

Key Points
SSG turns neutrophil trogocytosis of rituximab-opsonized malignant B cells into cell killing.
Neutrophil antibody–dependent killing of malignant B cells occurs primarily through FcγRI (CD64).

Abstract
Anti-CD20 antibodies such as rituximab are broadly used to treat B-cell malignancies. These antibodies can induce various effector functions, including immune cell-mediated antibody-dependent cellular cytotoxicity (ADCC). Neutrophils can induce ADCC toward solid cancer cells by trogoptosis, a cytotoxic mechanism known to be dependent on trogocytosis. However, neutrophils seem to be incapable of killing rituximab-opsonized B-cell lymphoma cells. Nevertheless, neutrophils do trogocytose rituximab-opsonized B-cell lymphoma cells, but this only reduces CD20 surface expression and is thought to render tumor cells therapeutically resistant to further rituximab-dependent destruction. Here, we demonstrate that resistance of B-cell lymphoma cells toward neutrophil killing can be overcome by a combination of CD47-SIRPα checkpoint blockade and sodium stibogluconate (SSG), an anti-leishmaniasis drug and documented inhibitor of the tyrosine phosphatase SHP-1. SSG enhanced neutrophil-mediated ADCC of solid tumor cells but enabled trogoptotic killing of B-cell lymphoma cells by turning trogocytosis from a mechanism that contributes to resistance into a cytotoxic anti-cancer mechanism. Tumor cell killing in the presence of SSG required both antibody opsonization of the target cells and disruption of CD47-SIRPα interactions. These results provide a more detailed understanding of the role of neutrophil trogocytosis in antibody-mediated destruction of B cells and clues on how to further optimize antibody therapy of B-cell malignancies.
Introduction
Neutrophils mediate antibody-dependent cellular cytotoxicity (ADCC) toward solid cancer cells and contribute to antibody-mediated destruction of cancer cells in vivo.1-3 However, human neutrophils are considered to be unable to destroy B-cell lymphoma cells opsonized with rituximab or other anti-CD20 antibodies.4-7 Instead, it has been reported that neutrophils mediate an antibody-dependent process called “antigen shaving” of the anti-CD20–opsonized B-cell lymphoma cells, also known as trogocytosis. By this process, neutrophils remove the CD20 antigen together with the anti-CD20 antibody from the target cell surface. This reduces target cell CD20 surface expression without causing B cell-lymphoma cell death.8 It is presumed that this creates a general resistance to anti-CD20 therapy, which contributes to tolerance against rituximab-mediated destruction.9-11
Recently, we described that neutrophils mediate ADCC toward solid cancer cells through trogocytosis, a unique mechanism of physical destruction of the target cell plasma membrane in which neutrophils take up pieces of tumor cell membrane ultimately resulting in target cell death. In this context, trogocytosis forms an essential requirement for killing.12 However, in the case of B-cell lymphoma cells, trogocytosis does not lead to killing. The reason for the discrepancy between neutrophil-mediated trogocytosis of B-cell lymphoma cells leading to antigen shaving and solid cancer cell trogocytosis leading to cancer cell death is not yet understood.
We have previously reported that CD47signal-regulatory protein α (CD47-SIRPα) checkpoint blockade typically enhances neutrophil-mediated killing of solid cancer cells.12,13 In this study, we hypothesized that neutrophil ADCC of B-cell lymphoma cells is controlled by these and additional immune checkpoints, which are known to commonly involve signaling via the tyrosine phosphatases SHP-1 and/or SHP-2.14 To block these checkpoints in a general way, we tested the effects of sodium stibogluconate (SSG). SSG is a pentavalent antimonial compound used to treat the parasitic disease leishmaniasis, which was also demonstrated to inhibit the tyrosine phosphatase SHP-1.15,16 In the last decade, SHP-1 has received attention as a potential target in cancer by having a tumor-promoting role17 but also by having an inhibitory role in hematopoietic cells.18,19 We found that SSG combined with CD47-SIRPα blockade induced neutrophil-mediated killing of anti-CD20 antibody-opsonized B-cell lymphoma cells, which creates opportunities for improving neutrophil-mediated killing and for antibody therapy for B-cell lymphomas specifically.
Methods
Primary cell isolation and culture
Experiments with blood from healthy donors were approved by the Sanquin Research Institutional Ethical Committee. Blood from 3 patients with chronic granulomatous disease (CGD) and 1 patient who was SHP-1 deficient, was collected after informed consent and according to the Declaration of Helsinki 1964. Human neutrophils were isolated as previously described20 by density gradient centrifugation using Percoll (1.076 g/mL; GE Healthcare) followed by erythrocyte lysis of the resulting pellet. Neutrophils (5 × 106 cells per mL) were cultured for 4 hours or overnight at 37°C and 5% CO2 with 10 ng/mL granulocyte colony-stimulating factor (G-CSF, or filgrastim) and 50 ng/mL IFN-γ (PeproTech) in RPMI 1640 medium (Gibco) supplemented with 10% (v/v) fetal calf serum, 2 mM L-glutamine, 100 U/mL penicillin, and 100 µg/mL streptomycin.
Monocytes were isolated from the peripheral blood mononuclear cell (PBMC) fraction using anti-CD14 MACS beads (Miltenyi Biotec), according to the manufacturer’s instructions. Then, 2 × 105 monocytes per well in 24-well plates were cultured for 1 week with 50 ng/mL macrophage colony-stimulating factor (PeproTech) in Iscove modified Dulbecco medium (IMDM) (Gibco) supplemented with 10% (v/v) fetal calf serum, 2 mM L-glutamine, 100 U/mL penicillin, and 100 µg/mL streptomycin.
Natural killer (NK) cells were isolated from the PBMC fraction using anti-CD56 MACS beads (Miltenyi Biotec), according to the manufacturer’s instructions. NK cells (0.5 × 106 cells per mL) were cultured overnight at 37°C and 5% CO2 in RPMI 1640 medium supplemented with 10% (v/v) fetal calf serum, 2 mM L-glutamine, 100 U/mL penicillin, and 100 µg/mL streptomycin.
After written informed consent from patients, blood samples were obtained from patients with chronic lymphocytic leukemia (CLL) during diagnostic or follow-up procedures at the Department of Hematology and the Department of Pathology of the Academic Medical Center (AMC) Amsterdam. This study was approved by the AMC Ethical Review Board and conducted in agreement with the Declaration of Helsinki. PBMCs from patients with CLL obtained after Ficoll density gradient centrifugation (Pharmacia Biotech, the Netherlands) were cryopreserved and stored. Expression of CD5 and CD19 (BD Biosciences, San Jose, CA) on leukemic cells was assessed by flow cytometry using a BD FACSCanto clinical flow cytometry system. CLL samples included in this study contained 85% to 99% CD5+/CD19+ cells. Cryopreserved cells (2 × 106 cells per mL) were thawed and allowed to recover for at least 1 hour or overnight in IMDM supplemented with 10% (v/v) fetal calf serum, 2 mM L-glutamine, 100 U/mL penicillin, and 100 µg/mL streptomycin at 37°C and 5% CO2. CLL cells were characterized for the fraction of CD5+CD19+ cells (CD5, clone REA782; Miltenyi Biotec; CD19, clone SJ25C1; BioLegend) and the expression of CD20 (clone 2H7; BioLegend) and CD47 (clone B6H12; Thermo Fisher Scientific) within this population. The remaining fraction was analyzed for the amount of T cells (CD3, clone UCHT1; Thermo Fisher Scientific), monocytes (CD14, clone M5E2; BD Pharmingen), and NK cells (CD56, clone B159; BD Pharmingen).
Cell lines
The Raji and Daudi cell lines (CD20+ human Burkitt lymphoma; American Type Culture Collection [ATCC]) and A431 (epidermal growth factor receptor–positive [EGFR+] human epidermoid carcinoma; ATCC) were cultured at 37°C and 5% CO2 in RPMI 1640 medium supplemented with 10% (v/v) fetal calf serum, 2 mM L-glutamine, 100 U/mL penicillin, and 100 µg/mL streptomycin. The SKBR3 cell line (human epidermal growth factor receptor 2/neu–positive [HER2/neu+] human breast cancer; ATCC) was cultured in IMDM (Gibco) supplemented with 20% (v/v) fetal calf serum, 2 mM L-glutamine, 100 U/mL penicillin, and 100 µg/mL streptomycin at 37°C and 5% CO2.
Genetic modification of cell lines
To generate Raji and A431 CD47 knockout (KO) cells (hereafter referred to as Raji CD47KO and A431 CD47KO), we generated a double-stranded oligo (Invitrogen) of several target sequences using the Zhang Laboratory Optimized CRISPR Design tool (http://crispr.mit.edu/). These were then cloned into pLentiCRISPR v2. The constructs were grown in Escherichia coli Stbl3, and the sequence was verified. Lentivirus was obtained through 293T cell culture (ATCC). Transduced cells were selected with 1 µg/mL puromycin (InvivoGen). Surviving cells were cultured at limiting dilution, and growing clones were routinely maintained. The knockdown of CD47 in SKBR3 cells was achieved using short hairpin RNA (5′-CCGGGCACAA- TTACTTGGACTAGTTCTCGAGAACTAGTCCAAGTAATTGTGCTTTTT-3′). SKBR3 scrambled cells were used as controls and were created using mock short hairpin RNA.
To reconstitute CD47 in Raji CD47KO cells, the human CD47 coding sequence with a 3′ 6×-histidine (His) tag was ordered from Thermo Fisher Scientific (5′ATGTGGCCCCTGGTAGCGGCGCTGTTGCTG). The hCD47-His sequence was cloned into pENTR1A, into which an internal ribosome entry site (IRES) and green fluorescent protein (GFP) were previously cloned. The resulting pENTR1A-hCD47-His IRES GFP was recombined with pLenti6.3-V5/DEST using LR Clonase II (Thermo Fisher Scientific), which generated pLenti6.3-hCD47-His IRES GFP. Lentiviral particles were grown by transient co-transfection of 293T cells with pLenti6.3-hCD47-His IRES GFP, pMDLg/pRRE, pRSVrev, and pCMV-VSVg. The cells were moved into Raji medium the day after transfection. Virus-containing supernatant was harvested on day 2 after transfection, filtered through 0.45 µM sterile filter and added to 5 × 105 Raji CD47KO cells. A second round of transduction was performed on day 3 after transfection. Transduced Raji cells were selected on 8-µg/mL blasticidin. CD47 expression of Raji cells was measured by using flow cytometry after staining with anti-CD47 (B6H1214 ) and anti-mouse Alexa Fluor 405 (Thermo Fisher Scientific), which remained stable over time.
Trogocytosis assay by flow cytometry
The uptake of tumor cell membrane by neutrophils (ie, trogocytosis) was quantified by using flow cytometry after labeling the tumor cells with lipophilic membrane dye DiD or DiO (Invitrogen) for 30 minutes at 37°C as described.12 Target cells were co-incubated for 4 hours with G-CSF/IFN-γ–stimulated neutrophils in a target:effector ratio of 1:5 together with the appropriate opsonizing antibodies and reagents. After 60 minutes (for Raji, Daudi, and A431 cells) or 90 minutes (for SKBR3 and CLL cells) of incubation at 37°C, cells were fixed and measured using flow cytometry (BD FACSCanto II). The neutrophil population was evaluated for the mean fluorescent intensity of DiD or DiO.
ADCC assay
ADCC was quantified in triplicate by a 51Cr release assay.13 Target cells were labeled with 100 µCi 51Cr (Perkin-Elmer) for 90 minutes at 37°C, followed by 3 washing steps with phosphate-buffered saline (Fresenius Kabi). Target cells were co-incubated overnight with neutrophils stimulated with G-CSF and/or IFN-γ at T:E 1:50 (1:5 for NK cells and 1:25 for macrophages) together with the appropriate opsonizing antibodies and reagents. For neutrophils that were stimulated overnight, viability was assessed using Annexin V (BD Biosciences) staining to correct for the amount of dead cells. After 4 hours of incubation at 37°C (6 hours when using CLL cells as targets and overnight when using macrophages as effector cells), the supernatant was harvested, and radioactivity was measured using a gamma counter (Wizard2, Perkin-Elmer). The percentage of cytotoxicity was calculated as [ (experimental cpm – spontaneous cpm) / (maximum cpm – spontaneous cpm)] x 100%.
SHP-1 phosphatase activity
The ability of SSG to inhibit the phosphatase SHP-1 was determined by the level of dephosphorylation of the fluorescent substrate 6,8-difluoro-4-methylumbelliferyl phosphate (DiFMUP; Thermo Fisher Scientific). Then 0.1 µg of human recombinant SHP-1 (BioVision) was co-incubated with increasing concentrations of SSG (12.5 or 100 µM) or cefsulodin (20, 40, or 60 µM [another inhibitor of SHP-1]) in 4-(2-hydroxyethylpiperazine′ethanesulfonic acid (HEPES) buffer in a 384-well plate (Corning). After 30 minutes of incubation, 2.6 µM of DiFMUP was added to initiate the phosphatase reaction for 1 hour at room temperature. Fluorescence was measured with an Infinite F200-Pro plate reader (Tecan) at an excitation wavelength of 358 nm and emission wavelength of 455 nm. The percentage of inhibition was calculated as follows: [1 – (experimental response – background) / (maximum response – background)] × 100%, where the background is DiFMUP only, and the maximum response of SHP-1 with DiFMUP only. As a positive control, 20 mM of 2-iodoacetamide (Calbiochem) was used to inactivate SHP-1 by alkylating the active site cysteine located in the phosphatase domain.
Antibodies and reagents
For target opsonisation, 5 µg/mL rituximab (a gift from Rien van Oers, AMC, Amsterdam), obinutuzumab (Roche), ofatumumab (Novartis), fragment crystallization (Fc) gamma receptor I (FcγRI)–selective rituximab (human immunoglobulin G1 [IgG1]-G237A21 ), trastuzumab (Roche), cetuximab (Merck KGaA), or 2.5 µg/mL anti-HLA-DR (clone L243, BioLegend) was added to the mixtures. To block CD47-SIRPα interactions, 5 µg/mL anti-CD47 f(ab′)2 or 5 µg/mL anti-SIRPα (clones B6H12 and 12C4, respectively13 ) was added to the target cells. SSG (Calbiochem) was used at a concentration of 100 µM for 5 × 106 neutrophils per mL, unless otherwise specified. Where indicated 1.6, 16, or 80 µM cefsulodin (Sigma-Aldrich) was preincubated with 5 × 106 neutrophils per mL. Anti-CD18 f(ab′)2 fragments (clone IB4, Ancell) were preincubated with neutrophils for 20 minutes to block CD18 integrins. For the diisopropyl fluorophosphate (DFP) experiments, 5 × 106 neutrophils in 1 mL were treated with 1 µL DFP (Sigma-Aldrich) for 10 minutes at room temperature and were washed once with phosphate-buffered saline before the cells were placed in HEPES with or without 100 µM SSG. For the FcγR-blocking experiments, f(ab′)2 fragments of anti-CD16 (clone 3G8; Ancell), anti-CD32 (clone 7.3; Ancell), or anti-CD64 (clone 10.1; Ancell) and combinations thereof were used; f(ab′)2 fragments of mouse IgG1 (mIgG1) (clone MOPC 31C; Ancell) were added as a control. Neutrophil FcγR expression was assessed on fresh neutrophils, or on neutrophilscultured 4 hours or overnight with G-CSF/IFN-γ, using fluorescein isothiocyanate (FITC)–labeled anti-CD16 (clone 3G8; BD Pharmingen), anti-CD32 (clone AT10; Bio-Rad), or anti-CD64 (clone 10.1; Bio-Rad).
Statistical analyses
Analyses were performed using Graphpad Prism. Statistical differences between groups were tested using an ordinary one-way analysis of variance and Sidak post hoc test, unless otherwise stated.
Results
Interfering with innate checkpoints such as CD47-SIRPα typically enhances neutrophil-mediated trogocytosis and killing, at least of cetuximab (anti-EGFR)- or trastuzumab (anti-HER2/neu)–opsonized solid cancer cells13,22 (supplemental Figure 1A-D). However, even inhibition of CD47-SIRPα interactions (using CD47KO cells) did not induce notable neutrophil-mediated killing of rituximab-opsonized B-cell lymphoma cells (Figure 1A). This was not limited to rituximab, because other therapeutic anti-CD20 antibodies, including obinutuzumab and ofatumumab (Figure 1B), were also not able to induce neutrophil ADCC (Figure 1A). Still, all of these antibodies did induce trogocytosis, and this was enhanced in the absence of CD47-SIRPα interactions (Figure 1C). Neutrophils are not the only cells that participate in antibody-mediated effector functions: NK cells are well-known for their ability to induce target cell apoptosis using ADCC,23,24 and macrophages are known to phagocytose their antibody-opsonized targets using antibody-dependent cellular phagocytosis (ADCP).25-29 Both NK cells and macrophages were capable of killing rituximab-opsonized B-cell lymphoma cells (Figure 1D-E), demonstrating that the lack of killing anti-CD20–opsonized B-cell lymphoma cells is confined to neutrophils. Nonetheless, neutrophils did not display an intrinsic defect in the killing of B-cell lymphoma cells, because opsonization with antibodies targeting different antigens such as anti-HLA-DR did induce strong killing (Figure 1A). Anti-HLA-DR–mediated killing was not only highly efficient, but was also accompanied by trogocytosis, albeit to a lesser extent than rituximab-mediated trogocytosis (Figure 1C). Together, this demonstrates that B-cell lymphoma cells are specifically resistant toward neutrophil-mediated anti-CD20–dependent killing, which supports the notion that neutrophil interaction with anti-CD20–opsonized targets somehow induces trogocytosis rather than trogoptosis, even during CD47-SIRPα checkpoint inhibition.
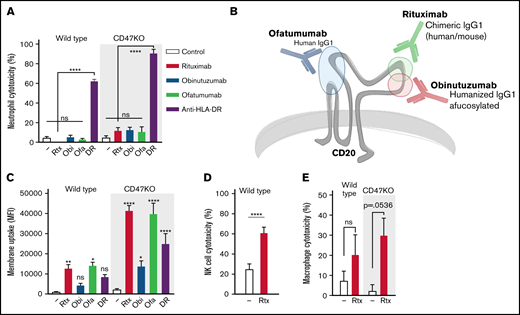
B-cell lymphoma cells display a specific resistance toward anti-CD20–mediated neutrophil killing. (A) Neutrophil ADCC of wild type and Raji CD47KO cells opsonized with rituximab (rtx), obinutuzumab (obi), ofatumumab (ofa), or anti-HLA-DR (DR) (n = 6-8 donors from 3-4 independent experiments); all anti-CD20 opsonized conditions were not significant (ns) compared with the control condition. (B) Epitopes and characteristics of rituximab, obinutuzumab, and ofatumumab. Rituximab and obinutuzumab have overlapping but distinct epitopes; ofatumumab binds to the 2 outer loops of CD20. (C) Neutrophil trogocytosis of wild type and Raji CD47KO cells opsonized with rituximab, obinutuzumab, ofatumumab, or anti-HLA-DR (n = 4-11 donors from 2-5 independent experiments); statistics indicate the comparison between opsonized and control conditions of the wild type or CD47KO cells. (D) NK cell ADCC of wild-type Raji cells (NK cells do not express SIRPα) opsonized with rituximab (n = 14 donors from 7 independent experiments). (E) Macrophage ADCC of wild type Raji cells opsonized with rituximab (n = 3 donors from 2 independent experiments). Data analysis used paired Student t test. Data are mean ± standard error of the mean (SEM). *P ≤ .05; **P ≤ .01; ****P ≤ .0001. MFI, mean fluorescent intensity.
B-cell lymphoma cells display a specific resistance toward anti-CD20–mediated neutrophil killing. (A) Neutrophil ADCC of wild type and Raji CD47KO cells opsonized with rituximab (rtx), obinutuzumab (obi), ofatumumab (ofa), or anti-HLA-DR (DR) (n = 6-8 donors from 3-4 independent experiments); all anti-CD20 opsonized conditions were not significant (ns) compared with the control condition. (B) Epitopes and characteristics of rituximab, obinutuzumab, and ofatumumab. Rituximab and obinutuzumab have overlapping but distinct epitopes; ofatumumab binds to the 2 outer loops of CD20. (C) Neutrophil trogocytosis of wild type and Raji CD47KO cells opsonized with rituximab, obinutuzumab, ofatumumab, or anti-HLA-DR (n = 4-11 donors from 2-5 independent experiments); statistics indicate the comparison between opsonized and control conditions of the wild type or CD47KO cells. (D) NK cell ADCC of wild-type Raji cells (NK cells do not express SIRPα) opsonized with rituximab (n = 14 donors from 7 independent experiments). (E) Macrophage ADCC of wild type Raji cells opsonized with rituximab (n = 3 donors from 2 independent experiments). Data analysis used paired Student t test. Data are mean ± standard error of the mean (SEM). *P ≤ .05; **P ≤ .01; ****P ≤ .0001. MFI, mean fluorescent intensity.
To investigate the hypothesis that neutrophil ADCC of B-cell lymphoma cells is controlled by additional immunoreceptors, we tested the effects of SSG, a documented inhibitor of SHP-1.15,16 We found that neutrophil ADCC of Raji lymphoma cells is strongly potentiated by SSG for the therapeutic anti-CD20 antibodies rituximab, obinutuzumab, and ofatumumab (Figure 2A). At 100 µM, SSG-mediated killing occurred only in the presence of opsonized antibodies, although higher concentrations of SSG did slightly increase the killing of nonopsonized CD47KO cells (supplemental Figure 2A). SSG enhanced neutrophil ADCC specifically, because no enhancement was observed for macrophage or NK cell–mediated lysis (supplemental Figure 2B-C). Notably, SSG-mediated killing occurred only in the absence of inhibitory CD47-SIRPα interactions (Figure 2A; supplemental Figure 2A), indicating the possibility of at least 2 inhibitory mechanisms seemingly acting in a hierarchical fashion. Trogocytosis itself was slightly but not significantly enhanced by SSG (Figure 2B-C), signifying a switch from antigen-shaving trogocytosis to killing trogocytosis (ie, trogoptosis12 ). These results were also observed for patient-derived CD20+ CLL cells (Figure 4C-D; supplemental Table 1). This is remarkable, because CLL cells express significantly less CD20 compared to other B-cell malignancies.30,31

SSG potentiates neutrophil ADCC and overcomes B-cell lymphoma cell resistance toward neutrophil killing. (A-B) Neutrophil ADCC and trogocytosis of wild type and Raji CD47KO cells opsonized with rituximab, obinutuzumab, or ofatumumab in the absence or presence of 100 µM SSG (n = 6-12 donors from 6 independent experiments for ADCC). There were no significant differences between control conditions and opsonized cells in absence of SSG (n = 6-10 donors from 5 independent experiments for trogocytosis). (C) Representative histograms of neutrophil DiD positivity (tumor cell membrane dye) in a trogocytosis experiment with Raji wild type or CD47KO cells opsonized with or without rituximab, in the absence or presence of SSG (dark gray–shaded histograms). (D) Neutrophil ADCC of Raji cells opsonized with or without rituximab in the absence or presence of SSG (purple bars). CD47-SIRPα signaling is inhibited by using Raji CD47KO cells or by blocking neutrophil SIRPα using SIRPα-blocking antibodies (n = 14 donors from 8 independent experiments). (E) Neutrophil ADCC of Raji wild type cells, Raji CD47KO cells, or Raji CD47KO cells with CD47 reconstitution, opsonized with or without rituximab in the absence or presence of SSG. The y-axis depicts the amount of cytotoxicity relative to the expression of CD47 on the Raji cells to highlight the role of CD47 on the tumor cells during ADCC by neutrophils (n = 6 donors from 3 independent experiments). (F) Representative histogram of the CD47 expression of Raji wild type cells, Raji CD47KO cells, or Raji CD47KO cells with CD47 reconstitution. Data are mean ± SEM. *P ≤ .05; **P ≤ .01; ***P ≤ .001; ****P ≤ .0001.
SSG potentiates neutrophil ADCC and overcomes B-cell lymphoma cell resistance toward neutrophil killing. (A-B) Neutrophil ADCC and trogocytosis of wild type and Raji CD47KO cells opsonized with rituximab, obinutuzumab, or ofatumumab in the absence or presence of 100 µM SSG (n = 6-12 donors from 6 independent experiments for ADCC). There were no significant differences between control conditions and opsonized cells in absence of SSG (n = 6-10 donors from 5 independent experiments for trogocytosis). (C) Representative histograms of neutrophil DiD positivity (tumor cell membrane dye) in a trogocytosis experiment with Raji wild type or CD47KO cells opsonized with or without rituximab, in the absence or presence of SSG (dark gray–shaded histograms). (D) Neutrophil ADCC of Raji cells opsonized with or without rituximab in the absence or presence of SSG (purple bars). CD47-SIRPα signaling is inhibited by using Raji CD47KO cells or by blocking neutrophil SIRPα using SIRPα-blocking antibodies (n = 14 donors from 8 independent experiments). (E) Neutrophil ADCC of Raji wild type cells, Raji CD47KO cells, or Raji CD47KO cells with CD47 reconstitution, opsonized with or without rituximab in the absence or presence of SSG. The y-axis depicts the amount of cytotoxicity relative to the expression of CD47 on the Raji cells to highlight the role of CD47 on the tumor cells during ADCC by neutrophils (n = 6 donors from 3 independent experiments). (F) Representative histogram of the CD47 expression of Raji wild type cells, Raji CD47KO cells, or Raji CD47KO cells with CD47 reconstitution. Data are mean ± SEM. *P ≤ .05; **P ≤ .01; ***P ≤ .001; ****P ≤ .0001.
SSG also enhanced the trogocytosis and killing of solid tumor cell lines opsonized with trastuzumab or cetuximab (supplemental Figure 1A-D), but the most prominent effects were seen with B-cell lymphoma cells. Here, the potentiating effects of SSG were strongest in the complete absence of CD47 (using CD47KO cells); hindering CD47-SIRPα interactions by blocking antibodies also resulted in enhanced killing but to a lesser extent (Figure 2D), possibly because of incomplete blockade of the CD47-SIRPα interaction. Of note, the binding capacity of the anti-SIRPα–blocking antibody to the different polymorphic variants of SIRPα was similar, because it enhanced ADCC with neutrophils with variant SIRPα1 or variant SIRPαBIT equally (supplemental Figure 3). Wild type CD47 reconstitution in Raji CD47KO cells reversed the potentiating effect of SSG, confirming the role of CD47 in resisting neutrophil ADCC and excluding off-target effects of the CRISPR/Cas9 KO technique (Figure 2E-F).
Neutrophil-mediated ADCC toward solid cancer cells requires the functional expression and activation of CD11b and CD18 integrins,32 whereas it is independent of the production of reactive oxygen species (ROS) or the release of proteases.12 We found that SSG-mediated killing of B-cell lymphoma cells displayed similar characteristics. When neutrophil CD18 integrins were blocked by using blocking antibodies, rituximab-mediated ADCC and trogocytosis were abolished (Figure 3A; supplemental Figure 4A). By using neutrophils from patients with chronic granulomatous disease that are unable to produce ROS, we demonstrate that SSG-mediated killing of B-cell lymphoma cells is independent of ROS (Figure 3B). Finally, by treating neutrophils with DFP to reduce proteolytic activity after neutrophil degranulation, we show that neutrophil-mediated ADCC of B-cell lymphoma cells is also independent of degranulation (Figure 3C; supplemental Figure 4B). Of note, SSG did not affect neutrophil degranulation and ROS production, nor did it affect adhesion or chemotaxis toward inflammatory cytokines (supplemental Figure 5A-D).
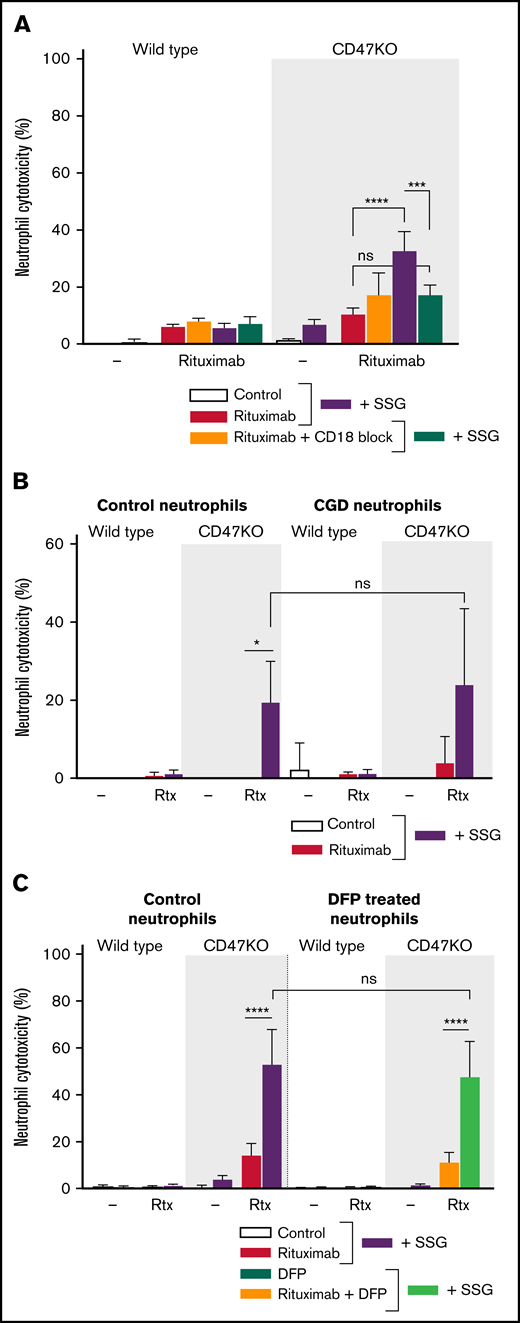
Neutrophil ADCC of B-cell lymphoma cells is dependent on CD11b/CD18 integrins and is independent of ROS and proteases. (A) Neutrophil ADCC of Raji wild type or CD47KO cells opsonized with rituximab in the absence or presence of SSG . Neutrophil CD18 integrin was blocked using anti-CD18 f(ab′)2 fragments (n = 4-6 donors from 3 independent experiments). (B) ADCC toward Raji wild type or CD47KO cells opsonized with rituximab in the absence or presence of SSG by neutrophils from 4 healthy donors (left) or from 3 patients with chronic granulomatous disease (CGD; right), which are unable to produce ROS. Note that ADCC is comparable between these 2 groups. Data shown were obtained from 3 independent experiments. (C) Neutrophil ADCC of Raji wild type or CD47KO cells opsonized with rituximab in the absence or presence of SSG. Neutrophils were treated with DFP which reduces proteolytic activity (n = 6 donors from 3 independent experiments). Data are mean ± SEM. *P ≤ .05; ***P ≤ .001; ****P ≤ .0001.
Neutrophil ADCC of B-cell lymphoma cells is dependent on CD11b/CD18 integrins and is independent of ROS and proteases. (A) Neutrophil ADCC of Raji wild type or CD47KO cells opsonized with rituximab in the absence or presence of SSG . Neutrophil CD18 integrin was blocked using anti-CD18 f(ab′)2 fragments (n = 4-6 donors from 3 independent experiments). (B) ADCC toward Raji wild type or CD47KO cells opsonized with rituximab in the absence or presence of SSG by neutrophils from 4 healthy donors (left) or from 3 patients with chronic granulomatous disease (CGD; right), which are unable to produce ROS. Note that ADCC is comparable between these 2 groups. Data shown were obtained from 3 independent experiments. (C) Neutrophil ADCC of Raji wild type or CD47KO cells opsonized with rituximab in the absence or presence of SSG. Neutrophils were treated with DFP which reduces proteolytic activity (n = 6 donors from 3 independent experiments). Data are mean ± SEM. *P ≤ .05; ***P ≤ .001; ****P ≤ .0001.
In addition to functional integrins, activating FcRs are essential for neutrophils to recognize cancer cells opsonized with intact antibodies (ie, containing functional Fc domains). Whereas neutrophils killed solid tumor cells through FcγRIIa (CD32a) and, to a somewhat lesser extent, through FcγRI (CD64; supplemental Figure 6),12,33 we found that FcγRI rather than FcγRIIa predominantly mediated ADCC of B-cell lymphoma cells (Figure 4A); only blockade of FcγRI or combinations thereof resulted in a significant decrease of ADCC. We confirmed this using an Fc-modified variant of rituximab (human IgG1-G237A) that selectively binds FcγRI. Because some residual binding of this Fc-modified variant of rituximab was still reported for the allotypic FcγRIIa variant 131RR,21 we used FcγRIIa-131HH donors in these experiments. As in the FcγR-blocking experiments (Figure 4A), neutrophil-mediated ADCC of B-cell lymphoma cells was mediated predominantly by FcγRI because SSG enhanced both wild type rituximab– and FcγRI rituximab–mediated ADCC (Figure 4B). Importantly, we observed the same for neutrophil-mediated killing of primary patient-derived CD20+ CLL cells. CLL cells were trogocytosed but not killed, except when SSG was combined with CD47 blockade (using F(ab′)2 fragments, to avoid opsonization), and no significant differences were found between wild type rituximab and FcγRI rituximab–mediated killing (Figure 4C-D). Finally, prestimulation of neutrophils with IFN-γ and G-CSF (consistently used in our experiments and known to enhance FcγRI expression)34,35 boosted FcγRI expression over time (Figure 4E) and formed a prerequisite for neutrophils to kill B-cell lymphoma cells (Figure 4F), in line with the observed importance of FcγRI in this process. Together, these data show that neutrophil-mediated killing of solid cancer cells and B-cell lymphoma cells is mediated through seemingly similar mechanisms. The FcγR used by neutrophils, however, is distinctly different. Whether FcγRI is the determinant factor in neutrophil-mediated killing of B-cell lymphoma cells or whether SSG orchestrates FcγRI-mediated killing remains unknown.
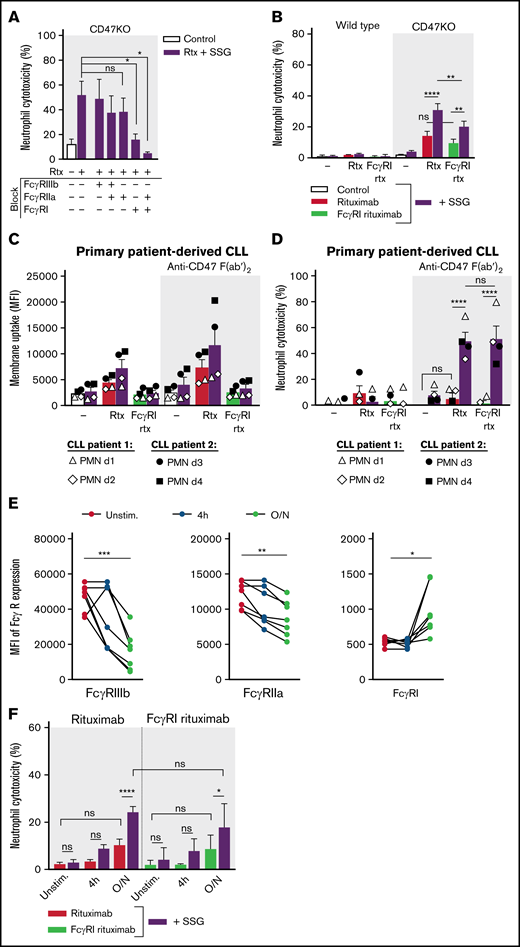
Neutrophil ADCC of B lymphoma cells is mainly dependent on FcγRI. (A) Neutrophil ADCC of Raji CD47KO cells opsonized with rituximab in the presence of SSG. FcγRI, FcγRIIa, FcγRIIIb or a combination thereof (indicated with “+” or “–”) were blocked on neutrophils using blocking f(ab′)2 fragments (n = 4-14 donors from 10 independent experiments). (B) Neutrophil ADCC of Raji wild type or CD47KO cells opsonized with rituximab or with FcγRI-selective rituximab in the absence or presence of SSG (n = 10 FcγRIIa-131HH–donors from 6 independent experiments). (C-D) Neutrophil trogocytosis and ADCC of 2 patient-derived primary CLL cells (patient 1, open symbols; patient 2, closed symbols) by neutrophils from healthy donors (2 allogeneic FcγRIIa-131HH neutrophil donors (d) per patient) with or without SSG. CLL cells were opsonized with rituximab or FcγRI-selective rituximab. CD47-SIRPα signaling was blocked by incubating the CLL cells with anti-CD47 f(ab′)2 fragments. For trogocytosis, SSG did not significantly enhance trogocytosis in the opsonized conditions. (E) Expression of FcγRIIIb, FcγRIIa, and FcγRI on neutrophils during stimulation with G-CSF and IFN-γ (n = 7 donors from 4 experiments). (F) Neutrophil ADCC of Raji CD47KO cells opsonized with rituximab or with FcγRI-selective rituximab in the absence or presence of SSG, by unstimulated (unstim) neutrophils, neutrophils stimulated for 4 hours, or neutrophils stimulated overnight (O/N) with G-CSF and IFN-γ (n = 7 donors from 3 independent experiments). Data are mean ± SEM. *P ≤ .05; **P ≤ .01; ***P ≤ .001; ****P ≤ .0001.
Neutrophil ADCC of B lymphoma cells is mainly dependent on FcγRI. (A) Neutrophil ADCC of Raji CD47KO cells opsonized with rituximab in the presence of SSG. FcγRI, FcγRIIa, FcγRIIIb or a combination thereof (indicated with “+” or “–”) were blocked on neutrophils using blocking f(ab′)2 fragments (n = 4-14 donors from 10 independent experiments). (B) Neutrophil ADCC of Raji wild type or CD47KO cells opsonized with rituximab or with FcγRI-selective rituximab in the absence or presence of SSG (n = 10 FcγRIIa-131HH–donors from 6 independent experiments). (C-D) Neutrophil trogocytosis and ADCC of 2 patient-derived primary CLL cells (patient 1, open symbols; patient 2, closed symbols) by neutrophils from healthy donors (2 allogeneic FcγRIIa-131HH neutrophil donors (d) per patient) with or without SSG. CLL cells were opsonized with rituximab or FcγRI-selective rituximab. CD47-SIRPα signaling was blocked by incubating the CLL cells with anti-CD47 f(ab′)2 fragments. For trogocytosis, SSG did not significantly enhance trogocytosis in the opsonized conditions. (E) Expression of FcγRIIIb, FcγRIIa, and FcγRI on neutrophils during stimulation with G-CSF and IFN-γ (n = 7 donors from 4 experiments). (F) Neutrophil ADCC of Raji CD47KO cells opsonized with rituximab or with FcγRI-selective rituximab in the absence or presence of SSG, by unstimulated (unstim) neutrophils, neutrophils stimulated for 4 hours, or neutrophils stimulated overnight (O/N) with G-CSF and IFN-γ (n = 7 donors from 3 independent experiments). Data are mean ± SEM. *P ≤ .05; **P ≤ .01; ***P ≤ .001; ****P ≤ .0001.
We hypothesized that SSG could prevent SHP-1–mediated inhibition of neutrophil effector functions. Although SSG did inhibit SHP-1 (Figure 5A), our results could not be explained by SHP-1 targeting only. First, cefsulodin (a known competitive and reversible SHP-1/2 inhibitor36 ) was not able to mimic the effect of SSG on ADCC (Figure 5B), whereas it did inhibit SHP-1 phosphatase activity similarly to SSG (Figure 5C). Second, neutrophils from an SHP-1–deficient patient could, similar to the result of SSG stimulation, kill rituximab-opsonized Raji cells, and although this indicates a role for SHP-1, SSG was still able to further potentiate killing, suggesting that SSG mediates effects independent of SHP-1 as well (Figure 5D).
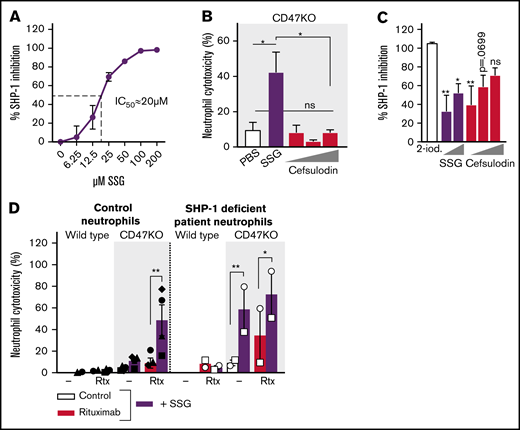
SSG does not potentiate neutrophil ADCC solely through the inhibition of SHP-1. (A) Dose-dependent inhibition of human recombinant SHP-1 by SSG (4 independent experiments were performed in triplicate). (B) Neutrophil ADCC toward Raji CD47KO cells opsonized with rituximab in the absence or presence of SSG or increasing concentrations (1.6, 16, or 80 µM) of cefsulodin (n = 4 donors from 2 independent experiments). (C) Inhibition of human recombinant SHP-1 by 20 µM 2-iodoacetamide (2-iod; maximum inhibition), increasing concentrations (12.5 or 100 µM) of SSG and increasing concentrations (20, 40, or 60 µM) of cefsulodin (6 independent experiments were performed in triplicate). Statistics indicate the comparison between 2-iodoacetamide and the different concentrations of SSG and cefsulodin. (D) Neutrophil ADCC of Raji wild type or CD47KO cells with 4 healthy neutrophil donors (closed symbols, left) and 1 SHP-1–deficient patient on 2 separate occasions (open symbols, right). Raji cells were opsonized with rituximab and neutrophils were incubated with or without SSG. Note that SHP-1–deficient neutrophils show high killing under basal conditions, which is further enhanced with SSG. Data shown is from 2 independent experiments. Data are mean ± SEM. *P ≤ .05; **P ≤ .01. IC50, 50% inhibitory concentration; PBS, phosphate-buffered saline.
SSG does not potentiate neutrophil ADCC solely through the inhibition of SHP-1. (A) Dose-dependent inhibition of human recombinant SHP-1 by SSG (4 independent experiments were performed in triplicate). (B) Neutrophil ADCC toward Raji CD47KO cells opsonized with rituximab in the absence or presence of SSG or increasing concentrations (1.6, 16, or 80 µM) of cefsulodin (n = 4 donors from 2 independent experiments). (C) Inhibition of human recombinant SHP-1 by 20 µM 2-iodoacetamide (2-iod; maximum inhibition), increasing concentrations (12.5 or 100 µM) of SSG and increasing concentrations (20, 40, or 60 µM) of cefsulodin (6 independent experiments were performed in triplicate). Statistics indicate the comparison between 2-iodoacetamide and the different concentrations of SSG and cefsulodin. (D) Neutrophil ADCC of Raji wild type or CD47KO cells with 4 healthy neutrophil donors (closed symbols, left) and 1 SHP-1–deficient patient on 2 separate occasions (open symbols, right). Raji cells were opsonized with rituximab and neutrophils were incubated with or without SSG. Note that SHP-1–deficient neutrophils show high killing under basal conditions, which is further enhanced with SSG. Data shown is from 2 independent experiments. Data are mean ± SEM. *P ≤ .05; **P ≤ .01. IC50, 50% inhibitory concentration; PBS, phosphate-buffered saline.
Discussion
CD20 antigen shaving as a result of anti-CD20 antibody therapy is a common problem in the treatment of patients with B-cell malignancies and probably contributes to the acquired resistance toward these anti-CD20 antibodies.9,11,37 In this study, we showed that antibody-dependent neutrophil trogocytosis of CD20+ B-cell lymphoma cells can be redirected toward neutrophil killing by a combination of SSG and CD47-SIRPα blockade. To our knowledge, we are the first to report that SSG enhanced neutrophil trogocytosis and killing of solid cancer cells in general but strongly potentiated the killing of B-cell lymphoma cells when combined with CD47-SIRPα checkpoint blockade. We were able to demonstrate this in cell lines of B cells as well as in patient-derived primary CLL cells.
One of our main findings indicated that SSG-mediated killing of B-cell lymphoma cells occurs through FcγRI rather than through FcγRIIa, the latter found to be the main FcγR in mediating neutrophil ADCC of solid cancer cells.12 Neutrophil FcγRI-mediated killing of B-cell lymphoma cells has been reported before, although this was killing mediated by anti-HLA-DR.38 We found that anti-HLA-DR–mediated killing is highly potent when compared with anti-CD20–mediated killing. It could be that the FcγR involved in neutrophil antibody–mediated effector functions decides the fate of the B-cell lymphoma cell: FcγRIIa mediates trogocytosis, whereas FcγRI mediates the killing of these anti-CD20–opsonized B cells. Whether or not SSG is involved in steering neutrophil ADCC through FcγRI or FcγRIIa, more insights into FcγR-specific effector functions may be crucial when aiming to reduce neutrophil trogocytosis and therefore acquired resistance during anti-CD20 therapy.
The exact mechanism by which SSG relieves inhibitory signaling in neutrophils still needs to be determined, but it does not seem to be dependent on SHP-1 phosphatase activity alone, as demonstrated by using neutrophils from an SHP-1–deficient patient. Still, our results suggest that SSG at least targets SHP-1, which has also been shown by Capuano et al.39 Here, SSG was used to reverse the hypo-responsiveness of NK cells, associated with long-lasting SHP-1 recruitment to FcγRIIIa after rituximab or ofatumumab ligation. This long-lasting recruitment resulted in attenuated levels of the tyrosine phosphorylation of downstream targets SLP-76, PLCγ2, and Vav1. In line with this, Stebbins et al40 demonstrated that SHP-1–mediated dephosphorylation of Vav1 was also shown to inhibit ADCC of NK cells. Because Vav1 and PLCγ2 are also involved in FcγR-mediated signaling in neutrophils, it is conceivable that a similar mechanism applies to neutrophil-mediated trogocytic killing.41-43 Moreover, research on leishmaniasis and pentavalent antimonial compounds (such as SSG) also demonstrated that SSG affects the immune system. Immunosuppressed patients with concurrent leishmaniasis do not seem to respond to treatment with SSG, for instance,44 suggesting that a well-functioning immune system is vital for SSG to exert its effects. A study that investigated the changes in expression profiles of lymph node aspirates from patients with leishmaniasis before and after treatment with SSG revealed changes in FcγR-mediated effector functions, including upregulation of protein kinase Syk and FcγRI.45
SSG is registered for the treatment of leishmaniasis, and a few phase 1/2 dose-escalating studies using combinations of SSG and IFNs in the context of cancer have been performed. However, apart from a variety of adverse effects, no effects on disease regression were reported.46,47 Still, these studies did not involve the use of therapeutic antibodies which, as we showed, is crucial for neutrophil SSG–mediated ADCC. Likewise, the effect of checkpoint blockade was not investigated. However, antibodies disrupting the CD47-SIRPα checkpoint are advancing in clinical development. Reported data from a phase 1b/2 trial (NCT02953509) show that anti-CD47 antibodies are well tolerated and also show promising efficacy in B-cell malignancies.48 The results of our study show that CD47-SIRPα checkpoint inhibition is needed to achieve neutrophil-mediated killing during anti-CD20 therapy of B-cell lymphomas, but it is only part of the solution. The findings presented here provide new insights into neutrophil ADCC of B-cell lymphoma cells and create opportunities for the further optimization of treatment of B-cell malignancies by combining SSG or alternative cell-permeable small molecule inhibitors with functional properties similar to those of CD47-SIRPα blockade. Therefore, future studies should involve a thorough investigation of the mechanism of action of SSG, which may be combined with the roles of FcγRIIa and FcγRI during neutrophil-mediated killing of malignant B cells.
Acknowledgments
The authors thank Paul Geurink and Huib Ovaa for helpful suggestions concerning the SHP-1 phosphatase activity experiments and Maarten Sijm for insights into the biochemistry of SSG.
This research was funded by a grant from the Dutch Cancer Society (11537).
Authorship
Contribution: D.J.v.R. designed and performed experiments and helped write the manuscript; M.B. provided FcγRI-selective rituximab and helped write the manuscript; P.V., A.T.J.T., B.K., K.S., L.W.T., and M.v.H. performed experiments; A.R.G. provided patient material; A.P.K. provided patient material and obinutuzumab and ofatumumab; G.V., T.W.K., and T.K.v.d.B. evaluated the data and the manuscript; and R.v.B. and H.L.M. designed the experiments and helped write the manuscript.
Conflict-of-interest disclosure: T.K.v.d.B. is the inventor of patent EP2282772, owned by Stichting Sanquin Bloedvoorziening, entitled “Compositions and Methods to Enhance the Immune System,” which describes targeting CD47-SIRPα interactions during antibody therapy in cancer. A.P.K. received research funding from Celgene, AbbVie, Roche, Janssen, and Astra-Zeneca and speaker’s fees from AbbVie and is active on the advisory boards of Janssen, Astra-Zeneca, and AbbVie. M.B. is funded by argenx. The remaining authors declare no competing financial interests.
Correspondence: Hanke L. Matlung, Sanquin Research and Landsteiner Laboratory, Plesmanlaan 125, 1066CX, Amsterdam, the Netherlands; email: h.matlung@sanquin.nl; and Dieke J. van Rees, Sanquin Research and Landsteiner Laboratory, Plesmanlaan 125, 1066CX, Amsterdam, the Netherlands; email: d.vanrees@sanquin.nl.

