

Key Points
Dual CAR strategy by low affinity IL-3–z and CD33.CCR minimizes on-target/off-tumor toxicity in CD123 and/or CD33 healthy cells.
Dual IL-3–z/CD33.CCR CIK cells demonstrate high efficacy and specificity for CD123+/CD33+ leukemic cells in preclinical models.

Abstract
Acute myeloid leukemia (AML) still represents an unmet clinical need for adult and pediatric patients. Adoptive cell therapy by chimeric antigen receptor (CAR)-engineered T cells demonstrated a high therapeutic potential, but further development is required to ensure a safe and durable disease remission in AML, especially in elderly patients. To date, translation of CAR T-cell therapy in AML is limited by the absence of an ideal tumor-specific antigen. CD123 and CD33 are the 2 most widely overexpressed leukemic stem cell biomarkers but their shared expression with endothelial and hematopoietic stem and progenitor cells increases the risk of undesired vascular and hematologic toxicities. To counteract this issue, we established a balanced dual-CAR strategy aimed at reducing off-target toxicities while retaining full functionality against AML. Cytokine-induced killer (CIK) cells, coexpressing a first-generation low affinity anti-CD123 interleukin-3–zetakine (IL-3z) and an anti-CD33 as costimulatory receptor without activation signaling domains (CD33.CCR), demonstrated a powerful antitumor efficacy against AML targets without any relevant toxicity on hematopoietic stem and progenitor cells and endothelial cells. The proposed optimized dual-CAR cytokine-induced killer cell strategy could offer the opportunity to unleash the potential of specifically targeting CD123+/CD33+ leukemic cells while minimizing toxicity against healthy cells.
Introduction
Despite substantial progresses in prognostic risk stratification1 and personalized treatment with novel targeted drugs,2 between 60% and 70% of adults with acute myeloid leukemia (AML) still relapse after initial responses and do not survive beyond 5 years.3 The graft-versus-leukemia effect elicited by donor T cells after allogeneic hematopoietic stem cell transplantation (allo-HSCT) is the best postremission treatment to eradicate residual chemoresistant leukemic cells.4 However, the majority of patients with AML are not eligible for allo-HSCT because of old age, comorbidities, infectious complications, or refractory disease.5 Furthermore, ∼40% of patients with AML relapse even after allo-HSCT6 and additional curative treatment options are lacking.
Chimeric antigen receptor (CAR) T cells have dramatically improved the outcome of patients with high-risk B-cell malignancies,7 whereas their successful application to other neoplasms, including AML, still remains an unmet need.8 A major barrier limiting the safe clinical translation of CAR T-cell therapy in the AML field is the lack of a selective target antigen overexpressed on AML blasts and leukemic stem cells but absent on normal tissues.9 CD33 and CD123 are 2 of the most attractive AML targets, because of their overexpression on both leukemic bulk and chemoresistant leukemic stem cells in most patients with AML, especially in NPM1 and/or FLT-3 mutated AML.10,11 Notably, CD123 overexpression has been associated with a negative prognosis with decreased overall survival and lack of clinical remission. Nevertheless, CD33 expression on normal hematopoietic stem/progenitor cells (HSPCs)12 and CD123 expression on both HSPCs and endothelial cells13 raise concerns for life-threatening toxicities. Dual targeting of AML with CAR T cells directed against both CD33 and C-type lectin-like molecule-1 resulted in prolonged grade 4 pancytopenia in all patients, requiring an allo-HSCT to rescue the induced myeloablation14 whereas a trial of CD123 CAR T-cell therapy was placed on hold after the occurrence of severe capillary leak syndrome (1 lethal) in 2 patients.15
Several strategies have been proposed to minimize off-target toxicity risks. CAR T cell–mediated myelotoxicity could be overcome by a subsequent allo-HCT, although such approach cannot be adopted in elderly or unfit patients.16 Infusion of messenger RNA “biodegradable” engineered CD123 CAR T cells has been used but is limited by inadequate efficacy.17 Alternative approaches include generation of CD33 knockout (KO) HSCs resistant to CD33 CAR T-cell myeloid toxicity,18 development of switchable CAR platforms,19 and identification of single or combined AML-restricted antigens.20-22 In this study, we fully exploited the therapeutic potential of the simultaneous targeting of CD33 and CD123, while limiting toxicity, by adopting and optimizing a dual CAR (DC)–trans-signaling strategy23 in which efficient T-cell activation was only granted by the simultaneous engagement of both antigens.
Materials and methods
RNA sequencing
The total RNA from 12 samples (triplicates of Ontario Cancer Institute (OCI)-AML3 wild type [WT], CD33KO, CD123KO, and CD33/CD123KO) was sequenced on an Illumina Novaseq 6000 platform, generating 150 base-paired end reads, and then analyzed.24
Generation of the DC CIK cells
The interleukin 3 (IL-3)-zetakine (IL-3z) (immunoglobulin G1 [IgG1]-Fc spacer and CD3z endodomain) and the CD33 chimeric costimulatory receptor (CCR) (IgG1-Fc spacer and CD28 and 4.1BB endodomains) were cloned in an SFG-retroviral construct (In-Fusion HD Cloning Kit). Then, the dual-CAR vector was cloned using a P2A system. Cytokine-induced killer (CIK) cells were subsequently transduced following a previously described protocol.25 For the nonviral CIK cell modification, the Sleeping Beauty (SB)-pT4 vector and SB100X-transposase were kindly provided by CoImmune, Inc (Durham, NC) and DC CIK cells were generated as previously described.26
In vitro assays
Operetta CLS image acquisition
Time-dependent apoptosis mediated by IL-3z WT CAR and IL-3z mutant CAR CIK cells in endothelial telomerase-immortalized human microvascular endothelium (TIME) cell line was evaluated using Operetta CLS (PerkinElmer). CIK cells were cocultured for 8 hours with the target cells at an effector-to-target ratio (E:T) of 5:1 and images were taken every 5 minutes. Custom-made MatLab-based (Mathworks Inc) software was used for image analysis to extract the populated area of the different dyes.
Methylcellulose colony-forming unit (CFU) assay
Peripheral blood (PB)-mobilized CD34+ cells were cocultured for 24 hours with or without CAR CIK cells (E:T, 1:1). Afterward, 1000 CD34+ cells were sorted on a fluorescence-activated cell sorter Aria II, resuspended in 3 mL MethoCult H4435, and 1 mL duplicates plated in 6-well SmartDish at 5 × 102 cells per well. Plates were incubated at 37°C and 5% CO2 for 14 days, after which different subtypes of CFUs were counted using an automated StemVision machine (STEMCELL Technologies).
Mice
For the OCI-AML3 model, 1 × 106 OCI-AML3 WT or KO clones expressing luciferase were transplanted in NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice. For the KG-1 model, NSG mice received a radiation dose of 0.9 Gy followed 24 hours later by an IV injection of 2.5 × 106 KG-1 cells expressing luciferase. CAR CIK cells were then injected via tail vein at a time and dose provided in the figure legends. For the patient-derived xenograft (PDX) model, 1 × 106 cells from a previously described26 tertiary AML PDX sample were injected IV. Procedures involving animal handling and care were conformed according to protocols approved by the Service center of Preclinical Research of Perugia’s animal house facility and by the Milano-Bicocca University, and were in accordance with national and international law.
Results
CD123 and/or CD33 KO impairs leukemia growth by modulating multiple cancer pathways in a model of NPM1-mutated AML
CD123 overexpression in AML has already been associated with increased proliferative potential and resistance to apoptosis,28 whereas the implication of CD33 overexpression remains elusive. To further define the functional contribution of these 2 antigens to AML, we analyzed the effects of their CRISPR–CRISPR-associated protein 9 KO (single and double) in the NPM1-mutated OCI-AML3 cell line (Figure 1A; supplemental Tables 1 and 2), as characterized by CD33 and CD123 overexpression29,30 and thus constituting a suitable model to investigate the effects of their genetic loss. As compared with the unedited parental cell line (OCI-AML3 WT), all the KO clones exhibited impaired proliferation in vitro (Figure 1B). Moreover, in growth competition assays against CD33- and/or CD123-edited clones, OCI-AML3 WT cells rapidly became the predominant population within 9 days of coculture (Figure 1C, P value <.0001 of each KO condition vs OCI-AML3 WT at day 9). Next, we assessed whether this in vitro evidence translated into different kinetics of tumor growth in vivo, exploiting luciferase-expressing genes in all OCI-AML3 clones. Although all the KO clones were still able to engraft in NSG mice (Figure 1D), the overall survival of mice injected with CD123 or CD123/CD33 KO cells was significantly longer than that of mice that had received transplantation with parental OCI-AML3 or CD33 KO cells (Figure 1E, P value <.05). To reveal which pathways linked to CD33 and CD123 overexpression could be involved in leukemic proliferation, we performed unbiased bulk RNA sequencing comparison between OCI-AML3 WT and KO clones. Gene ontology–term enrichment analyses of expressed genes in OCI-AML3 KO clones unveiled a massive downregulation of genes involved in cancer pathways (supplemental Figure 1). Among gene expression profiles (supplemental Tables 3-5), we found a significant upregulation of the cyclin-dependent kinase inhibitor 2A, a tumor suppressor gene fundamental for cell cycle entry,31 in all OCI-AML3 KO clones but especially in the CD123 KO ones. Moreover, CD33 KO clones showed an increased expression of major histocompatibility complex–related genes (HLA) and downregulated HOX genes (Figure 1F). Notably, HOX genes are characteristically upregulated in NPM1-mutated AML32 and represent a hallmark of this AML subgroup.33 Although restricted to a single, yet common, AML subgroup, these findings further support CD123 and CD33 targeting by CAR T cells as a viable approach not only for their overexpression profile but also for the major role played in leukemia biology, which potentially decreases the likelihood of antigen escape mechanisms.

CD33 and/or CD123 KO affects proliferation in NPM1 mutant OCI-AML3 cell line. (A) Representative histograms of CD33 and CD123 expression in WT and KO OCI-AML3 Luc/GFP+ clones, measured by flow cytometry after single-cell cloning. (B) Cell growth expansion curves of CD33 and/or CD123 KO Luc/GFP+ clones compared with the parental WT Luc/GFP+ OCI-AML3 cell line, monitoring luminescence emitted 2 hours after luciferine exposure for 3 days using a Spark plate reader (Tecan). Mean ± SD from biological triplicates is shown. (C) Luc/GFP+ OCI-AML3 KO cells were cocultured with GFP- OCI-AML3 WT cells at a 1:1 E:T ratio, and GFP expression was measured to assess clonal competition at the indicated time points by flow cytometry. Mean ± SD from representative biological triplicates is shown. P values from the Wilcoxon test are indicative of each KO clone condition compared with WT OCI-AML3 control at day 9. ∗∗∗∗P value <.0001. (D) Tumor burden of Luc/GFP+ OCI-AML3 WT and KO clones, measured by bioluminescent imaging at day 21 after NSG mice injection. (E) Kaplan-Meier curves of overall survival after Luc/GFP+ OCI-AML3 cell injection. P values adjusted for multiple comparisons are from the log-rank test and indicate comparisons between each KO clone condition and WT OCI-AML3 control. ∗P value <.05; ns, not significant (P value >.05). P values are indicative of each KO clone condition compared with WT OCI-AML3 control. (F) Gene expression variation of cyclin, HLA, and HOX family–related DEGs in OCI-AML3-CD33 KO, CD123 KO, and CD33-123 KO compared with WT. Upregulated and downregulated DEGs (log2 fold change) are colored in red and blue, respectively. DEGs, differentially expressed genes; GFP, green fluorescent protein; SD, standard deviation.
CD33 and/or CD123 KO affects proliferation in NPM1 mutant OCI-AML3 cell line. (A) Representative histograms of CD33 and CD123 expression in WT and KO OCI-AML3 Luc/GFP+ clones, measured by flow cytometry after single-cell cloning. (B) Cell growth expansion curves of CD33 and/or CD123 KO Luc/GFP+ clones compared with the parental WT Luc/GFP+ OCI-AML3 cell line, monitoring luminescence emitted 2 hours after luciferine exposure for 3 days using a Spark plate reader (Tecan). Mean ± SD from biological triplicates is shown. (C) Luc/GFP+ OCI-AML3 KO cells were cocultured with GFP- OCI-AML3 WT cells at a 1:1 E:T ratio, and GFP expression was measured to assess clonal competition at the indicated time points by flow cytometry. Mean ± SD from representative biological triplicates is shown. P values from the Wilcoxon test are indicative of each KO clone condition compared with WT OCI-AML3 control at day 9. ∗∗∗∗P value <.0001. (D) Tumor burden of Luc/GFP+ OCI-AML3 WT and KO clones, measured by bioluminescent imaging at day 21 after NSG mice injection. (E) Kaplan-Meier curves of overall survival after Luc/GFP+ OCI-AML3 cell injection. P values adjusted for multiple comparisons are from the log-rank test and indicate comparisons between each KO clone condition and WT OCI-AML3 control. ∗P value <.05; ns, not significant (P value >.05). P values are indicative of each KO clone condition compared with WT OCI-AML3 control. (F) Gene expression variation of cyclin, HLA, and HOX family–related DEGs in OCI-AML3-CD33 KO, CD123 KO, and CD33-123 KO compared with WT. Upregulated and downregulated DEGs (log2 fold change) are colored in red and blue, respectively. DEGs, differentially expressed genes; GFP, green fluorescent protein; SD, standard deviation.
DC CIK cells mediate high antileukemic efficacy through transacting costimulation
Single targeting of CD123 or CD33 by CAR T cells have raised safety concerns for potential severe on-target/off-tumor effects.34-36 Therefore, we conceived a DC design with transacting costimulation, in order to reduce off-target toxicities while retaining full functionality against AML. Target cells expressing different levels of CD123 and CD33 were used to test the efficacy and safety profile of DC CIK cells, namely the AML cell line KG-1, bone-marrow (BM) derived-CD34+ HSPCs, and the TIME endothelial cell line (Figure 2A-B).

DC CIK cells express both CAR and CCR efficiently and show potent and specific in vitro antileukemic activity against CD123+CD33+ targets. (A) Flow cytometric analysis of CD33 and CD123 expression on AML primary cells, KG-1 cell line, and on normal hCD34+ cells and endothelial TIME cell line. (B) CD33 and CD123 quantification on cell surface. The number of CD33 (n = 2 for KG-1, n = 4 for CD34+, n = 12 for AML blasts) and CD123 (n = 3 for KG-1, n = 5 for TIME, n = 4 for CD34+, n = 12 for AML blasts) molecules on the cell surface was quantified using a BD QuantiBRITE PE fluorescence quantitation kit. (C) DC IL-3z/CD33 vector scheme. Single CARs are included as controls. (i) single IL-3z CAR, (ii) single CD33 CAR with CD28-4-1BB in cis costimulation, and (iii) bicistronic DC IL-3z/CD33 CCR with a self-cleaving 2A peptide. (D) Representative dot plot of IL-3z CAR, CD33 CAR, and DC expression on CIK cells at the end of differentiation. Unmanipulated (NT) CIK cells were used as control. (E) Expression of IL-3 and scFv CD33 on the surface of IL-3z CAR (n = 4), CD33 CAR (n = 4), and DC CIK cells (n = 6) by flow cytometry at the end of differentiation. (F) Short-term (E:T ratio of 5:1; n = 4 in all groups) and (G) long-term (E:T ratio of 1:100; n = 4 in all groups) cytotoxicity and (H) cytokine release against CD123+/CD33+ KG-1, OCI-AML3, and THP-1 AML cell lines (n = 4 in all groups except for OCI-AML3 and MHH-CALL-4, n = 2). CD123−/CD33− CHH-CALL4 B-ALL cell line has been included as control. Summary from 4 independent CAR CIK cell donors is shown in panels F, G, and H. Paired comparisons were performed using the Tukey test and adjusted for multiple comparisons. ns, not significant (P value >.05); ∗∗P value <.01, ∗∗∗P value <.001, ∗∗∗∗P value <.0001. B-ALL, B-cell acute lymphoblastic leukemia; LS, leader sequence; SP, spacer; TM, transmembrane domain.
DC CIK cells express both CAR and CCR efficiently and show potent and specific in vitro antileukemic activity against CD123+CD33+ targets. (A) Flow cytometric analysis of CD33 and CD123 expression on AML primary cells, KG-1 cell line, and on normal hCD34+ cells and endothelial TIME cell line. (B) CD33 and CD123 quantification on cell surface. The number of CD33 (n = 2 for KG-1, n = 4 for CD34+, n = 12 for AML blasts) and CD123 (n = 3 for KG-1, n = 5 for TIME, n = 4 for CD34+, n = 12 for AML blasts) molecules on the cell surface was quantified using a BD QuantiBRITE PE fluorescence quantitation kit. (C) DC IL-3z/CD33 vector scheme. Single CARs are included as controls. (i) single IL-3z CAR, (ii) single CD33 CAR with CD28-4-1BB in cis costimulation, and (iii) bicistronic DC IL-3z/CD33 CCR with a self-cleaving 2A peptide. (D) Representative dot plot of IL-3z CAR, CD33 CAR, and DC expression on CIK cells at the end of differentiation. Unmanipulated (NT) CIK cells were used as control. (E) Expression of IL-3 and scFv CD33 on the surface of IL-3z CAR (n = 4), CD33 CAR (n = 4), and DC CIK cells (n = 6) by flow cytometry at the end of differentiation. (F) Short-term (E:T ratio of 5:1; n = 4 in all groups) and (G) long-term (E:T ratio of 1:100; n = 4 in all groups) cytotoxicity and (H) cytokine release against CD123+/CD33+ KG-1, OCI-AML3, and THP-1 AML cell lines (n = 4 in all groups except for OCI-AML3 and MHH-CALL-4, n = 2). CD123−/CD33− CHH-CALL4 B-ALL cell line has been included as control. Summary from 4 independent CAR CIK cell donors is shown in panels F, G, and H. Paired comparisons were performed using the Tukey test and adjusted for multiple comparisons. ns, not significant (P value >.05); ∗∗P value <.01, ∗∗∗P value <.001, ∗∗∗∗P value <.0001. B-ALL, B-cell acute lymphoblastic leukemia; LS, leader sequence; SP, spacer; TM, transmembrane domain.
DC CIK cells were generated by retroviral transduction with a first-generation anti-CD123 IL-3-zetakine37 (IL-3z CAR) linked to an anti-CD33 CCR (CD33 CCR) carrying CD28-4-1BB costimulatory moieties, through a 2A-sequence peptide. Single-transduced IL-3z CAR, CD33 CAR with CD28-4-1BB-z, and CD33 CCR CIK cells were included as controls (Figure 2C). In particular, CD33 CCR CIK cells were included to confirm the inability of the CD33 CCR to induce CIK cell effector functions per se (supplemental Figure 2A-D). A high transduction efficiency was achieved in all the conditions with a stoichiometric expression of the 2 receptors in the DC (Figure 2D-E). CD4, CD8, and memory phenotypes were similar in all CIK cell conditions (supplemental Figure 2B). All CAR CIK cells specifically induced in vitro target cell lysis (in short- and long-term cytotoxicity assays) and cytokine production when challenged with THP-1, KG-1, and OCI-AML3 CD123+/CD33+ AML cell lines (Figure 2F-H, P value <.0001 DC vs not-transduced (NT) CIK cells in THP-1 and KG-1; P value <.001 DC vs NT CIK cells in OCI-AML3). However, although the DC exhibited higher IL-2 production after antigen stimulation (Figure 2H), the benefit of a transacting stimulation has been blunted by the high in vitro activity displayed by the single IL-3z CAR. Therefore, in order to achieve limited T-cell activation upon single IL-3z CAR engagement and thus reducing potential DC CIK cell toxicities against CD123low healthy cells, we considered generating an IL-3z CAR with reduced binding affinity, to obtain a suboptimal signal 1.
Rational design through molecular dynamic (MD) simulations and validation of a low affinity IL-3-zetakine
MD simulation is a well-established computational method for investigating and understanding biomolecular behavior in atomic detail.38 Four IL-3 mutations (N18K, E22R, E43N, and F113A) known to have effects in the docking of the ligand to CD123,39,40 were tested alone or together (Mut4) by MD to predict IL-3 mutant binding affinity compared with IL-3 WT. Initially, the stability of the complexes was determined through 30 ns MD simulations by plotting the backbone (Cα atoms) root mean square deviation during the simulation, without significant differences between mutants and WT (supplemental Figure 3A). Moreover, hydrogen bond (HB) analysis did not show any significant changes (supplemental Figure 3B; supplemental Table 6). Together, these observations indicated that single and combined mutations do not affect the conformation of the complex, suggesting that unspecific binding to other molecules is unlikely to occur. Next, we investigated free binding energy (ΔGbinding) between IL-3Rα and IL-3 in WT complex and mutants of the last 10 ns of the trajectories, to envisage possible changes in binding affinity. Mut4 complex showed a lower calculated ΔGbinding value of approximately −24 Kcal/mol, indicating that the Mut4 is the least stable complex among all the mutants (supplemental Figure 3B). This result was confirmed also in extended simulation of 100 ns (supplemental Figure 3C), highlighting differences between WT and Mut4 systems in the HB occupancy analysis (supplemental Table 7). In particular, WT IL-3 showed more prolonged HBs during the simulation time, whereas the Mut4 complex lost Lys54/Glu43 and Arg234/Glu119 contact (Figure 3A-B). A low ΔGbinding in Mut4 complex was observed compared with WT also in the last 40 ns of the simulation (Figure 3C), in which the 2 systems showed their general stability, exhibiting a reduction of ∼43% (Figure 3D).

MD for the prediction of IL-3 mutants with low binding affinity to CD123. (A-B), WT and Mut4 interface interaction between IL-3 and IL-3Ra. HB is represented in yellow; salt bridge is represented in magenta; distance in Å for each interaction are also reported in magenta color. (C) ΔGbinding energies vs simulation time in WT and Mut4 systems. (D) Scatter plot of ΔGbinding energies of all analyzed frames in WT and Mut4 systems. (E) Time-course analysis of apoptosis measured by caspase-3 activation on target TIME cells cocultured for 8 hours with NT, IL-3z WT CAR-, and IL-3z mutant CAR–engineered CIK cells (E:T ratio of 5:1) using Operetta CLS. Custom-made MatLab-based software provided the image analyses for extraction of the populated area of the different dyes; n = 3 wells. (F) Live-cell imaging of the dynamic processes over time and space. First row represents the frame took at time 0, whereas the second row reports the frame taken after 8 hours.
MD for the prediction of IL-3 mutants with low binding affinity to CD123. (A-B), WT and Mut4 interface interaction between IL-3 and IL-3Ra. HB is represented in yellow; salt bridge is represented in magenta; distance in Å for each interaction are also reported in magenta color. (C) ΔGbinding energies vs simulation time in WT and Mut4 systems. (D) Scatter plot of ΔGbinding energies of all analyzed frames in WT and Mut4 systems. (E) Time-course analysis of apoptosis measured by caspase-3 activation on target TIME cells cocultured for 8 hours with NT, IL-3z WT CAR-, and IL-3z mutant CAR–engineered CIK cells (E:T ratio of 5:1) using Operetta CLS. Custom-made MatLab-based software provided the image analyses for extraction of the populated area of the different dyes; n = 3 wells. (F) Live-cell imaging of the dynamic processes over time and space. First row represents the frame took at time 0, whereas the second row reports the frame taken after 8 hours.
Finally, we biologically probed whether the low affinity IL-3z mutant CAR could reduce CIK cell activation upon challenge with the TIME endothelial cells by performing a dynamic live-cell imaging assay assessing microvascular cytotoxicity over time and space. Using caspase-3 as specific marker of early apoptosis, a lower caspase-3 activation was detected on target TIME cells when cocultured with NT and IL-3z mutant CAR-engineered CIK cells as compared with IL-3z WT CAR (Figure 3E-F; supplemental Video). Multiplexed cytokine profiling after long-term coculture showed that KG-1 cells but not TIME cells stimulated IL-3z WT CAR and IL-3z mutant CAR CIK cells to significantly release more Th1/Tc1 (granulocyte-macrophage [GM]-colony-stimulating factor, interferon gamma, tumor necrosis factor α, IL-2) cytokines (supplemental Figure 3D).
Low affinity DC CIK cells decrease on-target/off-tumor toxicity against endothelial and HSPCs in vitro
IL-3z mutant CAR was then coupled to the CD33 CCR in the newly generated low affinity DC (DC mutant) to test safety and antileukemic efficacy (Figure 4A). As compared with the DC WT, the DC mutant displayed less lytic activity, and therefore superior safety, when cocultured with the low–CD123-expressing TIME endothelial cell line at 2 different E:T ratios (10:1 and 5:1) (Figure 4B, P value <.05 DC WT vs NT; DC mutant vs NT, not significant). This low endothelial toxicity was further sustained by comparable low cytokine levels released by NT and DC mutant CIK cells after long-term cocultures with CD123low TIME endothelial cell line (Figure 4C). In contrast with cytotoxicity assays, no differences in cytokine production were observed between DC WT and mutant DC, probably because of very low CD123 density on TIME cells. This result is in line with previous studies27,41 reporting different levels of activation threshold for early cytotoxicity and later functions such as cytokine release. In contrast, in the presence of KG-1, a lower cytokine production (still higher than NT) by low affinity DC CIK cells has been observed, confirming prior findings with low affinity CARs.27
Low affinity IL-3z CAR displays lower cytotoxicity toward CD123+ endothelial cells and HSPCs. (A) Vector scheme of the DC construct. (B) Short-term cytotoxicity measured by adenylate kinase release (ToxiLightTM BioAssay Kit) by TIME cells after 4-hour coculture of NT CIK cells, IL-3z WT CAR CIK cells, IL-3z mutant CAR CIK cells, DC WT CIK cells, and DC mutant CIK cells (E:T ratio of 5:1 and 10:1). Median from 3 independent experiments (mean of triplicates) is shown for each condition. (C) Th1/Tc1 cytokine release after long-term cytotoxicity assays of KG-1 and TIME cells cocultured with NT, DC WT– and DC mutant–engineered CIK cells. Median from 3 independent experiments is shown for each condition. (D) Schematic diagram of the trans-acting activation whereby DC CIK cells can be fully activated only by CD123/CD33 overexpressing LSCs but not by CD123/CD33 low expressing HSPCs. (E) Representative pictures of STEMvision-scanned cultures showing duplicates of CFUs, generated from CD34+ cells previously cocultured with NT or CAR CIK cells. Red circles label erythroid (BFU-E), yellow circles label myeloid (CFU-GM), and blue labels mixed (CFU-GEMM) colonies. (F-G) Clonogenic efficiency of 500 resiDual CD34+ HSPCs after 24 hours coincubation with NT and CAR CIK cells (E:T 1:1). BFU-E, CFU-GM, and CFU-GEMM from NT and CAR CIK cell–treated conditions were quantified after 14 days and compared with CD34+ cells (n = 5 CAR CIK cell donors). (H-I), Absolute quantification by flow cytometry of CD34+/CD38+ residual cells after exposure to NT or CAR CIK cells (24 hours, E:T 1:1), separated by CMPs, GMPs, and MEPs. Gating strategy is detailed in supplemental Figure 5 (n = 3 CAR CIK cell donors). Paired comparisons were performed using the Tukey test and adjusted for multiple comparisons in panels B-C,F-I. ns, not significant (P value >.05); ∗P value <.05, ∗∗P value <.01, ∗∗∗P value <.001, ∗∗∗∗P value <.0001. CMP, common myeloid progenitors; GEMM, granulocyte, erythrocyte, monocyte, megakaryocyte; GMP, granulocyte–monocyte progenitors; LSCs, leukemic stem cells; MEP, megakaryocyte-erythroid progenitor.

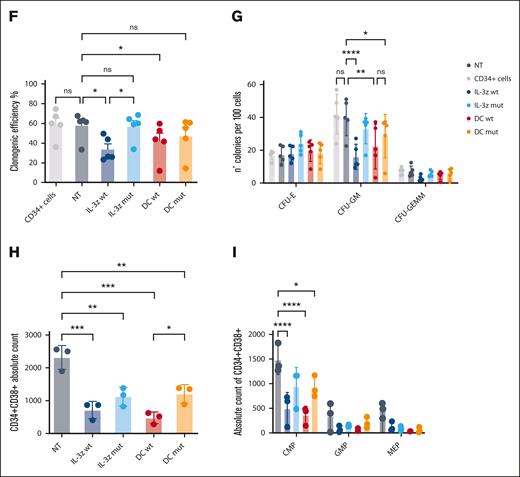
Considering the potential severe off-tumor effects on the hematopoietic compartment, we hypothesized that low affinity DC CIK cells would spare HSPCs by 2 distinct but complementary mechanisms: (i) avoiding cell activation against the majority of CD33+ hematopoietic cells because of a lack of stimulation through anti-CD33 CCR and (ii) decreasing reactivity toward low expressing CD123+ HSPCs because of reduced affinity of the anti-CD123 suboptimal CAR (Figure 4D). In vitro clonogenic efficiency and differentiation capacities of PB–derived healthy CD34+ HSPCs were tested after 24-hours coculture assay with different CIK cell conditions with similar CAR expression (supplemental Figure 4), through automated and standardized counting of all hematopoietic colony types after 14 days (Figure 4E). No significant differences have been observed between CD34+ HSPCs not exposed or preexposed to DC mutant CIK cells, suggesting that lowering the IL-3z affinity does not hamper the clonogenic capacity of CD34+ HSPCs and confirming the hypothesis that the CD33 CCR is not associated with enhanced myelotoxic potential (Figure 4F; DC mutant vs NT; P value, not significant). A significant reduction in clonogenic capacity was observed in IL-3z WT CAR and DC WT conditions, in particular in CFU-GM (Figure 4G, P value <.0001 for IL-3z vs NT; P value <.001 for DC WT vs NT). Moreover, the analysis of CD34+CD38+ myeloid progenitor subsets confirmed that common myeloid progenitors, granulocyte–monocyte progenitors, and megakaryocyte-erythroid progenitor proportions were less significantly affected by exposure to DC mutant CIK cells as compared with DC WT CIK cells (Figure 4H). Consistent with CD123 expression levels on different subsets of HSPCs,36 only IL-3z WT and DC WT CIK cells significantly reduced absolute counts of CD34+CD38+ common myeloid progenitors subpopulation (Figure 4I, P value <.0001 DC WT and IL-3z vs NT; P value <.05 DC mutant vs NT; and supplemental Figure 5). In conclusion, these data support that low affinity DC exhibits a lower off-target vascular and hematopoietic toxicity.
Low affinity DC CIK cells preserve antileukemic efficacy in vitro and improve antitumor control in vivo
Next, we evaluated whether low affinity DC CIK cells could match safety with an efficacy profile comparable with WT DC CIK cells. Phenotypic analysis performed after 21 days of culture showed similar CAR expression levels and percentage of T and natural killer (NK) cell–like populations as well as memory phenotype between all CAR CIK cell conditions (supplemental Figure 6A-B). Low affinity DC CIK cells retained a high in vitro cytotoxicity against CD123+/CD33+ KG-1 AML cell line in short- and long-term assays (Figure 5A-B, P value <.001 vs NT). DC mutant CIK cells also showed a significant control of primary blast growth in long-term cytotoxicity assays, comparable with DC WT CIK cells (Figure 5C, P value <.001 vs NT, supplemental Table 8).
Low affinity DC CIK cells retain high antileukemic efficacy. (A) Short-term (E:T ratio of 5:1, n = 5 for NT and DC mutant, n = 3 for DC WT) and (B) long-term (E:T ratio of 1:10, n = 6 for NT and DC mutant, n = 4 for DC WT) cytotoxicity against CD123+/CD33+ KG-1 cell line. Box and whiskers plot from independent CAR CIK cell donors is shown. (C) Long-term (E:T ratio of 1:10) cytotoxicity against primary AML cells (n = 4 different primary AMLs). Box and whiskers plot from independent CAR CIK cell donors is shown. (D) Schematic of the Luc KG-1 xenograft model. NSG mice were sublethally irradiated (0.9 Gy) on day −1 and injected via tail vein on day 0 with 2.5 × 105 KG-1 cells stably expressing GFP/luciferase. Mice were randomized to 5 treatment groups, each receiving 3 injections of vehicle or gene-modified CIK cells at day 3, 7, and 10 (n = 4 per group). (E) Bioluminescent imaging weekly measurements to quantify AML burden. (F) Longitudinal profile of the mean tumor burden imaged within the first 35 days showing suppression of leukemic growth in mice treated with DC CIK cells. (G) Kaplan-Meier curves of overall survival. P values adjusted for multiple comparisons are from the log-rank test and indicate comparisons between the KG-1 only cohort and the CAR CIK-treated cohorts. ∗P value <.05; ns, not significant (P value >.05). Paired comparisons were performed using the Tukey test and adjusted for multiple comparisons in panels A-C. ns, not significant (P value >.05); ∗P value <.05, ∗∗P value <.01, ∗∗∗∗P value <.0001.
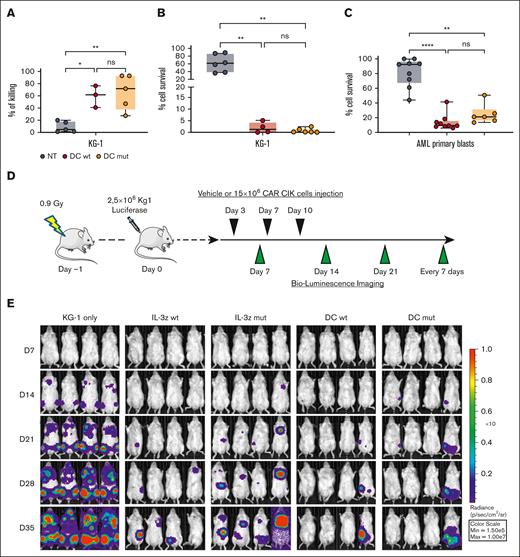
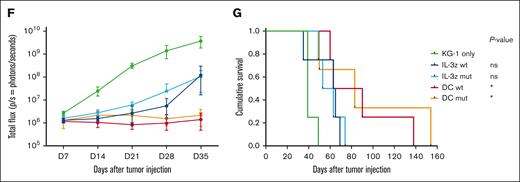
The antileukemic activity of DC mutant CIK cells was then confirmed in vivo in an early-treatment model of mice engrafted with the KG-1 AML cell line (Figure 5D). In the groups treated with IL-3z constructs, disease growth was delayed by the treatment without relevant impact on survival, whereas in the DC groups there was better disease control in terms of bioluminescence imaging (BLI) signal (Figure 5E-F) and significantly prolonged survival (Figure 5G, P value <.05 vs KG-1 only). This result correlates with the higher in vitro expansion exhibited by DC CIK cells in a long-term coculture assay with KG-1 cells, as compared with IL-3z CAR CIK cells (supplemental Figure 6C-D). The potency of DC mutant CIK cells was further confirmed in a more challenging AML treatment model, in which the treatment started 2 weeks after KG-1 injection (supplemental Figure 7A-D, P value <.05 vs KG-1 only). Together, these findings suggest that a low affinity DC CIK cell targeting approach preserves a high antileukemic activity.
Distinct spacer and transmembrane domains between chimeric receptors grant specificity to DC CIK cells
Because both IL-3z CAR and CD33 CCR carry the same spacer/transmembrane domains (IgG1-Fc spacer and a CD28 transmembrane), we aimed to refine the DC design to avoid any unwanted unspecific activation induced by a potential CAR-CCR dimerization. Indeed, we found that DC mutant CIK cells were significantly activated and produced cytokines not only when challenged with the WT KG1 (expressing both CD123 and CD33 antigens) but also in response to CD123- KG1 (supplemental Figure 8). This result is in line with a recent study by Hirabayashi et al42 that demonstrated that a single CD3z can mediate DC activation if both CAR constructs share the same spacer and/or transmembrane domain. Therefore, we generated a novel DC system with distinct spacers and transmembrane motifs (CD8 spacer and transmembrane for IL-3z CAR and CH3 spacer with CD28 transmembrane for CD33 CCR). Moreover, considering our extensive experience with the SB transposon system for the nonviral engineering of CIK cells, both at the preclinical and clinical level,43 we successfully cloned the DC construct within a pT4-transposon plasmid,26 reaching up to 50% of DC expression on CIK cells (Figure 6A).

Spacer and transmembrane optimization endow DC CIK cells of higher selectivity toward double-positive CD33/CD123 cells. (A) DC vector scheme. Bicistronic DC IL-3z/CD33 CCR carrying different spacers and transmembrane domains. (B-C), Immune synapse-binding avidity of NT and DC WT CIK cells to OCI-AML3 WT, OCI-AML3 CD33−, and OCI-AML3 CD123−/CD33− targets assessed via acoustic force microfluidic microscopy. Data represent mean ± SD and combined experiments from 2 separate donors. (D-E), Long-term (E:T ratio of 1:10) cytotoxicity against KG-1 cell line. Percentage of residual KG-1 cells after the 1-week coculture (D) and CD3 fold change after the 1-week coculture (E). Mean ± SEM from independent CIK cell donors is shown (n = 3 for all the conditions except for n = 1 against CD123−/CD33+ KG-1 cells). (F) Kaplan-Meier curves of overall survival. P value indicates comparison between the KG-1 only cohort and the DC CIK-treated cohorts. ns, not significant (P value >.05), ∗P value <.05. (G) Representative dot plot of PB analysis. (H) Analysis of hCD45+/CD33+/CD123+ cells in the PB of untreated and treated mice. (I) Analysis of hCD45+/CD3+ cells in the PB of DC CIK cell–treated mice. Paired comparisons were performed using the Tukey test and adjusted for multiple comparisons in panels D-E. ns, not significant (P value >.05); ∗P value <.05, ∗∗P value <.01, ∗∗∗P value <.001. SEM, standard error of the mean.
Spacer and transmembrane optimization endow DC CIK cells of higher selectivity toward double-positive CD33/CD123 cells. (A) DC vector scheme. Bicistronic DC IL-3z/CD33 CCR carrying different spacers and transmembrane domains. (B-C), Immune synapse-binding avidity of NT and DC WT CIK cells to OCI-AML3 WT, OCI-AML3 CD33−, and OCI-AML3 CD123−/CD33− targets assessed via acoustic force microfluidic microscopy. Data represent mean ± SD and combined experiments from 2 separate donors. (D-E), Long-term (E:T ratio of 1:10) cytotoxicity against KG-1 cell line. Percentage of residual KG-1 cells after the 1-week coculture (D) and CD3 fold change after the 1-week coculture (E). Mean ± SEM from independent CIK cell donors is shown (n = 3 for all the conditions except for n = 1 against CD123−/CD33+ KG-1 cells). (F) Kaplan-Meier curves of overall survival. P value indicates comparison between the KG-1 only cohort and the DC CIK-treated cohorts. ns, not significant (P value >.05), ∗P value <.05. (G) Representative dot plot of PB analysis. (H) Analysis of hCD45+/CD33+/CD123+ cells in the PB of untreated and treated mice. (I) Analysis of hCD45+/CD3+ cells in the PB of DC CIK cell–treated mice. Paired comparisons were performed using the Tukey test and adjusted for multiple comparisons in panels D-E. ns, not significant (P value >.05); ∗P value <.05, ∗∗P value <.01, ∗∗∗P value <.001. SEM, standard error of the mean.
To better investigate the target-specific transactivation of the CIK cells engineered with the optimized DC design, we used dynamic acoustic force measurements to evaluate binding avidity. WT, CD123 KO, and double CD123/CD33 KO OCI-AML3 cell clones were used as targets to check the contribution of the CD33-mediated engagement of the CCR in the synapse formation. We found that optimized DC CIK cells had a lower binding avidity when challenged with CD123-KO OCI-AML3 and a negligible avidity when challenged with double-negative targets, comparable with the binding avidity of NT CIK cells to all targets (Figure 6B-C). In long-term cytotoxic assays against WT and edited KG-1 cells, the newly optimized DC conferred CIK cells a high, specific antileukemic activity against CD123+ KG-1 cells (Figure 6D, P value <.001 vs NT in KG-1; P value <.01 vs NT in KG-1 CD123+CD33−). Notably, a preferential expansion was observed only when DC CIK cells were challenged against WT KG-1, demonstrating the gain in stimulation provided when the CCR is engaged (Figure 6E, P value <.05 vs NT). The newly designed DC also ensured tumor control and superior overall survival of engrafted KG-1 (Figure 6F-H, P value <.05 vs KG-1 only).
Nonviral-engineered low affinity DC CIK cells join safety and efficacy in a PDX AML mouse model
Envisaging a clinical translation of the low affinity DC CIK cells with distinct spacer and transmembrane domains, we verified the preserved safety against endothelial TIME cells in long-term cytotoxicity and cytokine production assays; a high antileukemic activity against KG-1 AML cell line and primary blasts was being maintained (Figure 7A-B). Moreover, in order to test DC mutant CIK cells against primary AML cells featuring an antigen density more reflective of a real-world population, we exploited a tertiary PDX mouse model established in our laboratory26 (Figure 7C). At sacrifice (day 35) the treated mice showed low tumor burden in PB and spleen, and tumor burden was almost cleared from the BM (0.062 ± 0.095, n = 4 vs 65.7 ± 6.7, n = 4 of untreated mice, P value <.05), as well as relevant CIK cell infiltration (1.2 ± 1.1 in the BM, 13.2 ± 18.3 in the PB, and 13.7 ± 15.4 in the spleen) (Figure 7E-H). These results support the potential of the proposed strategy of matching efficacy with safety, further encouraging its development for future clinical translation.

Optimized low affinity Dual CAR CIK cells control AML growth while reducing off-targeted toxicities. (A) Long-term (E:T ratio of 1:10, n = 2 for NT and DC mut) cytotoxicity against CD123+/CD33+ KG-1 cell line, primary AML cells and TIME cell line. Scatted plot (mean ± SD) from two independent CAR-CIK donors is shown. (B) Cytokine release against CD123+/CD33+ primary AML cells and TIME cell line (n = 2 for NT and DC mut). (C) Schematic of the AML patient xenograft mouse model. NSG mice were injected via tail vein on day 0 with 1 × 106 tertiary PDX AML cells. Mice were randomized to 2 treatment groups each receiving one injection of vehicle or gene-modified DC mut CIK cells at day 5 (n = 4 per group). (D) Analysis of hCD45+/CD33+/CD123+ cells in the PB, BM and spleen of untreated and treated mice. (E) Analysis of hCD45+/CD3+ cells in the PB, BM and spleen of Dual CAR-CIK treated mice. (F) Representative dot plot of PB analysis. P-values from the Wilcoxon test are referred to the comparison of DC mut-CIK-treated mice compared with untreated mice within PB, BM and spleen. ∗, p-value < 0.05.
Optimized low affinity Dual CAR CIK cells control AML growth while reducing off-targeted toxicities. (A) Long-term (E:T ratio of 1:10, n = 2 for NT and DC mut) cytotoxicity against CD123+/CD33+ KG-1 cell line, primary AML cells and TIME cell line. Scatted plot (mean ± SD) from two independent CAR-CIK donors is shown. (B) Cytokine release against CD123+/CD33+ primary AML cells and TIME cell line (n = 2 for NT and DC mut). (C) Schematic of the AML patient xenograft mouse model. NSG mice were injected via tail vein on day 0 with 1 × 106 tertiary PDX AML cells. Mice were randomized to 2 treatment groups each receiving one injection of vehicle or gene-modified DC mut CIK cells at day 5 (n = 4 per group). (D) Analysis of hCD45+/CD33+/CD123+ cells in the PB, BM and spleen of untreated and treated mice. (E) Analysis of hCD45+/CD3+ cells in the PB, BM and spleen of Dual CAR-CIK treated mice. (F) Representative dot plot of PB analysis. P-values from the Wilcoxon test are referred to the comparison of DC mut-CIK-treated mice compared with untreated mice within PB, BM and spleen. ∗, p-value < 0.05.
Discussion
In this study, we probe a novel strategy targeting both CD33 and CD123 without the risk of severe on-target/off-tumor toxicities, to improve the outcome of patients with AML. These 2 target antigens were chosen because of their broad expression in AML,44,45 especially in NPM1-mutated AML,10 and from the results of our transcriptome analysis on CD33 and/or CD123 KO AML cells. Indeed, in our model of NPM1-mutated AML, we found a close correlation between CD123 and/or CD33 overexpression and downregulation of cyclin dependent kinase inhibitor 2A. Moreover, we uncovered a correlation between CD33 overexpression and HOX genes upregulation, a hallmark of NPM1-mutated AML, and HLA downregulation. The importance of both CD123 and CD33 for AML pathogenesis and self-renewal is also consistent with the observation that loss of these antigens (antigen escape) has not previously been reported after targeted immunotherapies.46 Although NPM1-mutated AML has an overall good prognosis without allogeneic transplantation, AML with the cooccurrence of NPM1-mutation and FLT3-ITD or FLT3-ITD (representing 20%-30% of AML) are European LeukemiaNet 2022–defined as intermediate/adverse risk and frequently relapse after standard chemotherapy.47 Because CD123 and CD33 overexpression is characteristic of NPM1 and/or FLT3/ITD AML,10,30,48 we believe that the proposed DC mutant CIK cell strategy could be useful not only in patients with NPM1-mutated AML relapsing after chemoimmunotherapies and who are not eligible for allo-HSCT but also in the FLT3-ITD AML subset (with or without NPM1 mutations). Moreover, targeting CD123 could have a direct effect on all high-risk AMLs, as many studies have reported a direct correlation between CD123 levels and the number of leukemic blasts at the diagnosis,49,50 suggesting a role of CD123 in leukemic blast proliferation/survival. Notably, CD123 overexpression has been correlated to poor prognosis, resistance to apoptosis, and lower survival after standard treatments.51 A similar finding was recently reported in a pediatric setting from a prospective analysis on more than 1000 BM specimens, showing that high CD123 expression correlated with higher prevalence of KMT2A and FLT3-ITD mutations, a higher relapse risk, and lower survival.52
The simultaneous targeting of these antigens with T cells expressing 2 independent CARs for CD123 and CD33 have already allowed the elimination of either single- or double-positive target cells.53 However, CD33 and CD123 are also expressed on normal cells, including HSPCs and endothelial cells.54 Thus, the translation from preclinical studies to the clinic of CAR T-cell therapy directed to these antigens is associated with a serious risk of toxicity, which can be even higher when compared with a single targeting approach.
In order to fully exploit the therapeutic potential of simultaneously targeting these 2 antigens while limiting the concern for life-threatening toxicities, we generated and optimized a DC trans-signaling strategy in which efficient T-cell activation is only granted by the simultaneous engagement of both CD123 and CD33 antigens. CIK cells, that is, effector T lymphocytes with NK features acquired during ex vivo expansion,55 were genetically modified to coexpress a first-generation anti-CD123 CAR, providing the activation signal 1 (CD3ζ), and an anti-CD33 CCR delivering the costimulatory signal 2 (4-1BB and CD28 double costimulation). In the AML setting, an analogous approach has been preclinically described to enhance selectivity toward CD13 and TIM-3 double-positive AML cells.56 However, a moderate, yet significant, level of toxicity toward healthy CD13 single-positive cells, including HSCs, could not be avoided. Similarly, in our first prototype of a DC, a significant level of on-target/off-tumor toxicity toward endothelium and myeloid progenitors was still present after a single CD123 engagement in vitro. This prompted us to generate an anti-CD123 CAR with a reduced binding affinity to minimize the off-tumor CAR-induced activation.27,57 Instead of an anti-CD123 scFv-based CAR, we exploited the IL-3 sequence as the natural CD123 ligand to build an IL-3z. This allowed us to screen in silico potential low affinity IL-3 mutants by replacing already known IL-3 epitopes predicted to be crucial for CD123 binding, with the intent to minimize toxicities toward CD123+ normal cells. Four different point mutations were validated by MD simulation and trajectory analysis. The IL-3 mutant carrying 4 amino acid substitutions displayed the lowest ΔGbinding and was selected for subsequent in vitro and in vivo characterization. The selected IL-3z mutant significantly reduced CIK cell activation after encountering both CD123-low endothelial cells and myeloid progenitors, while allowing efficient AML targeting both in vitro and in vivo, when coupled to a CD33 CCR domain.
Decreasing signal 1 strength did not jeopardize the reactivity toward CD123+/CD33+ leukemic cells, as efficient the dual-CAR CIK cell activation threshold was reached only after the simultaneous engagement of both receptors, whereas a weaker or null CIK cell reactivity was observed in response to CD123+ endothelium and HSPCs, and CD33+ hematopoietic cells. This trans-signaling model was further improved by optimizing the hinge/transmembrane regions to avoid any possible homodimerization between the CAR and the CCR bearing the same CD28 transmembrane domains, as recently described.42 Different transmembrane domains were therefore introduced (CD8tm for anti-CD123 CAR and CD28tm for anti-CD33 CCR). This tactic allowed the avoidance of unwanted DC CIK cell activation against CD33 single-positive target cells. Binding avidity measurements showed how fine tuning of the DC system with distinct transmembrane motifs avoided CIK cell recognition of CD123−CD33+ cells, without compromising the binding and the subsequent full activation after double-antigen engagement on AML cells.
Envisaging a clinical translation of this immunotherapeutic strategy using a manufacturing technology more cost effective but still efficient in gene transfection,58 we transferred our DC from the retroviral vector to the SB transposon system, exploiting the already optimized nonviral CAR CIK cell platform we had developed for patients with B-cell acute lymphoblastic leukemia.43 The bicistronic construct was successfully expressed by CIK cells, confirming the great payload capacity of the transposon system. The ultimate nonviral-engineered low affinity DC CIK cell product preserved a beneficial safety profile and antileukemic activity in vitro. Furthermore, in a PDX established by our laboratory, expressing CD123 and CD33 antigens at levels more closely resembling physiological levels, high in vivo tumor control and robust CIK cell expansion has been observed.
Using engineered CIK cells as an allogeneic source for the DC approach would also overcome anti-AML CAR manufacture issues. In fact, the intensive induction/salvage chemotherapy schedules in AML can dramatically impair the quality and quantity of autologous T or NK cells at leukapheresis.59 Moreover, aggressiveness of relapsed/refractory (r/r) AML requires fast manufacturing to allow CAR T-cell infusion before progression or significant infectious complications. Among 10 patients enrolled in a recent phase 1 trial, evaluating autologous CD33 CAR T cells in r/r AML, apheresis could be performed in 8 patients, product release specification was met in 4, and infusion completed only in 3, who died from disease progression.60 Conversely, our first-in-human clinical trial exploring allogenic CD19 CAR CIK cells in r/r B-cell acute lymphoblastic leukemia after allo-HSCT showed feasibility to generate a CAR product from donor PB for all patients enrolled, with no reported graft-versus-host disease or immune effector cell–associated neurotoxicity syndrome, and only occasional grade 2 cytokine release syndrome.43 Such an impressive safety profile and encouraging activity of CAR CIK cells is particularly attractive in the AML setting, in which older age and comorbidities hamper adoptive cell therapy options because of the risk of severe cytokine release syndrome or immune effector cell–associated neurotoxicity syndrome.
In conclusion, we provide a rational approach to engineering a DC with trans-acting costimulation that provides advantages over single-targeting CARs, such as improved efficacy and high specificity for CD123 and CD33 recognition on leukemic cells while substantially reducing the risk of life-threatening toxicities.
Acknowledgments
The authors thank the parent committees Quelli Che…Con Luca Onlus, Comitato Maria Letizia Verga, Amici di Duccio, and Stefano Verri for their generous and constant support and CoImmune, Inc. for providing the SB plasmids. The authors thank Giusi Melita for technical support in the in vivo studies, and Simone Naso, Roberta Rossi, and Serena Donnini for technical support in the in vitro studies.
The work was supported by grants Ricerca Finalizzata-Giovani Ricercatori (GR-2016- 02363491), AIRC 5x1000 “Immunity in Cancer Spreading and Metastasis” (#21147), the Ministero della Salute Research project on CAR T cells for hematologic malignancies and solid tumors conducted under the aegis of Alliance Against Cancer network, the European Research Council (ERC Adv grant 2016 #740230 [B.F. and E.R.C.], Cons grant 2016 #725725 [M.P.M.]), and the Italian Association for Cancer Research (AIRC grant #23604 [B.F.]). The authors thank Quelli Che…Con Luca Onlus, which funded the project and Comitato per la vita “Daniele Chianelli.”
Authorship
Contribution: V.M.P. and M.C.R. were responsible for study conceptualization, methodology, data curation, formal analysis, validation, investigation, and visualization, writing of original draft, and review and editing the final manuscript; I.P. was responsible for data curation, validation, formal analysis, investigation, and methodology; S.G. was responsible for data curation, formal statistical analysis, and methodology; G.A. was responsible for data curation, validation, and methodology; G.P. performed data curation, validation, formal analysis, investigation, and visualization; V.C. was responsible for data curation, validation, formal analysis, investigation, visualization, and methodology; A.M. was responsible for formal analysis, investigation, visualization, and methodology; M. Sabino was responsible for formal analysis, visualization, and methodology; V.T. and G.S. were responsible for formal analysis, visualization, and methodology; F. Mezzasoma and F. Morena performed data curation, validation, formal analysis, and investigation; S.M. reviewed and edited the manuscript; D.S. performed data curation, validation, and formal analysis; J.F.A. was responsible for data curation, validation, methodology, and formal analysis; B.W. was responsible for data curation, validation, methodology, and formal analysis; M. Serafini reviewed and edited the manuscript; M.P.M. and B.F. provided supervision and resources and edited the manuscript; A.B. conceptualized the study, provided resources and supervision, acquired funding, and reviewed and edited the manuscript; and S.T. conceptualized the study, was responsible for data curation, formal analysis, validation, investigation, visualization, methodology, acquired funding, and reviewed and edited the manuscript.
Conflict-of-interest disclosure: Fondazione Tettamanti submitted a PCT application on 6 November 2015 (PCT/EP2015/075980), “Improved method for the generation of genetically modified cells,” Biondi, A., Biagi, E., Magnani, C.F., Tettamanti, S. The technology was licensed to CoImmune, Inc. for further development. M.P.M. reports honoraria from Rasna Therapeutics, Inc. for scientific advisor activities and serves as consultant for scientific advisory boards of AbbVie, Amgen, Celgene, Janssen, Novartis, Pfizer, and Jazz Pharmaceuticals. B.F. licensed a patent on NPM1 mutants (#102004901256449) and declares honoraria from Rasna Therapeutics, Inc. for scientific advisor activities. The remaining authors declare no competing financial interests.
Correspondence: Andrea Biondi, Pediatrics and Tettamanti Center, Fondazione IRCCS San Gerardo dei Tintori; School of Medicine and Surgery, University of Milano-Bicocca, Monza, Italy; e-mail: abiondi.unimib@gmail.com.

