mitochondrial priming sensitivity to venetoclax, navitoclax, and A-1331852. Venetoclax (P < .0001) and navitoclax (P < .0001) caused superior cytochrome c release compared with DMSO (Figures 2D-E). Similar to ETP-ALL, venetoclax elicited slightly higher priming across samples than navitoclax (P < .01; Figure 2D-E). Importantly, A-1331852 induced significant mitochondrial outer membrane permeabilization in T-ALL patient samples (P < .01) compared with DMSO. However, it did not outperform navitoclax (P = .13; Figure 2D-E), as we predicted from BH3 profiling using direct peptide sensitivity responses. To further test this, we compared mitochondrial priming induced by the BAD peptide to cytochrome c release induced by the corresponding BH3 mimetic, venetoclax, and the HRK peptide, compared with A-1331852. We observed a significant correlation between BAD and venetoclax (Spearman r = 0.66; P < .001) and between BAD and navitoclax (r = 0.72; P < .001), indicating that cytochrome c release caused by BH3 peptides is comparable to direct mitochondrial permeabilization caused by BH3 mimetics (supplemental Figure 1B).
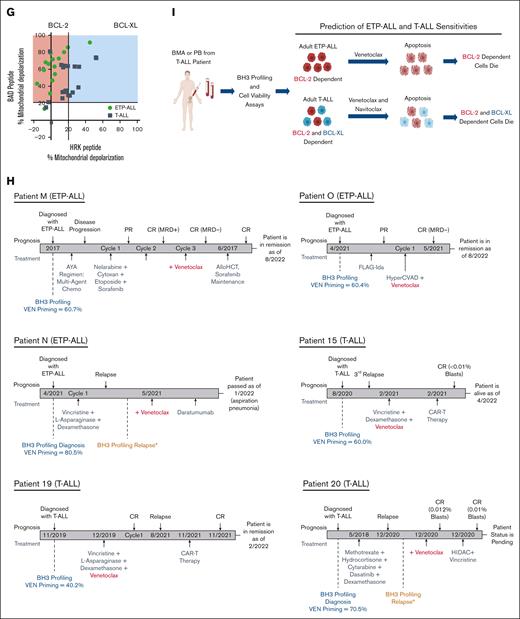
Adult T-ALL specimens are primarily dependent on BCL-2 and partially dependent on BCL-XL for survival. (A-B) FACS-based BH3 profiles obtained using the BAD (BCL-2 and BCL-XL dependence) and HRK (BCL-XL dependence) peptides. One-way ANOVA analysis of % cytochrome c release between BAD vs DMSO and HRK vs DMSO; BAD vs HRK. (C) Spearman correlation between % cytochrome c release for BAD vs HRK and BAD vs BAD-HRK. (D/E) FACS-based BH3 profiles for venetoclax, navitoclax, and A-1331852. One-way ANOVA analysis for % cytochrome c release between venetoclax vs DMSO, navitoclax vs DMSO, A-1331852 vs DMSO, venetoclax vs navitoclax, venetoclax vs A-1331852, and navitoclax vs A-1331852. (F) Cell death assays using annexin V in adult T-ALL primary samples treated with venetoclax, navitoclax, or A-1331852 for 8 hours. Refer to supplemental Figure 1 for the additional samples. Data are plotted as the percentage of live cells compared with DMSO controls. Mean ± SD of 3 replicates. (G) Dot plot of BAD peptide response vs HRK peptide response in adult ETP-ALL (green) and T-ALL (dark blue). Red indicates probable BCL-2 dependence and blue indicates probable BCL-XL dependence. (H) Clinical response timelines of patients M, O, N, 15, 19, and 20. (I) Proposed schematic for the prediction of venetoclax and navitoclax efficacy based on BCL-2 and BCL-XL dependence. Note: gating of adult T-ALL primary blast samples. Priming was normalized to that of DMSO. ∗P < .05; ∗∗P < .01; ∗∗∗P < .001; ∗∗∗∗P < .0001; CR, complete response; MRD, minimal residual disease; PR, partial response; UN, untreated; ns, no significance. (A,D) The dotted line represents the threshold for significant priming determined using DMSO ± 3xSD.

Because we observed robust mitochondrial depolarization in response to both BAD and HRK peptides, we hypothesized that T-ALL tumors would show enhanced cytotoxic response to BH3 mimetics drugs targeting BCL-2 and BCL-XL. Next, we performed cell death assays using annexin V on T-ALL primary tumors treated with venetoclax, navitoclax, and A-1331852. Despite heterogeneity, we observed sensitivity to venetoclax, and more than half of the samples showed higher cell death in response to navitoclax and/or A-1331852, reiterating that therapeutic antagonism in T-ALL targets both BCL-2 and BCL-XL proteins (Figure 2F; supplemental Figure 1C).
To further evaluate the mechanistic function, we performed western blotting and co-immunoprecipitation studies to assess protein expression and binding between pro- and antiapoptotic BCL-2 family proteins in 7 samples. Using PBMCs as a control, both ETP-ALL and T-ALL samples showed BIM:BCL-2 binding. However, BIM:BCL-XL binding was observed only in T-ALL tumors, suggesting that BIM was primarily sequestered by BCL-2 in ETP-ALL and co-sequestered by BCL-2 and BCL-XL in T-ALL (supplemental Figure 2A). Comparison of baseline BCL-2 family protein expression of BCL-2 revealed that patients with ETP-ALL had higher BCL-2/BCL-XL ratios than T-ALL (supplemental Figure 2B). ETP-ALL had lower levels of MCL-1 and p53 proteins than T-ALL (supplemental Figure 2B).
We next investigated whether there were age-dependent differences in mitochondrial priming between adult and pediatric ALL.1 We found that the mean of BAD-induced mitochondrial priming in pediatric T-ALL was higher than that in adult T-ALL counterparts (60.5%1 vs 55.5%; supplemental Figure 3A). The same was true for pediatric ETP-ALL vs adult ETP-ALL (71.4%1 vs 63.5%; supplemental Figure 3A). Collectively, ETP-ALL was more sensitive to BAD-induced mitochondrial depolarization than T-ALL (68.3% vs 57.5%; P < .01; Figure 2G; supplemental Figure 3B).1 Notably, we carried out a multicenter study providing us with a unique opportunity to evaluate the sensitivity of BH3 mimetics in adult patients with different demographics, with representation from both Western and Asian populations (supplemental Table). Irrespective of demographics, adult patients with ETP-ALL and T-ALL from both regions showed increased mitochondrial sensitivity to the BAD peptide compared with DMSO (supplemental Figure 3C). Further investigation extends beyond the scope of this study but suggests that vulnerabilities to BCL-2 family proteins may be conserved across different ethnicities.
Our previous findings on pediatric T-ALL led to the clinical testing of venetoclax in combination with hyperCVAD in patients with relapsed/refractory (R/R) T-ALL.1,19,23 However, whether adult patients with T-ALL also show a selective pattern of antiapoptotic dependence related to the differentiation stage of T-cell had not been previously reported. Although most samples accrued in this study were from the prevenetoclax era, we obtained clinical response data on venetoclax-based combinations from 3 patients with ETP-ALL and 3 patients with T-ALL. Four of the 6 patients achieved complete response to venetoclax-based therapy regimens as predicted from BH3 profiling, with 1 patient’s status pending and 1 patient passing from their disease (Figure 2H). Together, these results suggest that ETP-ALL and T-ALL both display dependency on BCL-2, which can be targeted therapeutically. This further emphasizes that combining BH3 mimetics with standard of care drugs for ALL may meaningfully improve patient responses. In addition, the results from a recent Phase I study (#NCT03181126) in R/R B-ALL and T-ALL using a combination of venetoclax and navitoclax showed that dual dependence on BCL-2 and BCL-XL is maintained in the relapse settings.10 The reported overall response rate in R/R ALL within all subgroups was 59.9% (n = 28/47), with pediatric subgroups showing a higher overall response rate of 75% (9/12) on combination therapy with venetoclax and navitoclax.10 An independent group also confirmed that 72.7% of patients with T-ALL (n = 8/11) showed predominant baseline BCL-2 dependency and switched to BCL-XL or BCL-2/BCL-XL dependence on treatment with BH3 mimetics.10
Because clinical data in adults with ETP-/T-ALL remain pending regarding whether the combination of venetoclax and navitoclax is superior to venetoclax alone, our study elaborates on this gap by elucidating discrete cellular dependencies on BCL-2 and BCL-XL, particularly in treatment-naïve samples. These details will help in the selection of BH3 mimetics therapy for adult patients with T-ALL (Figure 2I). Importantly, BCL-2 dependence is heterogeneous in terms of hematologic cancer type; CLL is homogeneously BCL-2 dependent,24 AML is co-dependent on BCL-2, and MCL-1,13 BCP-ALL is heterogeneously dependent on BCL-2,17 pediatric ETP-ALL is selectively BCL-2 dependent,1 and pediatric T-ALL is selectively BCL-XL dependent.1 Thus, these discrepancies emphasize the necessity of studying each cancer and age group independently to truly understand which BH3 mimetics may be clinically effective. Furthermore, despite the limitations of the small sample size due to the rarity of adult ALL occurrence and poor cell viability in some samples (<50% baseline viability), BH3 profiling distinctly identified BCL-2 and BCL-XL dependencies that correlated with BH3 mimetic sensitivity and clinical response, which is a major advantage compared with frank cell death or cell viability measurements requiring longer incubation times. Our study validated that BH3 profiling will continue to be a valuable functional tool for personalizing medicine by identifying protein dependencies and drug vulnerabilities in adult patients with ALL.
Acknowledgments: The authors thank all patients and families who consented to providing samples for this study at the Dana-Farber Cancer Institute, MD Anderson Cancer Center, National University Hospital Singapore, and Singapore General Hospital. The authors thank Triona Ni Chonghaile, Royal College of Surgeons in Ireland, for providing the editorial assistance. S.B. is a recipient of the Leukemia and Lymphoma Society Career Development Fellow Award. S.B. acknowledges support from the American Society of Hematology Global Research Award, NCIS and NUS Cancer Program Seed funding grant, and the National Medical Research Council of Singapore under award number OFIRG21NOV-0062. M.K. was supported by R01CA241191. J.S.G. was supported by the National Cancer Institute (NCI) of the National Institutes of Health (NIH) under award number K08CA245209, NIH/NCI SPORE in Myeloid Malignancies Grant Number 1P50CA206963, and the Ted and Eileen Pasquarello Tissue Bank in Hematologic Malignancies. A.L. acknowledges support from the NIH under award number P01CA066996. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH or NCI.
Contribution: S.B., M.K., and A.L. designed the study; J.S.G., N.J., J.S.M., W.Y.J., G.C.W., and M.O. coordinated the study, provided clinical samples and regulatory oversight, and collected the study data; S.B., E.A.O., K.S.B., and S.R. performed BH3 profiling and cell death assays, and analyzed the data; A.N.M. performed immunoprecipitation studies and analyzed the data; S.B. and E.A.O. wrote the manuscript; and all authors read, critically reviewed, and approved the final manuscript.
Conflict-of-interest disclosure: M.K. reports grants and/or other from AbbVie, F. Hoffman La-Roche, Stemline Therapeutics, Forty-Seven, Eli Lilly, Cellectis, Calithera, Ablynx, Agios, Ascentage, AstraZeneca, Reata Pharmaceutical, Rafael Pharmaceutical, Sanofi, Janssen, and Genentech. A.L. reports consultation and research support from AbbVie, Novartis, and AstraZeneca, and is an equity-holding member of the scientific advisory boards of Zentalis Pharmaceuticals, Flash Therapeutics, and Dialectic Therapeutics. J.S.G. received research funding from AbbVie, Genentech, Prelude, AstraZeneca, and Pfizer, and served on advisory boards for AbbVie, Genentech, Bristol Myers Squibb, and Servier. M.O. has served on advisory boards for Amgen, Pfizer, Janssen, and Bristol Myers Squibb. The remaining authors declare no competing financial interests.
Correspondence: Shruti Bhatt, Department of Pharmacy, Science Dr 3, Block S7 Room 02-16, Singapore 117543; e-mail: shruti_bhatt@nus.edu.sg; and Marina Konopleva, Department of Leukemia, University of Texas MD Anderson Cancer Center, 1400 Holcombe Blvd, FC3.3048, Houston, TX 77030; e-mail: mkonople@mdanderson.org.

