

TO THE EDITOR:
Multiple myeloma (MM) measurable residual disease (MRD) persisting after treatment is an adverse prognostic factor for progression-free survival (PFS) and overall survival.1 Genomic mutations occurring in the remaining clonal aberrant plasma cells (A-PCs) are linked to the development of drug resistance and disease relapse.2 Thus, personalized treatment based on the genomic profile of MRD could be highly beneficial and ultimately increase patients’ survival. However, although large-scale sequencing studies have characterized the genome of many malignancies, including MM,3-8 the genomic mutations present in MM MRD exist at the beginning of investigation.9 Here, we set up an exome sequencing analysis to identify genomic mutations characteristic for MM MRD and explore if they could mediate drug response, resistance, or disease progression.
Samples of peripheral blood and sorted clonal bone marrow A-PCs were collected from 22 patients after bortezomib-based treatment (supplemental Table 1; supplemental Figure 1) upon signing the informed consent form. The study was approved by the institutional ethics board of the University Hospital Ostrava (reference number 500/2016) and was conducted in accordance with the Declaration of Helsinki. All methodological details used in this study are provided in the supplemental methods; importantly, clonal A-PCs were sorted according to pathological immunophenotype using CD38, CD45, CD19, CD56 (CD117 when necessary)10 (supplemental Figure 2) with the purity exceeding 95%. This led to a median of 2000 cells per patient. DNA from those cells was amplified, and its exome was further analyzed. Here, we report only nonsynonymous somatic variants with frequency in human population <1% (supplemental Table 2; supplemental Figure 3; supplemental methods). In total, we identified 278 variants, with a median of 12.5 mutations per patient and a median coverage of 71. These variants were located in exons of 263 genes, which account for a median of 12.5 mutated genes per patient (Figure 1A). In the results (Figures 1 and 2), we focused only on genes expressed in our independent cohort of 10 MM patients’ A-PCs (D.Ž., A.A.S., T.Š., and T.J., unpublished data) and thus potentially playing a role in the MRD cells’ biology. From all analyzed MM MRD exomes, 8 genes were mutated in at least 2 patients (Figure 1A), which is consistent with high MM heterogeneity.3-7 Recurrently mutated genes included KRAS, DIS3, TRAF3, OGT, FRG1, UNC13C, FRMPD3, and TRAPPC8. Genes KRAS, DIS3, and TRAF3 are known MM drivers,6 OGT encodes a glycosyltransferase, and O-GlcNAcylation catalyzed by OGT is essential for stabilization of NRF1, a transcription factor of proteasome subunit genes, potentially linked to proteasome inhibitor resistance.11 FRG1 participates in messenger RNA processing, and its decreased expression promotes cancer progression, cell migration, invasion, and angiogenesis.12,13 UNC13C plays a role in vesicle maturation during exocytosis and acts as a tumor suppressor in solid cancers.14 TRAPPC8 is involved in endoplasmic reticulum to Golgi apparatus trafficking15 and was often mutated in solid cancers.16

Mutation profile of the MRD cohort. (A) Recurrently mutated genes and functionally important hits. Patients are depicted as columns; genes are depicted as rows. Previously identified MM associated genes (supplemental Table 3) are highlighted in red rectangles; star symbols indicate potentially actionable targets. Total number of single nucleotide variants (SNVs) in particular patients is given on the top. Driver frequencies from other studies were obtained from 5 papers.3–7 (B-C) Kaplan-Meier curves showing association of PFS with RAS-related pathways. Pathways KRAS.600_UP.V1_UP included synthetic lethal partners of oncogenic KRAS. Ras protein signal transduction pathway is a series of molecular signals within the cell that are mediated by a member of the Ras superfamily of proteins switching to a GTP-bound active state. List of genes included in respective pathways is provided below each graph. FDR, false discovery rate; mut, mutated; wt, wild type.
Mutation profile of the MRD cohort. (A) Recurrently mutated genes and functionally important hits. Patients are depicted as columns; genes are depicted as rows. Previously identified MM associated genes (supplemental Table 3) are highlighted in red rectangles; star symbols indicate potentially actionable targets. Total number of single nucleotide variants (SNVs) in particular patients is given on the top. Driver frequencies from other studies were obtained from 5 papers.3–7 (B-C) Kaplan-Meier curves showing association of PFS with RAS-related pathways. Pathways KRAS.600_UP.V1_UP included synthetic lethal partners of oncogenic KRAS. Ras protein signal transduction pathway is a series of molecular signals within the cell that are mediated by a member of the Ras superfamily of proteins switching to a GTP-bound active state. List of genes included in respective pathways is provided below each graph. FDR, false discovery rate; mut, mutated; wt, wild type.
Comprehensive analysis of driver genes is not feasible in such small MRD cohorts; thus, we compared our results with a list of known drivers and other MM-associated genes to better understand MRD pathogenesis.4-7 Our data set contained 9 MM genes from 8 patients (supplemental Table 3), including KRAS, NRAS, DIS3, TRAF3, SF3B1, NKFBIA, MYC, IKZF3, and BTG1. Interestingly, NRAS mutations were undetectable in a recently published MRD cohort.9 In 12 patients (55%), we did not identify any mutations in the above-mentioned genes, nor did they share some other common mutations; however, several of those patients relapsed. Thus, the malignant characteristics of plasma cells are likely caused by different mechanisms.
To uncover possible common patterns underlying the heterogenous mutation profile in the MRD cohort, we ran pathway analysis for each patient using 7 gene set collections, together including 7627 gene sets (supplemental Table 4). The results showed no pathways significantly enriched and simultaneously commonly mutated among patients (supplemental Table 5). Simple overlap with pathways typical for MM17 revealed mutations in the MAPK18 pathway (7 patients; 32%), NF-κB18 pathway (3 patients; 14%), P53 pathway18 (0 patients), proteasome subunits19 (1 patient; 5%), and cereblon20 (2 patients; 9%) (supplemental Table 6). The most commonly shared pathways with at least 1 affected gene are shown in supplemental Figures 4-11.
To depict novel genes or pathways important for our MM MRD cohort, we performed survival analysis for genes and pathways (supplemental Tables 7-8; supplemental Figures 12-18). Only mutations in FRMPD3 present in 2 patients were associated with shorter PFS (Figure 2B-C). This gene is involved in signal transduction, and it was not found frequently mutated in previous MM studies.3,16 Interestingly, 2 RAS related pathways, “KRAS.600_UP.V1_UP,” including synthetic lethal partners of oncogenic KRAS (FDR 0.015), and “GO_RAS_PROTEIN_SIGNAL_TRANSDUCTION” (FDR 0.049), including 4 and 6 samples, showed significant difference in PFS (Figure 1B-C). This finding is consistent with known frequent impairment of RAS signaling in MM and offers new genes with potential applicability of RAS inhibitors.21
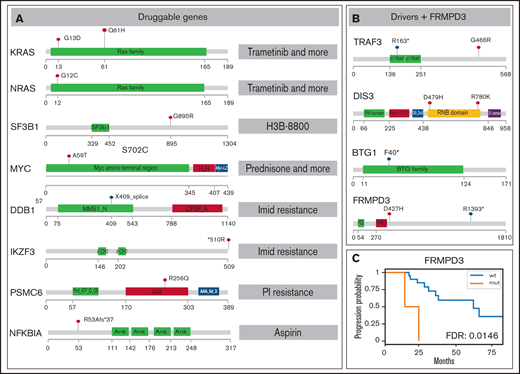
SNV overview of important MM MRD genes. Functional domains are shown for each gene; mutated positions are represented by colored lollipop marks (red, single nucleotide change; blue, splice site/nonsense mutation). (A) Mutations in genes potentially useful in clinics are suggested for preclinical studies. Interacting drugs are given on the right. (B) Genes identified as drivers without assigned treatment and gene FRMPD3 are schematically shown. (C) Kaplan-Meier curve showing gene FRMPD3 that is the only shared gene with significant PFS association. PI, proteasome inhibitor.
SNV overview of important MM MRD genes. Functional domains are shown for each gene; mutated positions are represented by colored lollipop marks (red, single nucleotide change; blue, splice site/nonsense mutation). (A) Mutations in genes potentially useful in clinics are suggested for preclinical studies. Interacting drugs are given on the right. (B) Genes identified as drivers without assigned treatment and gene FRMPD3 are schematically shown. (C) Kaplan-Meier curve showing gene FRMPD3 that is the only shared gene with significant PFS association. PI, proteasome inhibitor.
Our ultimate goal was to identify novel genes that could be targeted by available drugs. Therefore, we annotated the gene set with known pharmacological information. We used The Drug-Gene Interaction Database22 covering a broad spectrum of drugs and diseases, Precision Oncology Knowledge Base23 summarizing druggable mutations, and the literature search to retrieve genes important for myeloma drug resistance. Overall, we have generated a set of mutations in 8 drug-interacting genes with evidence of expression in plasma cells that were mutated in 7 patients (Figure 2A; supplemental Tables 9-11). The most interesting hit was a mutation in the PSMC6 gene (R256Q), coding subunit of 19S proteasome complex, present in a patient treated with bortezomib, who reached VGPR and MRD depth 10e−3 and had one of the shortest PFS (18 months). Mutations in this gene were previously found only in 4 patients in the CoMMpass study,16 but the gene was already shown to be important in bortezomib resistance.24 The effect of the specific substitution R256Q was confirmed by in vitro functional tests (M.Š., K.G., T.J., M.Z., Z.C., and T.Š., manuscript in preparation). Of note, mutation of the BCMA gene, a frequent target of chimeric antigen receptor T-cell immunotherapy, was detected in 1 case. Mutations in this gene could be potentially important for the binding between the BCMA epitope and the antibody. KRAS was the only druggable gene mutated in >1 patient (Figure 1A) and also possessed affected amino acids exactly fitting with the positions for selective treatment (G13D, Q61H).
In summary, we performed whole-exome analysis of somatic variants in a pure population of sorted MM MRD samples with low A-PC infiltration to describe its mutation pattern and to reveal its further utilization in clinics. A limited number of aberrant cells present at the MRD stage and application of whole-genome amplification did not allow the analysis of larger genomic changes than SNVs and short indels. Copy number variant analysis revealed ambiguous results without a clear pattern (supplemental Figure 19). In the heterogeneous spectrum of mutated genes, we did not reveal any unifying feature of MRD clones. In context of that, there is very interesting exposure of the mutation in the proteasome subunit PSMC6 that, despite being scarcely mutated in myeloma population, it was confirmed in cell lines as a bortezomib resistance causing mutation; thus, it may still be useful for the patient’s treatment design. The survival analysis revealed mutations in 2 RAS-associated pathways that were linked to shorter PFS and thus can be important for disease progression. Discovery of new genetic aberrations with a yet unknown role in MM opens new avenues for further investigation in preclinical studies and can provide new targets for treatment upon validation in the laboratory and clinics.
Acknowledgments: This work was supported by the Czech Health Research Council grant (AZV 17-30089A), by the European Regional Development Fund, Project ENOCH (CZ.02.1.010.00.016_0190000868), and by Institutional Development Plan MH CZ-DRO-FNOs/2019 and MH CZ-DRO-FNOs/2020. This work was supported by the Ministry of Education, Youth and Sports of the Czech Republic through the e-INFRA CZ (ID:90140).
Contribution: M.Z. contributed to the research by fluorescence activated cell sorting, DNA processing and amplification, bioinformatic, and by following data analysis and wrote the manuscript; G.S. designed and led the bioinformatic analysis; T.Š. designed the research, consulted results, and wrote the manuscript; V.F. performed the pathway analysis; Z.C., K.G., and L.B. contributed to bone marrow preparation and DNA processing; T.J., L.Ř., and R.B. performed flow cytometry assessment of samples; J. F. and L.Č. contributed with fluorescence activated cell sorting; J.S., J.M., V.M., L.H., L.P., and A.J. provided patient samples for the research; F.K., T.P., J.R.B., M.H., and M.Š. consulted results and contributed to completing the manuscript; R.H. designed the research; D.Ž. and A.A.S. provided expression data; all authors approved the manuscript.
Conflict-of-interest disclosure: R.H. has had a consultant or advisory relationship with Janssen, Amgen, Celgene, AbbVie, BMS, Novartis, PharmaMar, and Takeda; has received honoraria from Janssen, Amgen, Celgene, BMS, PharmaMar, and Takeda; has received research funding from Janssen, Amgen, Celgene, BMS, Novartis, and Takeda. V.M. has had a consultant relationship, received honoraria, and is member of an entity’s Board of Directors or advisory committees in Janssen, Takeda, Amgen, BMS/Celgene, Sanofi, and The Binding Site. The remaining authors declare no competing financial interests.
Correspondence: Roman Hajek, University Hospital Ostrava, Faculty of Medicine, University of Ostrava, 17 listopadu 1790, 708 52 Ostrava, Czech Republic; e-mail: roman.hajek@fno.cz.

