

Key Points
A substantial fraction of patients with relapsed/refractory B-ALL will have non-CNS EMD.
CAR T cells may have limited efficacy in multifocal non-CNS EMD, and serial imaging is needed to identify and monitor EMD.

Abstract
Chimeric antigen receptor (CAR) T cells effectively eradicate medullary B-cell acute lymphoblastic leukemia (B-ALL) and can traffic to and clear central nervous system (CNS) involvement. CAR T-cell activity in non-CNS extramedullary disease (EMD) has not been well characterized. We systematically evaluated CAR T-cell kinetics, associated toxicities, and efficacy in B-ALL non-CNS EMD. We conducted a retrospective review of B-ALL patients with non-CNS EMD who were screened for/enrolled on one of three CAR trials (CD19, CD22, and CD19/22) at our institution. Non-CNS EMD was identified according to histology or radiographic imaging at extramedullary sites excluding the cerebrospinal fluid and CNS parenchyma. Of ∼180 patients with relapsed/refractory B-ALL screened across multiple early-phase trials over an 8-year period, 38 (21.1%) presented with isolated non-CNS EMD (n = 5) or combined medullary/non-CNS EMD (n = 33) on 18-fluorodeoxyglucose positron emission tomography/computed tomography (FDG PET/CT) imaging. A subset receiving CAR T cells (18 infusions) obtained FDG PET/CT scans preinfusion and postinfusion to monitor response. At best response, 72.2% (13 of 18) of patients showed a medullary minimal residual disease–negative complete remission and complete (n = 7) or partial (n = 6) non-CNS EMD response. Non-CNS EMD responses to CAR T cells were delayed (n = 3), and residual non-CNS EMD was substantial; rarely, discrepant outcomes (marrow response without EMD response) were observed (n = 2). Unique CAR-associated toxicities at non-CNS EMD sites were seen in select patients. CAR T cells are active in B-ALL non-CNS EMD. Still, non-CNS EMD response to CAR T cells may be delayed and suboptimal, particularly with multifocal disease. Serial FDG PET/CT scans are necessary for identifying and monitoring non-CNS EMD.
Introduction
Chimeric antigen receptor (CAR) T-cell therapy is effective in heavily pretreated patients with B-cell acute lymphoblastic leukemia (B-ALL). Complete remission (CR) rates after CD19- and CD22-directed CAR T-cell therapies range from 60% to 90% in children and young adults with multiply relapsed/refractory (r/r) disease.1-6 Studies of anti-CD19 CAR T-cell therapies in adults with B-cell non-Hodgkin lymphomas, while also effective, show lower response rates, with 40% to 50% of patients exhibiting a response to therapy.7,8 Characterization of response to CAR T cells in patients with B-ALL extramedullary disease (EMD), who may present with combined medullary and lymphomatous disease, has been limited.
Studies of B-ALL EMD have historically focused on identification and treatment of “sanctuary sites” in the central nervous system (CNS) and testes.9,10 An estimated 10% to 20% of patients with newly diagnosed B-ALL present with combined medullary/EMD.11,12 Because screening for EMD beyond lumbar puncture and testicular examination is not routine and has not been standardized in B-ALL disease assessment, this estimation likely underrepresents the true incidence of EMD, especially at sites outside the cerebrospinal fluid and CNS parenchyma (non-CNS EMD) and in those with r/r disease. Manifestations of non-CNS EMD are highly heterogeneous and may evade early detection.13 At disease recurrence, nearly one-half of patients with B-ALL present with isolated medullary disease, yet a substantial proportion (15%-25%) relapse with some combination of medullary/extramedullary involvement.9 Approximately 20% present with isolated CNS disease and roughly 5% with isolated testicular relapse,9,14 but the incidence of non-CNS EMD outside these well-established sites for B-ALL relapse is unknown.
Post-hematopoietic stem cell transplantation (HSCT) relapse with non-CNS EMD is also a relatively frequent occurrence with heterogeneous manifestations. Associated with dismal outcomes, non-CNS EMD relapse after HSCT potentially represents a limitation of surveillance of immunotherapy in the posttransplant setting.15-17 Patients who proceed to HSCT with unrecognized non-CNS EMD may have especially poor outcomes given the importance of achieving pre-HSCT minimal residual disease (MRD) negativity.18 Although the use of whole-body imaging is well established in adult solid tumors and lymphomas, its role remains poorly defined in acute leukemias.19,20 A growing body of literature suggests that 18-fluorodeoxyglucose positron emission tomography/computed tomography (FDG PET/CT) imaging is feasible and may be high-yield for detection of non-CNS EMD in children and young adults with leukemia.
Although the majority of CAR T-cell outcomes in B-ALL emerge from treatment of medullary disease,1-6 CAR T cells targeting CD19 and CD22 have shown efficacy in clearing CNS disease, and experience is still being gained to optimize this approach.21,22 A limited number of case studies have reported success in eradicating non-CNS EMD of the lymph nodes, breast, cervix, kidney, and skin with anti-CD19 CAR T cells.23-26 Although preliminary results from large trials have offered some insights, published experience with CAR T-cell therapy in patients with combined medullary/non-CNS EMD or isolated non-CNS EMD is still lacking.27,28 Thus, understanding of CAR T-cell kinetics, associated toxicities, and efficacy in patients with non-CNS EMD warrants further investigation. We evaluated the response of non-CNS EMD to CAR T-cell therapy in patients enrolled on our phase 1 CAR T-cell trials. We also characterized sites of occult non-CNS EMD in patients referred to our center, analyzed CAR T-cell kinetics, and explored unique attributes of CAR T-cell toxicity in the setting of non-CNS EMD.
Methods
Study population
We conducted an institutional review board–approved retrospective review (#NCT03827343) of patients with B-ALL screened for and/or enrolled on 1 of 3 institutional review board–approved CAR T-cell trials at the National Cancer Institute (#NCT01593696, #NCT02315612, and #NCT03448393) between July 1, 2012, and May 1, 2020. Patients included in this analysis had at least one 18-fluorodeoxyglucose positron emission tomography/computed tomography (FDG PET/CT) scan and were evaluated for treatment with an anti-CD19, anti-CD22, or anti-CD19/22 bispecific CAR T-cell construct.
Objectives
The primary objective of this study was to systematically evaluate the response of non-CNS EMD to CAR T cells in relation to bone marrow response. Secondary objectives included reporting on the presentation of non-CNS EMD in patients with r/r B-ALL, identifying optimal time to best response of non-CNS EMD with CAR T cells, characterizing peripheral blood CAR T-cell expansion and persistence in patients with non-CNS EMD vs those without EMD, and describing unique CAR T cell–associated toxicities in patients with non-CNS EMD.
Disease assessments
Restaging was performed at day 28 post–CAR T-cell infusion and continued until best response or progressive disease (PD). Bone marrow aspirate and biopsy samples were assessed by morphology, with marrow classified as M1 (<5%), M2 (5%-25%), or M3 (>25%) using standard definitions. MRD using flow cytometry, performed by NCI Flow Cytometry, had a validated limit of detection of blasts of 0.002% of mononuclear cells.29,30 Cerebrospinal fluid was analyzed by routine cytopathology in addition to flow cytometry, which had a lower limit of detection of ∼1.0%.
Non-CNS EMD, including testicular involvement, was assessed by using FDG PET/CT imaging. A single nuclear medicine physician with 12 years of experience in oncology imaging performed a retrospective unblinded central review of all scans included in this study. Deauville score and maximum standardized uptake values were used to identify and serially characterize sites of non-CNS EMD. Response of non-CNS EMD to CAR T cells was centrally graded in terms of quantity and quality of sites resolved using the following designations: complete response (CR), which required full disease eradication; partial response (PR); stable disease; and PD (supplemental Table 1).
Statistical analysis
Standard descriptive statistics were used to describe patient disease and CAR T-cell response characteristics. All statistical tests were performed in GraphPad Prism (GraphPad Software) version 9.3.1 using a threshold of significance of P < .05.
Results
All screened patients with non-CNS EMD (N = 38)
Of ∼180 patients with r/r B-ALL screened for early-phase clinical trials during the study period, 38 (21.1%) had isolated non-CNS EMD (n = 5) or combined medullary/non-CNS EMD (n = 33) detectible on FDG PET/CT imaging upon presentation to our institution (Figure 1A). The median age was 18.6 years (range, 4.7-30.7 years). In this heavily pretreated cohort, 27 (71.0%) had undergone prior HSCT, 23 (60.5%) had prior blinatumomab or inotuzumab ozogamicin exposure, and 13 (34.2%) had received prior CD19- or CD22-directed CAR T cells. Review of the medical history revealed that at initial diagnosis, a small number of patients had isolated non-CNS EMD (n = 5), combined CNS/non-CNS EMD (n = 2), or CNS-only EMD (n = 2); this information, however, was not documented in most cases (68.4%) (Table 1).
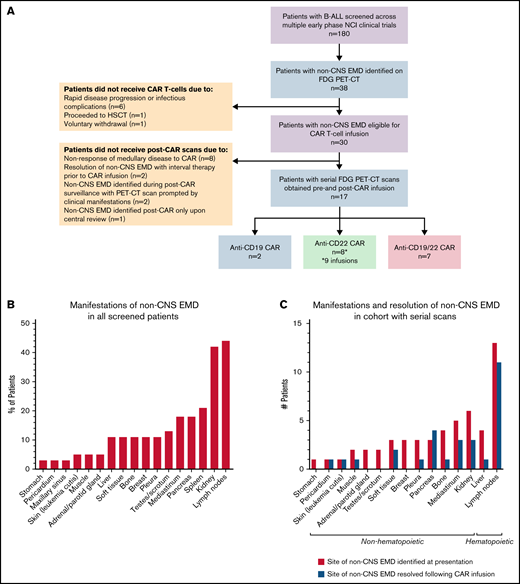
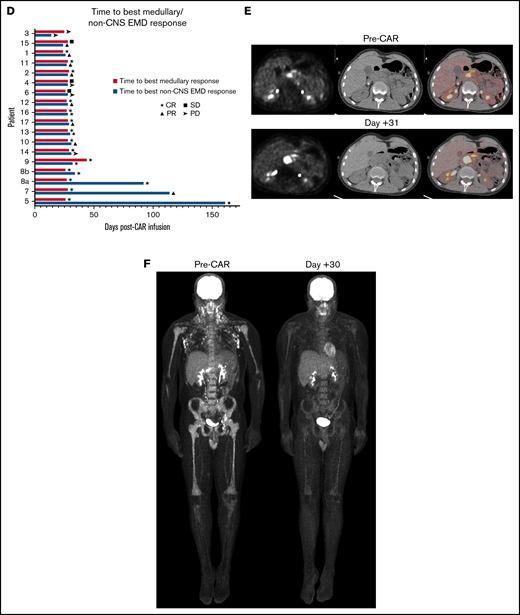
Sites of non-CNS EMD and response to CAR T cells. (A) Patients with non-CNS EMD identifiable during retrospective central review. (B) Manifestations of non-CNS EMD (by percentage of patients) identified during central review of FDG PET/CT imaging from 38 patients screened across multiple early-phase trials at our institution over an 8-year period. (C) Non-CNS EMD in the cohort of 17 patients (18 infusions) who obtained serial FDG PET/CT images pre– and post–CAR T-cell infusion, with sites of non-CNS EMD exhibiting a CR to CAR T cells represented in blue. (D) Time to best response of medullary/non-CNS EMD in the cohort of 17 patients (18 infusions) who obtained serial FDG PET/CT images pre– and post–CAR T-cell infusion. (E) FDG PET/CT scans obtained pre– and post–CD19/22 CAR T-cell infusion showing a discrepant medullary/non-CNS EMD response. Patient 14 attained a medullary MRD-negative CR but exhibited non-CNS EMD PD, with new and worsening sites of non-CNS EMD identified in the adrenal gland, retroperitoneal lymph node, pancreas, and testes ∼1 month post–CAR T-cell infusion. (F) FDG PET/CT scans obtained pre– and post–CD19/22 CAR T-cell infusion showing a concurrent CR of medullary/non-CNS EMD at best response. Patient 13 presented to our institution with multifocal non-CNS EMD involving the lymph nodes, mediastinum, kidneys, and pancreas. NCI, National Cancer Institute; SD, stable disease.
Sites of non-CNS EMD and response to CAR T cells. (A) Patients with non-CNS EMD identifiable during retrospective central review. (B) Manifestations of non-CNS EMD (by percentage of patients) identified during central review of FDG PET/CT imaging from 38 patients screened across multiple early-phase trials at our institution over an 8-year period. (C) Non-CNS EMD in the cohort of 17 patients (18 infusions) who obtained serial FDG PET/CT images pre– and post–CAR T-cell infusion, with sites of non-CNS EMD exhibiting a CR to CAR T cells represented in blue. (D) Time to best response of medullary/non-CNS EMD in the cohort of 17 patients (18 infusions) who obtained serial FDG PET/CT images pre– and post–CAR T-cell infusion. (E) FDG PET/CT scans obtained pre– and post–CD19/22 CAR T-cell infusion showing a discrepant medullary/non-CNS EMD response. Patient 14 attained a medullary MRD-negative CR but exhibited non-CNS EMD PD, with new and worsening sites of non-CNS EMD identified in the adrenal gland, retroperitoneal lymph node, pancreas, and testes ∼1 month post–CAR T-cell infusion. (F) FDG PET/CT scans obtained pre– and post–CD19/22 CAR T-cell infusion showing a concurrent CR of medullary/non-CNS EMD at best response. Patient 13 presented to our institution with multifocal non-CNS EMD involving the lymph nodes, mediastinum, kidneys, and pancreas. NCI, National Cancer Institute; SD, stable disease.
Demographic characteristics of 38 patients with non-CNS EMD identifiable by FDG PET/CT imaging at presentation to our institution
| Characteristic | Value |
|---|---|
| Age at initial diagnosis, median (range), y | 11.5 (2.5-27.4) |
| Age at presentation to our institution, median (range), y | 18.6 (4.7-30.7) |
| Sex, n (%) | |
| Male | 27 (71.1) |
| Female | 11 (28.9) |
| EMD at initial diagnosis, n (%) | |
| CNS EMD | 3 (7.8) |
| Non-CNS EMD | 5 (13.2) |
| Combined CNS/non-CNS EMD | 2 (5.3) |
| Unknown | 28 (68.4) |
| Prior number of lines of therapy, median (range) | 5 (2-9) |
| Prior HSCT (n = 27), n (%) | |
| 1 | 22 (60.5) |
| >1 | 5 (13.2) |
| Prior immunotherapy (n = 23), n (%) | |
| Prior blinatumomab | 16 (42.1) |
| Prior inotuzumab | 7 (18.4) |
| Prior CAR T-cell therapy (n = 13), n (%) | |
| Prior anti–CD19 CAR | 12 (31.6) |
| Prior anti–CD22 CAR | 1 (2.6) |
| Medullary disease at presentation, n (%)* | |
| MRD-negative | 5 (13.2) |
| Low burden | 13 (34.2) |
| High burden | 20 (52.6) |
| CNS status at presentation, n (%)† | |
| CNS1/CNS2 | 35 (92.1) |
| CNS3 | 3 (7.9) |
| Non-CNS EMD at presentation, n (%) | |
| Single site | 12 (31.6) |
| Multiple sites | 26 (68.4) |
| Indication for FDG PET/CT imaging, n (%) | |
| Documented history of non-CNS EMD | 23 (60.5) |
| Incidental finding on other imaging | 8 (21.1) |
| Abnormal physical examination | 4 (10.5) |
| Isolated CNS relapse with suspected non-CNS EMD | 3 (7.9) |
| Characteristic | Value |
|---|---|
| Age at initial diagnosis, median (range), y | 11.5 (2.5-27.4) |
| Age at presentation to our institution, median (range), y | 18.6 (4.7-30.7) |
| Sex, n (%) | |
| Male | 27 (71.1) |
| Female | 11 (28.9) |
| EMD at initial diagnosis, n (%) | |
| CNS EMD | 3 (7.8) |
| Non-CNS EMD | 5 (13.2) |
| Combined CNS/non-CNS EMD | 2 (5.3) |
| Unknown | 28 (68.4) |
| Prior number of lines of therapy, median (range) | 5 (2-9) |
| Prior HSCT (n = 27), n (%) | |
| 1 | 22 (60.5) |
| >1 | 5 (13.2) |
| Prior immunotherapy (n = 23), n (%) | |
| Prior blinatumomab | 16 (42.1) |
| Prior inotuzumab | 7 (18.4) |
| Prior CAR T-cell therapy (n = 13), n (%) | |
| Prior anti–CD19 CAR | 12 (31.6) |
| Prior anti–CD22 CAR | 1 (2.6) |
| Medullary disease at presentation, n (%)* | |
| MRD-negative | 5 (13.2) |
| Low burden | 13 (34.2) |
| High burden | 20 (52.6) |
| CNS status at presentation, n (%)† | |
| CNS1/CNS2 | 35 (92.1) |
| CNS3 | 3 (7.9) |
| Non-CNS EMD at presentation, n (%) | |
| Single site | 12 (31.6) |
| Multiple sites | 26 (68.4) |
| Indication for FDG PET/CT imaging, n (%) | |
| Documented history of non-CNS EMD | 23 (60.5) |
| Incidental finding on other imaging | 8 (21.1) |
| Abnormal physical examination | 4 (10.5) |
| Isolated CNS relapse with suspected non-CNS EMD | 3 (7.9) |
MRD-negative indicates no disease detectable by flow cytometry. Low burden includes M1 marrow (<5% blasts); high burden indicates M2 (5%-25% blasts) and M3 (>25% blasts) marrow.
CNS1 indicates 0 blasts detectable on cytospin; CNS2, white blood cell counts <5/μL, cytospin positive for blasts; and CNS3, white blood cell counts ≥5 μL, cytospin positive for blasts.
Sixty-five percent (n = 25) of screened patients presented with multifocal non-CNS EMD. Sites of disease, described in detail in Table 2 and Figure 1B, were highly heterogeneous and in some cases very extensive. Particularly unique sites of involvement included the breast (n = 3 [10.5%]), pancreas (n = 8 [18.4%]), kidney (n = 16 [42.1%], with bilateral involvement in 13 patients), and skin (n = 2 [5.3%]). The majority of those screened had concurrent medullary relapse, although 5 (13.2%) patients exhibited MRD negativity in the bone marrow upon presentation to our institution (Table 1). More than one-half (n = 20 [52.6%]) had high-burden medullary disease (M3); few patients (n = 3) had active CNS involvement (white blood cell count ≥5 µL, cytospin positive for blasts) at the time of referral.
Manifestations of non-CNS EMD in all subjects with-CNS EMD identifiable by FDG PET/CT imaging (N = 38) at presentation to our institution
| Patient | Sites of non-CNS EMD identified on FDG PET/CT imaging |
|---|---|
| 1 | Retroperitoneal lymph node |
| 2 | Scalp soft tissue, mesenteric, periaortic, retroperitoneal lymph nodes, right kidney, left kidney, liver |
| 3 | Liver |
| 4 | Right kidney, left kidney |
| 5 | Mesenteric, retroperitoneal lymph nodes, right kidney, left kidney, extrusion from vertebral bone marrow into psoas |
| 6 | Mesenteric, peritoneal lymph nodes, mediastinum, pericardium, pleura, intramuscular lesion |
| 7 | Orbital bone, parotid gland, cervical, supraclavicular, axillary, peritoneal lymph nodes, pancreas |
| 8a* | Temporal bone, subcutaneous tissue surrounding external auditory canal, cervical, supraclavicular lymph nodes, pancreas |
| 8b* | Thoracic neural foramen, lumbar neural foramen (vertebral bodies) |
| 9 | Right kidney, left kidney, pleura |
| 10 | Supraclavicular, mesenteric lymph nodes, pleura, left kidney |
| 11 | Parotid gland, maxillary sinus, cervical, supraclavicular, mesenteric lymph nodes, mediastinum, liver, stomach, scrotum |
| 12 | Breast, subcutaneous left lower extremity lesions, extrusion from right extremity bone marrow to surrounding soft tissue |
| 13 | Cervical, axillary, retroperitoneal, mesenteric, inguinal, pelvic lymph nodes, mediastinum, right kidney, left kidney, pancreas |
| 14 | Retroperitoneal, pelvic wall lymph nodes, pancreas, testes |
| 15 | Breast, cervical, axillary lymph nodes, mediastinum |
| 16 | Liver |
| 17 | Inguinal, pelvic wall, popliteal lymph nodes, deep thigh lymph nodes, skin (leukemia cutis), bone |
| 18 | Right kidney, left kidney, testes |
| 19 | Right kidney, left kidney, spleen, pancreas |
| 20 | Right kidney, left kidney, spleen |
| 21 | Testes |
| 22 | Maxillary sinus, right kidney |
| 23 | Inguinal lymph nodes, spleen |
| 24 | Mesenteric, retroperitoneal lymph nodes, mediastinum, right kidney, left kidney, spleen, pancreas |
| 25 | Breast, retroperitoneal lymph nodes, spleen, pancreas |
| 26 | Extrusion from sternum to surrounding soft tissues |
| 27 | Left kidney |
| 28 | Spleen |
| 29 | Pancreas |
| 30 | Right kidney, left kidney |
| 31 | Inguinal lymph node |
| 32 | Testes |
| 33 | Right kidney, left kidney, pleura |
| 34 | Right kidney, left kidney |
| 35 | Spleen |
| 36 | Skin (leukemia cutis) |
| 37 | Spleen |
| 38 | Mediastinum, right kidney, left kidney |
| Patient | Sites of non-CNS EMD identified on FDG PET/CT imaging |
|---|---|
| 1 | Retroperitoneal lymph node |
| 2 | Scalp soft tissue, mesenteric, periaortic, retroperitoneal lymph nodes, right kidney, left kidney, liver |
| 3 | Liver |
| 4 | Right kidney, left kidney |
| 5 | Mesenteric, retroperitoneal lymph nodes, right kidney, left kidney, extrusion from vertebral bone marrow into psoas |
| 6 | Mesenteric, peritoneal lymph nodes, mediastinum, pericardium, pleura, intramuscular lesion |
| 7 | Orbital bone, parotid gland, cervical, supraclavicular, axillary, peritoneal lymph nodes, pancreas |
| 8a* | Temporal bone, subcutaneous tissue surrounding external auditory canal, cervical, supraclavicular lymph nodes, pancreas |
| 8b* | Thoracic neural foramen, lumbar neural foramen (vertebral bodies) |
| 9 | Right kidney, left kidney, pleura |
| 10 | Supraclavicular, mesenteric lymph nodes, pleura, left kidney |
| 11 | Parotid gland, maxillary sinus, cervical, supraclavicular, mesenteric lymph nodes, mediastinum, liver, stomach, scrotum |
| 12 | Breast, subcutaneous left lower extremity lesions, extrusion from right extremity bone marrow to surrounding soft tissue |
| 13 | Cervical, axillary, retroperitoneal, mesenteric, inguinal, pelvic lymph nodes, mediastinum, right kidney, left kidney, pancreas |
| 14 | Retroperitoneal, pelvic wall lymph nodes, pancreas, testes |
| 15 | Breast, cervical, axillary lymph nodes, mediastinum |
| 16 | Liver |
| 17 | Inguinal, pelvic wall, popliteal lymph nodes, deep thigh lymph nodes, skin (leukemia cutis), bone |
| 18 | Right kidney, left kidney, testes |
| 19 | Right kidney, left kidney, spleen, pancreas |
| 20 | Right kidney, left kidney, spleen |
| 21 | Testes |
| 22 | Maxillary sinus, right kidney |
| 23 | Inguinal lymph nodes, spleen |
| 24 | Mesenteric, retroperitoneal lymph nodes, mediastinum, right kidney, left kidney, spleen, pancreas |
| 25 | Breast, retroperitoneal lymph nodes, spleen, pancreas |
| 26 | Extrusion from sternum to surrounding soft tissues |
| 27 | Left kidney |
| 28 | Spleen |
| 29 | Pancreas |
| 30 | Right kidney, left kidney |
| 31 | Inguinal lymph node |
| 32 | Testes |
| 33 | Right kidney, left kidney, pleura |
| 34 | Right kidney, left kidney |
| 35 | Spleen |
| 36 | Skin (leukemia cutis) |
| 37 | Spleen |
| 38 | Mediastinum, right kidney, left kidney |
8a and 8b represent a single patient who received 2 separate infusions of the same CAR product at initial treatment and subsequent disease recurrence.
Evaluation of non-CNS EMD by using whole-body imaging was prompted by a recorded history of non-CNS EMD at some point in the disease course for most patients (n = 23 [60.5%]). Imaging was also indicated in cases of an incidental finding on an alternative imaging modality performed for another indication (n = 8), abnormal physical examination (n = 4), and isolated CNS relapse with suspected non-CNS EMD involvement (n = 3) (Table 1). Of 38 screened patients referred specifically for CAR therapy, 30 were eligible for infusion on one of our CAR T-cell trials.
Cohort with serial FDG PET/CT scans (n = 17 [18 infusions])
Serial FDG PET/CT imaging before and after CAR T-cell infusion was obtained for 17 of 30 patients who proceeded to CAR infusion. Among the 13 patients who did not receive additional scans, 10 did not undergo post–CAR imaging due to resolution of non-CNS EMD with interval radiation therapy (n = 1) or chemotherapy (n = 1) before cell infusion or lack of cytokine release syndrome (CRS) with evidence of medullary nonresponse to CAR (n = 8). Two patients who did not receive serial scans had non-CNS EMD identified on post–CAR imaging prompted by clinical manifestations, whereas 1 patient had non-CNS EMD identified post–CAR imaging only upon central review (Figure 1A). Results for the cohort with serial scans have been calculated of 18 scan/patient pairs as 1 patient was analyzed for 2 separate infusions of the same CAR product administered 1 year apart at initial therapy and disease recurrence.
Two patients were infused with anti-CD19 CAR T cells, 8 received anti-CD22 CAR T cells (8 patients, 9 infusions), and 7 received an anti-CD19/22 bispecific CAR construct. The pretreatment scan was performed a median of 17.5 days before CAR T-cell infusion (range, 5-40 days). The first posttreatment scan was obtained a median of 28 days after infusion (range, 14-34 days). FDG was administered intravenously at a median dose 6.95 mCi (interquartile range, 1.54 mCi), with images obtained a median of 63.5 minutes after radiotracer injection. Median blood glucose level before injection was 88.5 mg/dL.
Medullary/non-CNS EMD response to CAR T cells
All patients with non-CNS EMD assessed by using serial imaging (n = 18) had active medullary disease at CAR T-cell infusion. The majority (n = 14) had multifocal non-CNS EMD. Seventy-two percent (n = 13) simultaneously exhibited a medullary CR to CAR T cells and a CR or PR of non-CNS EMD at best response (Table 3). Best response was achieved at 28 days in 15 (83.3%) of 18 cases (Figure 1D). Ongoing responses were seen in 3 patients who received CD22 CAR T cells, all of whom had detectible CAR T cells at 3, 4, and 6 months’ postinfusion. Fourteen patients in this cohort attained a medullary MRD-negative CR at best response. Of this subgroup, one-half (n = 7) concurrently exhibited a non-CNS EMD CR (Figure 1F), whereas 42.9% (n = 6) showed only PR of non-CNS EMD at best response. One patient reported non-CNS EMD PD at follow-up despite attaining a medullary CR (Figure 1E). Of the 3 patients with stable medullary disease at follow-up, 1 had non-CNS EMD PR and 2 reported non-CNS EMD PD. A single patient in this cohort showed PD of medullary and non-CNS EMD at best response.
Response of medullary/non-CNS EMD to CAR T cells in 17 patients with 18 sets of FDG PET/CT scans pre- and post–CAR infusion
| Pre-CAR, at presentation to our institution | Post–CAR infusion, best response | Overall outcome | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Patient | CAR | Non-CNS EMD at pre-CAR FDG PET/CT | maxSUV, non-CNS EMD | maxSUV, focal BM disease | BM by morphology (% of MNCs) | Non-CNS EMD at best response FDG PET/CT | SUV, Non-CNS EMD | Non-CNS EMD response | BM response (day +28) | |
| 1 | CD19 | Retroperitoneal lymph node* | 17.31 | 3.25† | 0.04 | No residual EMD | 2.63 | CR | CR | Not eligible for second HSCT and experienced initial relapse with disseminated disease (CNS/non-CNS EMD/medullary). Died of PD 222 d post–CAR infusion |
| 2 | CD19 | Scalp soft tissue, mesenteric, periaortic, retroperitoneal lymph nodes, right kidney,* left kidney, liver | 15.20 | Not available | 0 | Right kidney* | 4.08 | PR | CR | Not eligible for second HSCT and experienced initial relapse with medullary/non-CNS EMD. Died of PD 730 d post–CAR infusion |
| 3 | CD22 | Liver | 6.7 | 10.91 | 2.20 | Liver* | 9.18 | PD | PD | Died of PD 47 d post–CAR infusion |
| 4 | CD22 | Right kidney,* left kidney | 8.70 | 4.91 | 0.05 | Right kidney,* left kidney, vertebral bodies, rib, left humerus | 8.93 | PD | Stable disease | Died of PD 913 d post–CAR infusion |
| 5 | CD22 | Mesenteric, retroperitoneal lymph nodes, right kidney,* left kidney, extrusion from vertebral BM into psoas muscle | 13.51 | 8.7 | 27.0 | No residual EMD | 2.20 | CR (6 mo) | CR | Not eligible for second HSCT and experienced initial relapse with medullary disease. Died of PD 622 d post–CAR infusion6 |
| 6 | CD22 | Mesenteric, peritoneal lymph nodes, mediastinum, pericardium, pleura,* intramuscular lesion | 8.97 | 6.80 | 44.0 | Mesenteric, peritoneal, supraclavicular lymph nodes, pleura,* spleen, liver | 6.93 | PD | Stable disease | Died of PD 157 d post–CAR infusion |
| 7 | CD22 | Orbital bone, parotid gland, cervical, supraclavicular, axillary, peritoneal lymph nodes, pancreas* | 12.09 | 7.9 | 93.3 | Supraclavicular, axillary lymph nodes, orbital bone, parotid gland | 2.50 | PR (4 mo) | CR | Not eligible for second HSCT and experienced initial relapse with medullary/non-CNS EMD. Died of PD 1063 d post–CAR infusion54 |
| 8a‡ | CD22 | Temporal bone,* subcutaneous tissue surrounding external auditory canal, cervical, supraclavicular lymph nodes, pancreas | 63.27 | 2.99† | 0.50 | No residual EMD | 4.50 | CR (3 mo) | CR | Not eligible for second HSCT and monitored. Received subsequent CAR infusion after initial relapse with antigen-positive CNS/non-CNS EMD. See 8b |
| 8b‡ | CD22 | Thoracic, lumbar neural foramen* | 19.96 | 2.75† | 0 | No residual EMD | 3.82 | CR | CR | Not eligible for second HSCT and experienced initial relapse with isolated CNS disease and a myeloid sarcoma. Died of progressive myeloid disease 424 d post–CAR infusion55 |
| 9 | CD22 | Right kidney, left kidney, pleura* | 4.18 | 2.56† | 56.6 | No residual EMD | 1.19 | CR | CR | Proceeded to HSCT and experienced initial posttransplant relapse with disseminated disease (CNS/non-CNS EMD/medullary). Died of PD 330 d post-HSCT (395 d post–CAR infusion) |
| 10 | CD22 | Supraclavicular, mesenteric lymph nodes, pleura, left kidney* | 5.43 | 2.23† | 53.3 | Pleura, left kidney* | 3.67 | PR | CR | Proceeded to HSCT and experienced posttransplant relapse (sites unknown). Died of PD 174 d post-HSCT (240 d post–CAR infusion) |
| 11 | CD19/22 | Parotid gland, maxillary sinus, cervical, supraclavicular, mesenteric lymph nodes, mediastinum, liver,* stomach, scrotum | 22.5 | 15.40 | 0.35 | Parotid gland, maxillary sinus, stomach, scrotum | 2.9 | PR | CR | Not eligible for second HSCT and experienced initial relapse with medullary/non-CNS EMD. Died of PD 457 d post–CAR infusion |
| 12 | CD19/22 | Breast,* subcutaneous left lower extremity lesions, extrusion from right lower extremity BM to soft tissue | 11.70 | 8.30 | 0.004 | Breast,* subcutaneous left lower extremity lesions | 4.74 | PR | CR | Proceeded to second HSCT and experienced initial relapse with isolated non-CNS EMD. Died of PD 668 d post-HSCT (834 d post–CAR infusion) |
| 13 | CD19/22 | Cervical, axillary,* retroperitoneal, mesenteric, inguinal, pelvic lymph nodes, mediastinum, right kidney, left kidney, pancreas | 11.66 | 9.80 | 90.1 | No residual EMD | 2.09 | CR | CR | Proceeded to HSCT and experienced initial relapse with medullary disease. Died of PD 593 d post-HSCT (720 d post–CAR infusion) |
| 14 | CD19/22 | Retroperitoneal, pelvic wall lymph nodes, pancreas,* testes | 8.29 | 3.2 | 4.96 | Retroperitoneal lymph node, pancreas,* adrenal gland, testes | 11.67 | PD | CR | Died of PD 346 d post–CAR infusion |
| 15 | CD19/22 | Breast,* cervical, axillary lymph nodes, mediastinum | 34.93 | 38.8 | 0.49 | Breast* | 11.58 | PR | Stable disease | Received additional therapies. Experienced medullary disease progression followed by CNS/non-CNS EMD relapse. Remains alive with disseminated disease 682 d post–CAR infusion |
| 16 | CD19/22 | Liver* | 6.05 | 2.57† | 0 | No residual EMD | 1.99 | CR | CR | Proceeded to HSCT and experienced initial relapse with medullary/non-CNS EMD. Died of PD 335 d post-HSCT (390 d post–CAR infusion) |
| 17 | CD19/22 | Inguinal, pelvic wall,* popliteal, deep thigh lymph nodes, skin, left foot periosteum | 10.39 | 2.07† | 0.01 | Inguinal lymph node,* left foot periosteum | 3.18 | PR | CR | Not eligible for second HSCT and experienced initial relapse with medullary/non-CNS EMD. Died of PD 302 d post–CAR infusion |
| Pre-CAR, at presentation to our institution | Post–CAR infusion, best response | Overall outcome | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Patient | CAR | Non-CNS EMD at pre-CAR FDG PET/CT | maxSUV, non-CNS EMD | maxSUV, focal BM disease | BM by morphology (% of MNCs) | Non-CNS EMD at best response FDG PET/CT | SUV, Non-CNS EMD | Non-CNS EMD response | BM response (day +28) | |
| 1 | CD19 | Retroperitoneal lymph node* | 17.31 | 3.25† | 0.04 | No residual EMD | 2.63 | CR | CR | Not eligible for second HSCT and experienced initial relapse with disseminated disease (CNS/non-CNS EMD/medullary). Died of PD 222 d post–CAR infusion |
| 2 | CD19 | Scalp soft tissue, mesenteric, periaortic, retroperitoneal lymph nodes, right kidney,* left kidney, liver | 15.20 | Not available | 0 | Right kidney* | 4.08 | PR | CR | Not eligible for second HSCT and experienced initial relapse with medullary/non-CNS EMD. Died of PD 730 d post–CAR infusion |
| 3 | CD22 | Liver | 6.7 | 10.91 | 2.20 | Liver* | 9.18 | PD | PD | Died of PD 47 d post–CAR infusion |
| 4 | CD22 | Right kidney,* left kidney | 8.70 | 4.91 | 0.05 | Right kidney,* left kidney, vertebral bodies, rib, left humerus | 8.93 | PD | Stable disease | Died of PD 913 d post–CAR infusion |
| 5 | CD22 | Mesenteric, retroperitoneal lymph nodes, right kidney,* left kidney, extrusion from vertebral BM into psoas muscle | 13.51 | 8.7 | 27.0 | No residual EMD | 2.20 | CR (6 mo) | CR | Not eligible for second HSCT and experienced initial relapse with medullary disease. Died of PD 622 d post–CAR infusion6 |
| 6 | CD22 | Mesenteric, peritoneal lymph nodes, mediastinum, pericardium, pleura,* intramuscular lesion | 8.97 | 6.80 | 44.0 | Mesenteric, peritoneal, supraclavicular lymph nodes, pleura,* spleen, liver | 6.93 | PD | Stable disease | Died of PD 157 d post–CAR infusion |
| 7 | CD22 | Orbital bone, parotid gland, cervical, supraclavicular, axillary, peritoneal lymph nodes, pancreas* | 12.09 | 7.9 | 93.3 | Supraclavicular, axillary lymph nodes, orbital bone, parotid gland | 2.50 | PR (4 mo) | CR | Not eligible for second HSCT and experienced initial relapse with medullary/non-CNS EMD. Died of PD 1063 d post–CAR infusion54 |
| 8a‡ | CD22 | Temporal bone,* subcutaneous tissue surrounding external auditory canal, cervical, supraclavicular lymph nodes, pancreas | 63.27 | 2.99† | 0.50 | No residual EMD | 4.50 | CR (3 mo) | CR | Not eligible for second HSCT and monitored. Received subsequent CAR infusion after initial relapse with antigen-positive CNS/non-CNS EMD. See 8b |
| 8b‡ | CD22 | Thoracic, lumbar neural foramen* | 19.96 | 2.75† | 0 | No residual EMD | 3.82 | CR | CR | Not eligible for second HSCT and experienced initial relapse with isolated CNS disease and a myeloid sarcoma. Died of progressive myeloid disease 424 d post–CAR infusion55 |
| 9 | CD22 | Right kidney, left kidney, pleura* | 4.18 | 2.56† | 56.6 | No residual EMD | 1.19 | CR | CR | Proceeded to HSCT and experienced initial posttransplant relapse with disseminated disease (CNS/non-CNS EMD/medullary). Died of PD 330 d post-HSCT (395 d post–CAR infusion) |
| 10 | CD22 | Supraclavicular, mesenteric lymph nodes, pleura, left kidney* | 5.43 | 2.23† | 53.3 | Pleura, left kidney* | 3.67 | PR | CR | Proceeded to HSCT and experienced posttransplant relapse (sites unknown). Died of PD 174 d post-HSCT (240 d post–CAR infusion) |
| 11 | CD19/22 | Parotid gland, maxillary sinus, cervical, supraclavicular, mesenteric lymph nodes, mediastinum, liver,* stomach, scrotum | 22.5 | 15.40 | 0.35 | Parotid gland, maxillary sinus, stomach, scrotum | 2.9 | PR | CR | Not eligible for second HSCT and experienced initial relapse with medullary/non-CNS EMD. Died of PD 457 d post–CAR infusion |
| 12 | CD19/22 | Breast,* subcutaneous left lower extremity lesions, extrusion from right lower extremity BM to soft tissue | 11.70 | 8.30 | 0.004 | Breast,* subcutaneous left lower extremity lesions | 4.74 | PR | CR | Proceeded to second HSCT and experienced initial relapse with isolated non-CNS EMD. Died of PD 668 d post-HSCT (834 d post–CAR infusion) |
| 13 | CD19/22 | Cervical, axillary,* retroperitoneal, mesenteric, inguinal, pelvic lymph nodes, mediastinum, right kidney, left kidney, pancreas | 11.66 | 9.80 | 90.1 | No residual EMD | 2.09 | CR | CR | Proceeded to HSCT and experienced initial relapse with medullary disease. Died of PD 593 d post-HSCT (720 d post–CAR infusion) |
| 14 | CD19/22 | Retroperitoneal, pelvic wall lymph nodes, pancreas,* testes | 8.29 | 3.2 | 4.96 | Retroperitoneal lymph node, pancreas,* adrenal gland, testes | 11.67 | PD | CR | Died of PD 346 d post–CAR infusion |
| 15 | CD19/22 | Breast,* cervical, axillary lymph nodes, mediastinum | 34.93 | 38.8 | 0.49 | Breast* | 11.58 | PR | Stable disease | Received additional therapies. Experienced medullary disease progression followed by CNS/non-CNS EMD relapse. Remains alive with disseminated disease 682 d post–CAR infusion |
| 16 | CD19/22 | Liver* | 6.05 | 2.57† | 0 | No residual EMD | 1.99 | CR | CR | Proceeded to HSCT and experienced initial relapse with medullary/non-CNS EMD. Died of PD 335 d post-HSCT (390 d post–CAR infusion) |
| 17 | CD19/22 | Inguinal, pelvic wall,* popliteal, deep thigh lymph nodes, skin, left foot periosteum | 10.39 | 2.07† | 0.01 | Inguinal lymph node,* left foot periosteum | 3.18 | PR | CR | Not eligible for second HSCT and experienced initial relapse with medullary/non-CNS EMD. Died of PD 302 d post–CAR infusion |
Patient 5 was previously reported in an article by Fry et al.6 Patient 7 was reported as a case by Shah et al.54 Patient 8a/8b was reported as a case by Mo et al.55 BM, bone marrow; maxSUV, maximum standardized uptake; MNCs, mononuclear cells.
Site of non-CNS EMD corresponding to maxSUV.
BM maxSUV calculated as average maxSUV of vertebral bodies (L3, L4, and L5) in the absence of focal BM disease. In patients with no residual non-CNS EMD, maxSUV value at best response reflects physiological FDG uptake at site of non-EMD identified on pre-CAR scan.
8a and 8b represent outcomes for a single patient who received 2 separate infusions of the same CAR product at initial treatment and subsequent disease recurrence.
Of the 7 patients with a non-CNS EMD CR, 3 had focal or loco-regional involvement only. Thus, 3 (75%) of 4 patients with focal non-CNS EMD achieved a CR, whereas only 4 (28.6%) of 14 patients with multifocal EMD had a CR of non-CNS EMD (P = .26). Regarding site-specific response, lymph node involvement responded to CAR T cells more frequently than other sites: 11 (84.6%) of 13 patients with non-CNS EMD of the lymph nodes exhibited a CR of some or all of the disease identified at presentation (Figure 1C).
Discrepant responses were observed on occasion: 1 patient attained a medullary MRD-negative CR but had non-CNS EMD PD at follow-up; another showed stable disease in the marrow and PR of non-CNS EMD. Two additional patients exhibited non-CNS EMD PD with stable MRD-positive medullary disease after CAR T-cell infusion.
Among 17 patients (18 infusions) with non-CNS EMD, 5 proceeded to HSCT (HSCT-naive, n = 4; second HSCT, n = 1), and 8 were not eligible for second HSCT or had PD (n = 4). With a median follow-up of 440.5 days (range, 47-1063 days) post–CAR infusion, 1 patient remains alive with residual disease. Others have died of complications of PD (Table 3).
CAR T-cell expansion in non-CNS EMD
CAR T-cell expansion in patients with non-CNS EMD (n = 11) vs without non-CNS EMD (n = 87) differed by CAR construct in those who exhibited symptoms of CRS. Patients with non-CNS EMD treated with anti-CD22 CAR T cells exhibited substantially higher absolute CAR T-cell expansion (n = 7; median, 2167 cells/mL; range, 105.3-13 653 cells/mL) than their counterparts without non-CNS EMD (n = 51; median, 573.8 cells/mL; range, 0.65-11 345 cells/mL) (P = .04). Peak CAR T-cell expansion did not differ in those with vs without non-CNS EMD for either the CD19 or CD19/22 CAR constructs, although patient numbers were limited (Figure 2A-D).
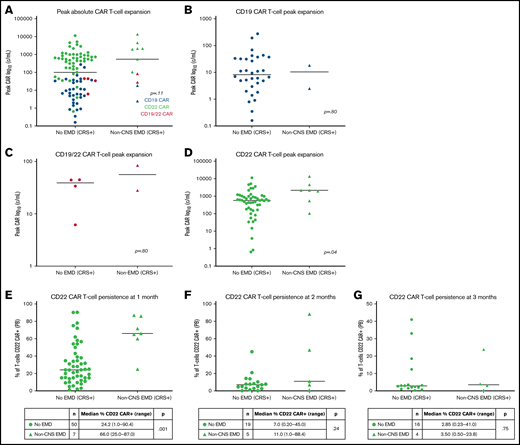
CAR T-cell kinetics and CD22 CAR T-cell persistence in patients with non-CNS EMD. (A) Peak absolute CAR T-cell expansion in patients with non-CNS EMD vs those without non-CNS EMD treated with anti-CD19 (B), anti-CD19/22 (C), and anti-CD22 (D) CAR T cells. For CD22 CAR patients, substantially higher peak CAR T-cell expansion was shown in those with non-CNS EMD (n = 7; median, 2167 cells/mL; range, 105.3-13 653 cells/mL) compared with those without non-CNS EMD (n = 51; median, 573.8 cells/mL; range, 0.65-11 345 cells/mL) (P = .04). CD22 CAR T-cell persistence: (E) ∼1 month (median, 26 days; range, 18-30 days), (F) ∼2 months (median, 58 days; range, 41-69 days), and (G) ∼3 months (median, 94 days; range 84-129 days) after CAR T-cell infusion.
CAR T-cell kinetics and CD22 CAR T-cell persistence in patients with non-CNS EMD. (A) Peak absolute CAR T-cell expansion in patients with non-CNS EMD vs those without non-CNS EMD treated with anti-CD19 (B), anti-CD19/22 (C), and anti-CD22 (D) CAR T cells. For CD22 CAR patients, substantially higher peak CAR T-cell expansion was shown in those with non-CNS EMD (n = 7; median, 2167 cells/mL; range, 105.3-13 653 cells/mL) compared with those without non-CNS EMD (n = 51; median, 573.8 cells/mL; range, 0.65-11 345 cells/mL) (P = .04). CD22 CAR T-cell persistence: (E) ∼1 month (median, 26 days; range, 18-30 days), (F) ∼2 months (median, 58 days; range, 41-69 days), and (G) ∼3 months (median, 94 days; range 84-129 days) after CAR T-cell infusion.
CAR T-cell persistence in non-CNS EMD
Given the limited persistence of our CD19 and CD19/22 CAR constructs, analysis of CAR T-cell persistence was performed exclusively in CD22 CAR patients who had confirmed non-CNS EMD at CAR infusion and subsequently experienced CRS.31,32 In 57 patients with CD22 CAR+ T cells detectible in the peripheral blood by flow cytometry 1 month post-infusion, those with non-CNS EMD (n = 7) had a higher proportion of T cells that were CD22 CAR+ (median, 66.0%; range, 25.0%-87.0%) than those without EMD (median, 24.2%; range, 1.0%-90.4%) (P = .0010) (Figure 2E). A substantial proportion of CD22 CAR patients proceeded directly to HSCT, limiting the availability of CAR T-cell persistence data >1 month post-infusion. For those with evaluable data 2 or 3 months’ postinfusion, there was no statistically significant difference in CAR+ T-cell persistence among patients with or without non-CNS EMD (Figure 2F-G).
Unique CAR T cell–associated toxicities in non-CNS EMD
CRS occurred in 11 (61.1%) patients, with 8 experiencing maximum CRS grade 1 to 2. Site-specific CAR T cell–associated inflammatory toxicities manifested as swelling and erythema in patients with non-CNS EMD involving the orbit, breast, and lymph nodes (Table 4). Development of substantial lymphedema prompted subsequent evaluation to rule out a deep venous thrombosis in 1 patient. In those with pleural-based disease, pulmonary toxicity was evidenced by worsening or new development of pleural effusions, ground-glass opacities, and new oxygen requirement (Figure 3A,C). Notably, 1 patient with a recent history of pleural disease, and full resolution after interim chemotherapy immediately before CAR T-cell therapy, presented with these unique pulmonary toxicities post–CAR infusion with evidence of CAR T-cell trafficking (Figure 3D). CAR T cells were additionally identified in the pleural fluid of 1 patient during CRS (Figure 3B). Elevated serum creatinine levels suggestive of renal toxicity in patients with known leukemic infiltration of the kidneys could have been in part related to CRS (Table 4). All toxicities were transient and reversible with the exception of those in 1 patient, in whom disease progression and CAR response occurred simultaneously, and pulmonary toxicity was in part due to progressive leukemia.
Unique CAR T cell–associated toxicities in a subset of patients (n = 7) who obtained serial scans (n = 17 [18 infusions])
| Patient | CRS maximum grade (ASTCT) | Site-specific toxicities | Peak peripheral blood CAR % (% of T cells) | Peak site-specific CAR+ % (% of T cells) |
|---|---|---|---|---|
| 1 | 2 | Possibility of inflammation at site of retroperitoneal disease with transient appendiceal thickening identified on CT imaging. Etiology of CT findings could not be confirmed with imaging alone | 1.6 | − |
| 5 | 1 | Possibility of pain from inflammation at psoas site of disease with focal abnormality in paravertebral soft tissues potentially related to inflammatory process. Etiology of CT findings could not be confirmed with imaging alone | 60.0 | − |
| 6 | 2 | Increased work of breathing and oxygen requirement with onset of CRS. Worsening bilateral pleural effusions and ground-glass opacities demonstrated on CT imaging | 89.5 | Pleural fluid: 72, day +27; BAL: 74, day +33 |
| 7 | 3 | Oxygen requirement with onset of CRS and inflammation of orbital mass with eyelid swelling. Swelling of right upper extremity associated with inflammation in lymph nodes manipulated in prior mastectomy | 97.8 | BAL: 90, day +28 |
| 9 | 2 | Oxygen requirement during CRS. Bilateral pleural effusions and ground-glass opacities consistent with inflammation at site of pleural-based disease seen on CT imaging. Rising serum creatinine with onset of CRS (peak, 0.88 mg/dL, day +11; baseline, 0.50-0.66 mg/dL) and acute kidney injury | 88.0 | − |
| 15 | 0 | Pain and swelling associated with breast erythema during CRS and pain associated with swelling of axillary lymph node | 3.3 | − |
| 39* | 1 | Respiratory distress with oxygen requirement during CRS. Malignant bilateral pleural effusions with alveolar infiltrates revealed on CT imaging during CRS | 90.2 | BAL: 85.0, day +54 |
| Patient | CRS maximum grade (ASTCT) | Site-specific toxicities | Peak peripheral blood CAR % (% of T cells) | Peak site-specific CAR+ % (% of T cells) |
|---|---|---|---|---|
| 1 | 2 | Possibility of inflammation at site of retroperitoneal disease with transient appendiceal thickening identified on CT imaging. Etiology of CT findings could not be confirmed with imaging alone | 1.6 | − |
| 5 | 1 | Possibility of pain from inflammation at psoas site of disease with focal abnormality in paravertebral soft tissues potentially related to inflammatory process. Etiology of CT findings could not be confirmed with imaging alone | 60.0 | − |
| 6 | 2 | Increased work of breathing and oxygen requirement with onset of CRS. Worsening bilateral pleural effusions and ground-glass opacities demonstrated on CT imaging | 89.5 | Pleural fluid: 72, day +27; BAL: 74, day +33 |
| 7 | 3 | Oxygen requirement with onset of CRS and inflammation of orbital mass with eyelid swelling. Swelling of right upper extremity associated with inflammation in lymph nodes manipulated in prior mastectomy | 97.8 | BAL: 90, day +28 |
| 9 | 2 | Oxygen requirement during CRS. Bilateral pleural effusions and ground-glass opacities consistent with inflammation at site of pleural-based disease seen on CT imaging. Rising serum creatinine with onset of CRS (peak, 0.88 mg/dL, day +11; baseline, 0.50-0.66 mg/dL) and acute kidney injury | 88.0 | − |
| 15 | 0 | Pain and swelling associated with breast erythema during CRS and pain associated with swelling of axillary lymph node | 3.3 | − |
| 39* | 1 | Respiratory distress with oxygen requirement during CRS. Malignant bilateral pleural effusions with alveolar infiltrates revealed on CT imaging during CRS | 90.2 | BAL: 85.0, day +54 |
Of the patients (66.6%) without evidence of site-specific toxicity after CAR T-cell infusion, six did not show symptoms of CRS, four experienced CRS grade 1 to 2, and two experienced CRS grade 3. ASTCT, American Society of Transplantation and Cellular Therapy; BAL, bronchoalveolar lavage.
Patient 39 presented with unique pulmonary toxicities but did not have FDG-avid non-CNS EMD at the time of treatment and was not included in the serial scan study cohort.
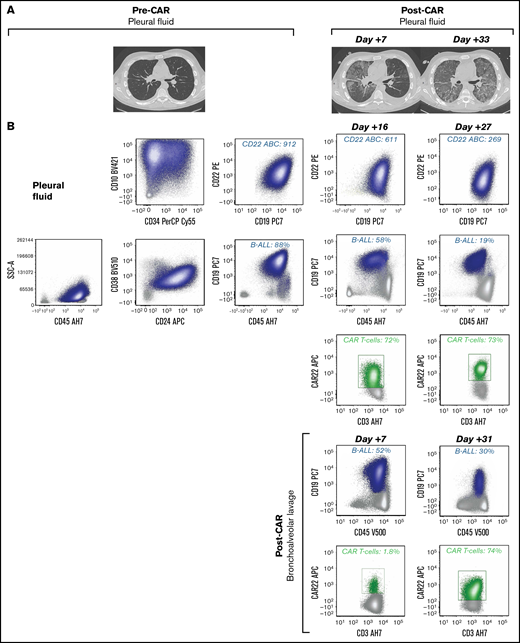
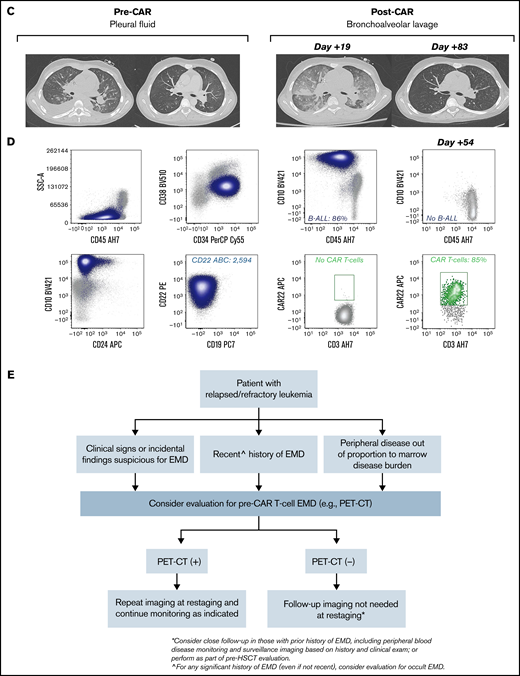
Unique CAR T cell–associated toxicities and CAR T-cell expansion in select patients (n = 2) with non-CNS EMD. (A) CT scans obtained pre– and post–CD22 CAR infusion showing CAR T cell–associated pulmonary toxicity in a subject (Patient 6) with pleural-based non-CNS EMD. Clinically, CAR T-cell trafficking to non-CNS EMD was evidenced by development of new pleural effusions, ground-glass opacities, and oxygen requirement. (B) Corresponding flow cytometry of pleural fluid before CD22 CAR infusion identified B-ALL comprising 88% of mononuclear cells (MNCs). B-ALL blasts (navy blue) expressed slightly dim CD45, CD19, CD10, partial CD34, CD22, dim CD38, and CD24. CD22 antibody-binding capacity (ABC) was 912, a quantitative measure of antigen site density on the blast cell surface. Subsequently, flow cytometry performed at day +16 post–CD22 CAR infusion showed an expansion of CD22 CAR T cells (green), comprising 72% of T cells, which persisted at day +27. The amount of B-ALL disease decreased to 58% of MNCs and 19% of MNCs at day +16 and day +27, respectively. CD22 ABC decreased posttherapy from 912 to 611 and 269 at day +16 and day +27, respectively. Flow cytometry was also performed on bronchoalveolar lavage post–CAR infusion. The amount of B-ALL disease decreased from 52% of MNCs at day +7 to 30% of MNCs at day +31. At day +7, 1.8% of T cells were CD22 CAR T cells (green); they expanded to comprise 74% of T cells at day +31. (C) CT scans obtained pre– and post–CD22 CAR infusion in a subject (Patient 39) with pleural-based disease exhibiting delayed resolution (day +83) of CAR T cell–associated inflammatory toxicities. (D) Corresponding flow cytometry of pleural fluid before CD22 CAR T-cell therapy identified B-ALL comprising 86% of MNCs. B-ALL blasts (navy blue) expressed a spectrum of CD45 from dim to negative, bright CD10, CD34, CD22, and dim CD38; they were negative for CD19 and CD24. The CD22 ABC was 2594. As expected, no CAR T cells were detected by using the flow cytometry assay. Subsequently, flow cytometry was performed on a bronchoalveolar lavage specimen at day +54 post–CD22 CAR infusion. Expansion of CD22 CAR T cells was detected (green), comprising 85% of T cells, and there was no evidence of B-ALL. Notably, Patient 39 did not have non-CNS EMD identifiable on FDG PET/CT imaging during central review and was not included in the initial study cohort. (E) Generalized approach to indications for evaluation of non-CNS EMD in the peri–CAR T-cell setting. SSA, side scatter area.
Unique CAR T cell–associated toxicities and CAR T-cell expansion in select patients (n = 2) with non-CNS EMD. (A) CT scans obtained pre– and post–CD22 CAR infusion showing CAR T cell–associated pulmonary toxicity in a subject (Patient 6) with pleural-based non-CNS EMD. Clinically, CAR T-cell trafficking to non-CNS EMD was evidenced by development of new pleural effusions, ground-glass opacities, and oxygen requirement. (B) Corresponding flow cytometry of pleural fluid before CD22 CAR infusion identified B-ALL comprising 88% of mononuclear cells (MNCs). B-ALL blasts (navy blue) expressed slightly dim CD45, CD19, CD10, partial CD34, CD22, dim CD38, and CD24. CD22 antibody-binding capacity (ABC) was 912, a quantitative measure of antigen site density on the blast cell surface. Subsequently, flow cytometry performed at day +16 post–CD22 CAR infusion showed an expansion of CD22 CAR T cells (green), comprising 72% of T cells, which persisted at day +27. The amount of B-ALL disease decreased to 58% of MNCs and 19% of MNCs at day +16 and day +27, respectively. CD22 ABC decreased posttherapy from 912 to 611 and 269 at day +16 and day +27, respectively. Flow cytometry was also performed on bronchoalveolar lavage post–CAR infusion. The amount of B-ALL disease decreased from 52% of MNCs at day +7 to 30% of MNCs at day +31. At day +7, 1.8% of T cells were CD22 CAR T cells (green); they expanded to comprise 74% of T cells at day +31. (C) CT scans obtained pre– and post–CD22 CAR infusion in a subject (Patient 39) with pleural-based disease exhibiting delayed resolution (day +83) of CAR T cell–associated inflammatory toxicities. (D) Corresponding flow cytometry of pleural fluid before CD22 CAR T-cell therapy identified B-ALL comprising 86% of MNCs. B-ALL blasts (navy blue) expressed a spectrum of CD45 from dim to negative, bright CD10, CD34, CD22, and dim CD38; they were negative for CD19 and CD24. The CD22 ABC was 2594. As expected, no CAR T cells were detected by using the flow cytometry assay. Subsequently, flow cytometry was performed on a bronchoalveolar lavage specimen at day +54 post–CD22 CAR infusion. Expansion of CD22 CAR T cells was detected (green), comprising 85% of T cells, and there was no evidence of B-ALL. Notably, Patient 39 did not have non-CNS EMD identifiable on FDG PET/CT imaging during central review and was not included in the initial study cohort. (E) Generalized approach to indications for evaluation of non-CNS EMD in the peri–CAR T-cell setting. SSA, side scatter area.
Use of pembrolizumab to augment CAR expansion and response to non-CNS EMD
Based on the preliminary safety and efficacy of combining programmed cell death protein 1 (PD-1)–directed immune checkpoint inhibition with CAR T cells in other B-cell lymphomas,33 this combination has been explored in B-ALL.34 In 3 patients (2 after the data cutoff for the systematic review) with particularly difficult-to-treat non-CNS EMD and suboptimal CAR response, we attempted to augment CAR T-cell activity with this rational combination. Our patients tolerated therapy without serious immune-related adverse events but also without response (supplemental Table 2).
Discussion
Given the tremendous successes of CAR T cells in eradication of leukemia and non-Hodgkin lymphomas,35 the current study sought to both explore outcomes of non-CNS EMD in patients with r/r B-ALL receiving CAR T-cell therapy and ascertain the frequency and distribution of non-CNS EMD in r/r B-ALL. Our results highlight the importance of monitoring for occult non-CNS EMD and on the potential efficacy and limitations of CAR T cells in non-CNS EMD.
In evaluating the incidence of non-CNS EMD in patients referred to our trials, we found that ∼21% of 180 patients with B-ALL screened at our institution had non-CNS EMD identifiable on FDG PET/CT imaging, the majority of whom had relapsed after prior HSCT. Although this value was obtained from a heavily pretreated population with r/r disease in which FDG PET/CT imaging was prompted by patient history, clinical examination, or findings on alternative imaging, it potentially underrepresents the incidence of non-CNS EMD in B-ALL, as FDG PET/CT imaging is not routinely used in acute leukemia disease assessment. Notably, a relatively high percentage of patients with non-CNS EMD received prior immunotherapy. Although patients referred to our center are generally more refractory, introducing a selection bias, close monitoring of non-CNS EMD recurrence post-immunotherapy is warranted, particularly given the experience with post-HSCT relapse. A limited number of case reports and retrospective reviews have examined the utility of FDG PET/CT imaging in assessing medullary disease36,37 and non-CNS EMD, with sites identified in the soft tissue, head and neck lymph nodes, liver, and pancreas.38-40 Cistaro et al40 describe a striking case of pediatric B-ALL with isolated lymph node involvement that evaded all routine monitoring and in which PET/CT imaging was ultimately required to facilitate a diagnosis, revealing an increased sensitivity for non-CNS EMD detection compared with other imaging modalities.
Few studies have systematically evaluated the sensitivity of FDG PET/CT imaging for non-CNS EMD detection in acute leukemia. In patients with newly diagnosed or relapsed B-ALL or acute myeloid leukemia (n = 79), Zhou et al41 found FDG PET-CT imaging to be highly sensitive (93.3%), although not extremely specific (71.4%) in diagnosing EMD. The authors nonetheless underscore the importance of whole-body imaging in this population. Cunningham and Kohno20 concur, concluding from a review of 124 cases of FDG PET/CT use in leukemia that the extent of EMD was significantly underestimated in the absence of total body scanning. Collectively, the benefits of early, comprehensive disease assessment with noninvasive imaging technology may potentially outweigh the risks associated with additional radiation exposure in children.19 This may be particularly true in patients in whom identification of non-CNS EMD would inform the treatment approach or critically change risk (eg, pre-HSCT). Continued technological advances in FDG PET/CT imaging have allowed for reductions in radiation exposure, which may facilitate easier incorporation into the pediatric B-ALL population. PET/magnetic resonance imaging, an alternative whole-body imaging modality with reduced radiation exposure, has shown comparable sensitivity and specificity to FDG PET/CT imaging in limited studies of pediatric lymphomas and solid tumors.42-44 Its role in B-ALL has not been studied but is worth exploring, acknowledging limitations both of cost and accessibility. Beyond imaging, experience using circulating tumor DNA with clonoSEQ (Adaptive Biotechnologies) to predict post–CAR relapse risk in adult lymphomas45 may inform future studies in B-ALL seeking to evaluate the role of next-generation sequencing for detecting non-CNS EMD. Although most patients have progression of non-CNS EMD during routine follow-up, incorporation of next-generation sequencing, already used as a proxy of peripheral blood disease clearance, as a part of routine surveillance in B-ALL may facilitate early detection of non-CNS EMD relapse before presentation with fulminant disease.
In the setting of CAR T-cell therapy, we determined that clinical response of medullary and non-CNS EMD to CAR T cells was concordant at best response in most patients, with nearly one-half of this heavily pretreated cohort ultimately exhibiting a medullary/non-CNS EMD CR. However, non-CNS EMD responses to CAR T cells still varied: delayed non-CNS EMD response was substantial, as was residual non-CNS EMD after CAR T-cell infusion. Best non-CNS EMD response lagged behind medullary response until 3 to 6 months after treatment in several cases, and a considerable proportion of patients exhibited only a non-CNS EMD PR at best response despite attaining a medullary CR. This suggests that optimal time to best response may differ in the bone marrow and at extramedullary sites and, furthermore, that the CR rate of non-CNS EMD is likely lower than that of isolated medullary disease. Patients with multifocal non-CNS EMD also seemed less likely to achieve a non-CNS EMD CR, but additional study in a larger data set is needed to confirm this association. By systematically reviewing the role for FDG PET/CT imaging in assessing non-CNS EMD response to CAR T cells, with centralized radiology review, our results show that serial monitoring of non-CNS EMD in r/r B-ALL is essential to ensure that patients truly achieve an MRD-negative remission after novel cellular therapies.
The current study additionally describes unique presentations of site-specific CAR T-cell toxicities in non-CNS EMD. Inflammatory complications seen in several patients were believed to correlate with CAR T-cell expansion. Specifically, we were able to identify CAR T cells in the pleural fluid of select patients with pulmonary toxicities. Cases reporting significant local inflammation of non-CNS EMD involving the optic nerve and skin have similarly confirmed CAR T-cell capability of trafficking to sites of B-ALL non-CNS EMD.46,47 Although we were limited in our ability to look at site-specific CAR T-cell trafficking in most patients, our experience provides insights into the toxicity profile of CAR T cells in non-CNS EMD and the impact of CAR T-cell kinetics and trafficking on clinical presentation.
Regarding CAR T-cell kinetics, patients with non-CNS EMD receiving CD22 CAR T cells exhibited substantially higher CAR T-cell expansion than those without EMD. Our results also suggest that CAR persistence is enhanced in patients with non-CNS EMD compared with those without any EMD, although further study in a larger cohort is needed to explore this association across alternative constructs. The generalizability of these findings may be limited because the CD22 CAR construct was known to be more persistent than either the CD19 or CD19/22 CAR constructs; however, our observations illustrate that differences in CAR T-cell construct may critically affect outcomes, and further study of outcomes of non-CNS EMD with US Food and Drug Administration–approved constructs is warranted. Investigating the mechanisms of CAR T-cell trafficking and exhaustion in non-CNS EMD will be essential to optimizing responses to CAR T cells and may elucidate factors leading to the rare discrepant medullary/extramedullary responses to therapy seen in our study. Analysis of CAR T-cell product characteristics and markers of T-cell exhaustion may provide further insights into the variability between individual responses to non-CNS EMD, and additional studies are ongoing.
Incorporation of checkpoint blockade to reduce T-cell exhaustion and augment CAR T-cell efficacy in the immunosuppressive tumor microenvironment has been investigated in several small studies combining PD-1 inhibition with CD19-directed CAR T cells for B-cell malignancies.33 Although the experience with PD-1 inhibitors in adult B-cell lymphomas is more robust,48-51 Li et al34 have reported limited success in using pembrolizumab or nivolumab after CD19 CAR T-cell therapy in children with relapsed ALL/lymphoma. In our limited experience, pembrolizumab (albeit incorporated at differing time points and for varying indications) was not effective in inducing CAR T-cell re-expansion or optimizing non-CNS EMD response. This combination warrants prospective study to gain a better understanding of the optimal time point for incorporation of immune checkpoint inhibition in relation to CAR T-cell infusion and an appropriate duration of therapy with the checkpoint inhibitor. Other bridging strategies, such as radiation therapy (used by 1 patient in our series to eradicate disease pre-CAR), may have a unique role in debulking EMD pre– and/or post–CAR T-cell therapy, but reports are limited,47,52 and further study is needed.53
In addition to the limitations of heterogeneity across patients and CAR T-cell constructs, thus restricting the generalizability of our results, our heavily pretreated cohort may be skewed for a higher incidence of non-CNS EMD. Nonetheless, our analysis provides important insights into disease metrics for this population and may indicate a need to revise the current paradigm in B-ALL disease surveillance. In this regard, we provide our proposal for when specific evaluation of non-CNS EMD may be indicated (Figure 3E). The role of insurance coverage for FDG PET/CT imaging in patients at high risk of EMD (eg, history of EMD, clinical findings suspicious for EMD) must additionally be considered, as detection of occult non-CNS EMD may be particularly critical for those with r/r disease or in the peri-HSCT setting. Our analysis of CAR T-cell persistence and trafficking to sites of non-CNS EMD was also limited by availability of patient samples. Future studies incorporating serial biopsies to evaluate CAR T-cell trafficking and antigen expression in non-CNS EMD are needed to evaluate mechanisms of suboptimal response to therapy.
In summary, we raise awareness about the need to assess non-CNS EMD in B-ALL and highlight the potential strengths and limitations of CAR T-cell therapy in the setting of non-CNS EMD. Our results show that CAR T cells are active against B-ALL non-CNS EMD and constitute a promising option for patients with isolated or combined medullary/non-CNS EMD. Still, non-CNS EMD response to CAR T-cell therapy may lag behind medullary response, and the CR rate of non-CNS EMD is likely lower than that of medullary disease with CAR T cells, leading to worse outcomes overall. These findings reaffirm that serial monitoring of non-CNS EMD with FDG PET/CT imaging is essential, and further investigation is warranted to best incorporate whole-body imaging in the B-ALL treatment paradigm. Elucidating factors leading to diminished CAR T-cell efficacy in the non-CNS EMD microenvironment will be crucial to optimization strategies for therapy in the future.
Acknowledgments
The authors gratefully acknowledge the study participants and their families, referring medical care teams, the faculty and staff of the NIH Clinical Center who provided their expertise in the management of the study participants, and the data managers and research nurses and patient care coordinators involved with this work.
This work was supported in part by the Intramural Research Program, Center of Cancer Research, National Cancer Institute and NIH Clinical Center, National Institutes of Health (ZIA BC 011823, N.N.S.) and the Warren Grant Magnuson Clinical Center.
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Authorship
Contribution: E.M.H. and N.N.S. wrote the first version of the manuscript; E.M.H., B.Y., M.L., D.A.L., and N.N.S. performed primary data analysis and evaluated correlative studies; C.M.Y., H.-W.W., and M.S.-S. performed flow cytometry and analyzed results; A.L. and M.A.A. provided radiology review and images for submission for publications; and B.Y., J.C.M., D.W.L., J.A.L., H.S., and N.N.S. provided patient care and contributed critically to the manuscript. No non-author wrote the first draft or any part of the paper. All authors contributed to reviewing the final manuscript and have agreed to be co-authors.
Conflict-of-interest disclosure: D.W.L. consults for Harpoon Therapeutics and Juno Therapeutics; and his institution receives clinical trial support from Kite Pharma. All funding was provided by the NIH Intramural Research Program with the exception of the St. Baldrick’s Foundation Scholar award provided to D.W.L. for work done involving the CD19 CAR T-cell trial.
Correspondence: Nirali N. Shah, Pediatric Oncology Branch, CCR, NCI, NIH, Building 10, Room 1W-3750, 9000 Rockville Pike MSC 1104, Bethesda, MD 20892; e-mail: nirali.shah@nih.gov.

