

Key Points
Ibrutinib suppresses CLL cell CTLA4 expression in vitro and in vivo.
CTLA4 expression on CLL is regulated by non-BTKs that differ from T-cell CTLA4 regulation.

Abstract
Cytotoxic T lymphocyte antigen 4 (CTLA4) is a major immune checkpoint and target for cancer immunotherapy. Although originally discovered and primarily studied on T cells, its role on other cell types has also been recognized in recent years. Here we describe an unexpected interaction between ibrutinib (a targeted inhibitor of Bruton tyrosine kinase [BTK]) and CTLA4 expression on malignant chronic lymphocytic leukemia (CLL) cells. Although BTK itself does play a role in CTLA4 expression in CLL, we demonstrate that ibrutinib’s main suppressive effect on CTLA4 protein expression and trafficking occurs through non-BTK targets influenced by this drug. This suppression is not seen in T cells, indicating a different mechanism of CTLA4 regulation in CLL vs T cells. Appreciating this distinct mechanism and the beneficial non-BTK effects of ibrutinib may contribute to understanding the immune benefits of ibrutinib treatment and lead to therapeutic approaches to improve immune function in patients with CLL by suppressing CTLA4 expression.
Introduction
Chronic lymphocytic leukemia (CLL) is the most prevalent lymphoid malignancy in the United States. It causes significant morbidity and mortality, largely through its suppressive effects on the immune system. Treatments have progressed significantly in recent years, but for most patients, CLL is incurable. Throughout the entire disease course of CLL, there is a high risk of morbidity and mortality owing to immune suppression and its associated complications, including infections, which are one of the most common causes of death in patients with CLL.1,2 This risk begins in patients with monoclonal B-cell lymphocytosis (a precursor to CLL), increases during the “watch-and-wait” period before patients require treatment, and is often exacerbated by CLL-directed therapies.1,3,4 In addition to infection, patients with CLL are at a high risk of secondary malignancies, and CLL is associated with decreased survival from these cancers.5-8 These increased risks are largely attributed to ineffective immune surveillance.
The T-cell dysfunction seen in patients with CLL is one of the most apparent complications of this disease. Patients with CLL have changes in both T-cell numbers and function, including decreased CD4/CD8 ratio, Th2 bias, high regulatory T-cell number, a shift from naive toward memory subsets, and an increased expression of inhibitory receptors and exhaustion markers, including PD-1, CTLA4, BTLA, LAG3, CD160, and CD244.9-14 These correlate with decreased T-cell functional activity, including proliferation, formation of the immunological synapse, and cytotoxicity.10 Despite this profound dysfunction, T-cell–mediated immunotherapy is a promising strategy for CLL treatment. Leukemia-reactive T cells can be detected in most patients, and successes with allogeneic stem cell transplant indicate that an effective anti–leukemia immune response is possible.15,16
Cytotoxic T lymphocyte antigen 4 (CTLA4) is an immune checkpoint molecule that is an established target for stimulating anti–cancer immune responses. CTLA4 has most commonly been associated with activated T cells, where it functions to suppress the costimulatory signal from CD80 and CD86. Although less studied in other contexts, CTLA4 expression has been detected on various other cell types, including normal B cells, monocytes, dendritic cells, and various tumor-derived cell lines and tumor cells.17-24 Notably, CTLA4 expression on tumor cells has been associated with poor prognosis in breast, nasopharyngeal, and esophageal carcinomas.25-27 However, its expression patterns and functions are poorly understood in non–T-cell contexts on both normal and tumor cells.
Our group and others have previously demonstrated that CLL tumor cells express CTLA4.28,29 CTLA4 is intracellular in resting CLL cells but is brought to the leukemic cell surface after interaction with a CLL-reactive activated T cell.29 It subsequently functions to remove CD80 and CD86 from the same or neighboring cells through trans-endocytosis, thereby decreasing T-cell activation through loss of the costimulatory signal.29 Furthermore, tumor-specific CTLA4 blockade can slow disease progression in a murine CLL model.29 Nonspecific CTLA4 blockade (affecting both CLL and T cells) can enhance in vitro cytotoxicity using a bispecific antibody.30 Other work has demonstrated that CLL CTLA4 is a key regulator of CLL cell proliferation and survival in vitro.31-33 CTLA4 expression on CLL cells correlates with disease progression and is increased in lymph nodes (where CLL proliferates and interacts with T cells).28,34 In B-cell lymphoma, CTLA4 has also been ascribed multiple functions, including CD86 internalization and proliferative and immunoregulatory signaling, with CTLA4 blockade/silencing slowing tumor growth in vivo.23
Ibrutinib is a small molecule irreversible inhibitor of Bruton tyrosine kinase (BTK) that has become a preferred first-line treatment for CLL based on positive phase 3 studies comparing it with chemoimmunotherapy.35,36 We and others have demonstrated that ibrutinib inhibits CLL proliferation, diminishes bulk disease, and improves immune function in patients with CLL.37,38 It is unknown how much of this effect is attributable to direct effects on CLL cells vs BTK inhibition in other cells or ITK inhibition in T cells (driving them toward T-helper cell 1 polarization).37,39 This immune normalization and proliferation block are seen even in patients with persistent leukemic burden, indicating that the immune/proliferative benefits involve mechanisms beyond the direct reduction of leukemic burden. Previous data demonstrated a decrease in CTLA4 expression on T cells in patients treated with ibrutinib or acalabrutinib (another irreversible BTK inhibitor), but the CLL cell CTLA4 expression was not measured.37 Another recent paper demonstrated a decrease in CTLA4 messenger RNA (mRNA) and intracellular CTLA4 expression in ibrutinib- or acalabrutinib-treated CLL cells.30
Here, we expand our understanding of CTLA4 regulation in CLL and demonstrate that ibrutinib treatment directly suppresses CTLA4 expression on CLL cells while indirectly modulating T-cell CTLA4 expression. We furthermore demonstrate that interferon-γ (IFN-γ, which may also stimulate CLL during a T-cell response) induces CTLA4 mRNA expression, which is likewise inhibited by ibrutinib. Our data provide a key signal toward understanding the regulation of CTLA4 in non–T cells as well as the beneficial immune effects of this important drug.
Methods
Patients and samples
Patient samples were collected after written informed consent from an institutional review board–approved protocol. CLL cells were isolated using RosetteSep Human B Cell Enrichment Cocktail, following the manufacturer’s recommended protocol. Normal donor samples were isolated from buffy coats (Versiti Blood Center of Ohio or American Red Cross) or whole blood collected from healthy volunteers after written informed consent. T cells were isolated from healthy donors using RosetteSep Human T Cell Enrichment Cocktail, RosetteSep Human CD4+ T Cell Enrichment Cocktail, or RosetteSep Human CD8+ T Cell Enrichment Cocktail, following the manufacturer’s recommended protocol. CLL and T-cell purity was confirmed using flow cytometry.
Cell culture
All cells (including primary T cells and CLL cells) were cultured in RPMI-1640 media with 10% fetal bovine serum, 2 mM l-glutamine, 56 U/mL penicillin, and 56 μg/mL streptomycin (all from Gibco). Cells were maintained at 37°C in a 5% CO2 incubator.
CLL–T-cell coculture
Anti-CD3 (10 μg/mL; eBioscience, clone OKT3) in phosphate-buffered saline was adsorbed onto a plate surface for 4+ hours, and then the solution was removed. CLL and T cells were added at a 1:1 ratio, and media were supplemented with 1 μg/mL anti-CD28 (eBioscience, clone CD28.2). Where applicable, drug treatments consisted of 1 μM treatment for 1 hour before washing with fresh media and coculture. IFN-γ stimulation consisted of 100 IU/mL IFN-γ (Peprotech). CTLA4 was measured after 48 hours by flow cytometry when CTLA4 peaks in both CLL and primary B cells.17,29
Flow cytometry
Samples were stained for surface markers (CTLA4, Live/Dead) in phosphate-buffered saline + 10% fetal bovine serum for 20 minutes on ice, permeabilized using BD Cytofix/Cytoperm solution on ice for 20 minutes, and then stained for intracellular markers (CTLA4, CD19, CD3) in BD Perm/Wash buffer on ice for 30 minutes. Samples were run on a Beckman Coulter Gallios, collecting at least 10 000 events of interest, and analyzed using Kaluza software. CTLA4 positivity was determined vs isotype control. The following antibodies were used: CTLA4-PE (BD, clone BNI3), CTLA4-APC (BD, clone BNI3), mouse IgG2a,κ isotype control-PE (BD, clone G155-178), mouse IgG2a,κ isotype control-APC (BD, clone G155-178), CD19-PerCP-Cy5.5 (Biolegend, clone HIB19), CD3-AF700 (BD, clone UCHT1), and Live/Dead Fixable Violet (Invitrogen). CLL cells were gated as live/CD19+/CD3− and then analyzed for CTLA4-PE (surface) and CTLA4-APC (intracellular). Intracellular staining of CD19 and CD3 provided better gating separation of B- vs T-cell populations after activation in coculture.
qPCR
RNA was isolated from CLL cells using TRIzol extraction (Ambion 15596018) following the manufacturer’s recommended protocol. Complementary DNA (cDNA) was produced from 2 μg RNA using 1 μL random hexamers (Invitrogen 100026484), incubating at 65°C for 5 minutes, and then adding 1 μL deoxyribose nucleotide triphosphate mix (Invitrogen 18427-013), 4 μL 5× first-strand buffer (Invitrogen Y02321), 2 μL dithiothreitol (Invitrogen Y00147), 1 μL M-MLV reverse transcriptase (Invitrogen 28025-021), and 1 μL ribonuclease inhibitor (Invitrogen 100000840). cDNA was generated on a thermocycler at 37°C for 5 minutes, 25°C for 10 minutes, 37°C for 50 minutes, and then 70°C for 15 minutes. Quantitative polymerase chain reaction (qPCR) was run using 1 μL cDNA, 10 μL Taqman Fast Advanced Master Mix, and 1 μL Gene Expression Primer (from Applied Biosystems: glyceraldehyde-3-phosphate dehydrogenase [GAPDH] #4352665, TBP #4325803, 18S rRNA #4318839; from Taqman: total CTLA4 #Hs03044418_m1, soluble CTLA4 #Hs03044419_m1, membrane-bound CTLA4 #Hs01011591_m1). The plate was run for 2 minutes at 95°C followed by 40 cycles of 95°C for 1 second and 60°C for 20 seconds. Results were quantified using the ΔΔCt method vs control genes and expressed as fold change vs the unstimulated condition.
Soluble CTLA4 ELISA
Plasma was obtained from whole blood of healthy donors, patients with CLL, or patients with acute myeloid leukemia (AML) by centrifugation. Soluble CTLA4 was measured using LEGEND MAX Human Soluble CTLA-4 enzyme-linked immunosorbent assay (ELISA) Kit (Biolegend), following the manufacturer’s recommended protocol.
Immunoblot analysis
CLL cell lysates were analyzed using standard sodium dodecyl sulfate–polyacrylamide gel electrophoresis methodology, probed with primary antibodies and horseradish peroxidase (HRP) –conjugated secondary antibodies, and visualized with Super Signal West Pico substrate (Thermo) and a Bio-Rad ChemiDoc system. The following antibodies were used: anti-BLK (Cell Signaling Technology 3262), anti-phospho-BLK (Invitrogen PA5-64566), anti-GAPDH (Millipore MAB374), anti-mouse HRP (Bio-Rad 1706516), and anti-rabbit HRP (Cell Signaling Technology 7074).
Statistics
Flow cytometry data were analyzed using Kaluza 2.1. Densitometry analysis was conducted using AlphaView. Data were exported to GraphPad Prism for plotting. Statistical analyses were completed using mixed effects models, analysis of variance (ANOVA), and paired t tests using SAS (SAS, Inc, Cary, NC) at the Center for Biostatistics at the OSU Comprehensive Cancer Center.
Results
Nonactivated CLL cells from peripheral blood express little to no CTLA4 on the cell surface. Therefore, we used coculture with activated T cells to induce CTLA4 surface expression on CLL cells.29 To measure the effects of ibrutinib on this expression, we treated CLL cells with 1 μM ibrutinib or acalabrutinib for 1 hour before washing them with fresh media and adding them to coculture. Acalabrutinib is a more selective, irreversible BTK inhibitor, whereas ibrutinib also inhibits several additional kinases irreversibly.40 CTLA4 expression was measured by flow cytometry after 48 hours. As shown in Figure 1A, ibrutinib treatment suppressed surface and intracellular CTLA4 expression in CLL cells. In contrast, acalabrutinib has no effect on CTLA4 expression in these cells. We additionally measured CTLA4 mRNA expression in unstimulated CLL cells, finding that either ibrutinib or acalabrutinib treatment could inhibit this expression (Figure 1B). Collectively, these data suggest that CTLA4 surface protein (and therefore CTLA4 function) is regulated by non-BTK “alternative-target” effects of ibrutinib, whereas transcription is at least partly BTK signaling-dependent.
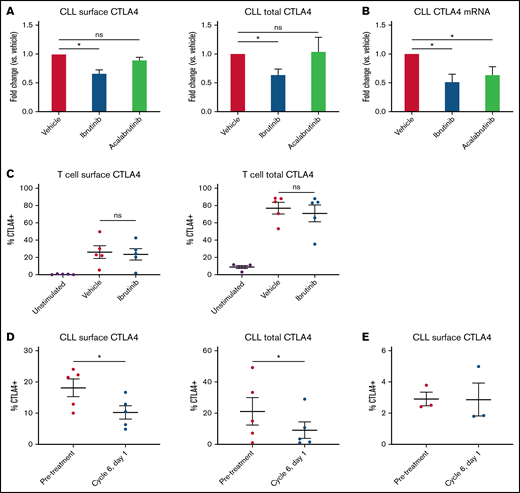
Ibrutinib suppresses CLL-expressed CTLA4. (A) CLL cells were treated with 1 μM ibrutinib or acalabrutinib for 1 hour before washing and coculture with activated T cells. CTLA4 expression on CLL cells was measured 48 hours later by surface and intracellular flow cytometry (n = 10 ibrutinib, 7-8 acalabrutinib). (B) CLL cells were treated with 1 μM ibrutinib or acalabrutinib for 1 hour before washing. CTLA4 mRNA levels were measured 48 hours later by qPCR (n = 18 ibrutinib, 14 acalabrutinib). (C) T cells were treated with 1 μM ibrutinib or acalabrutinib for 1 hour before washing and stimulation with 10 μg/mL anti-CD3 adsorbed onto the plate surface and 1 μg/mL anti-CD28. CTLA4 expression was measured by surface and intracellular flow cytometry after 48 hours of stimulation (n = 5). (D) Matched primary CLL leukemia samples were obtained from patients before treatment and after 5 months of ibrutinib treatment. CLL cells were cocultured with allogeneic activated T cells. CTLA4 expression was measured after 48 hours using surface and intracellular flow cytometry (n = 5). (E) Matched primary CLL leukemia samples were obtained from patients before treatment and after 5 months of acalabrutinib treatment. CLL cells were cocultured with allogeneic activated T cells. CTLA4 expression was measured after 48 hours using surface and intracellular flow cytometry (n = 3). Graphs show mean ± standard error of the mean. *P < .05 by mixed effect modeling (A-C) or paired t-test (D-E). ns, not significant.
Ibrutinib suppresses CLL-expressed CTLA4. (A) CLL cells were treated with 1 μM ibrutinib or acalabrutinib for 1 hour before washing and coculture with activated T cells. CTLA4 expression on CLL cells was measured 48 hours later by surface and intracellular flow cytometry (n = 10 ibrutinib, 7-8 acalabrutinib). (B) CLL cells were treated with 1 μM ibrutinib or acalabrutinib for 1 hour before washing. CTLA4 mRNA levels were measured 48 hours later by qPCR (n = 18 ibrutinib, 14 acalabrutinib). (C) T cells were treated with 1 μM ibrutinib or acalabrutinib for 1 hour before washing and stimulation with 10 μg/mL anti-CD3 adsorbed onto the plate surface and 1 μg/mL anti-CD28. CTLA4 expression was measured by surface and intracellular flow cytometry after 48 hours of stimulation (n = 5). (D) Matched primary CLL leukemia samples were obtained from patients before treatment and after 5 months of ibrutinib treatment. CLL cells were cocultured with allogeneic activated T cells. CTLA4 expression was measured after 48 hours using surface and intracellular flow cytometry (n = 5). (E) Matched primary CLL leukemia samples were obtained from patients before treatment and after 5 months of acalabrutinib treatment. CLL cells were cocultured with allogeneic activated T cells. CTLA4 expression was measured after 48 hours using surface and intracellular flow cytometry (n = 3). Graphs show mean ± standard error of the mean. *P < .05 by mixed effect modeling (A-C) or paired t-test (D-E). ns, not significant.
We then decided to measure the effects of ibrutinib on T-cell CTLA4 expression in vitro, to see if this treatment similarly inhibits T-cell CTLA4 expression. This additionally allowed us to see if the decreased T-cell CTLA4 expression seen in ibrutinib-treated patients is caused by a direct effect of the drug on T cells. We pretreated T cells from healthy donors for 1 hour with 1 μM ibrutinib and then stimulated them with plate-adsorbed anti-CD3 and soluble anti-CD28. Forty-eight hours later, CTLA4 expression on T cells was measured by flow cytometry. In contrast to its in vivo effects on T cells and direct in vitro suppression on CLL cells, in vitro ibrutinib treatment failed to suppress T-cell expression of CTLA4 (Figure 1C). This indicates that the T-cell CTLA4 modulation seen in treated patients is due to indirect effects and points to different regulatory mechanisms in normal T cells vs CLL cells.
To test the in vivo relevance of ibrutinib’s inhibition of leukemic CTLA4 suppression, we used primary CLL leukemia samples from untreated vs ibrutinib-treated patients. Comparing patient samples obtained before treatment vs after 5 months on ibrutinib, we demonstrate that CTLA4 expression on activated CLL cells is suppressed following in vivo treatment (Figure 1D). We followed this with a similar analysis of patients before vs after acalabrutinib treatment, finding that in accordance with its lack of in vitro efficacy, acalabrutinib treatment in vivo did not decrease CTLA4 surface expression by CLL cells, although the pretreatment CTLA4 expression of these samples was already quite low (Figure 1E).
Although activated T cells have previously been demonstrated to stimulate CLL CTLA4 expression, the specific T-cell subset mediating this effect has not yet been elucidated. To understand this, we isolated total T cells, CD4+ helper T cells, and CD8+ cytotoxic T cells from the same healthy donors and compared their ability to stimulate CTLA4 expression on CLL cells. We demonstrate that CD4+ T cells stimulate significantly greater CTLA4 surface expression on CLL cells than total T cells or CD8+ T cells alone, suggesting that CD4+ T cells are the primary cell type stimulating CTLA4 expression on CLL cells (Figure 2). Intracellular CTLA4 expression did not vary by the type of T cells used, although intracellular CTLA4 expression by CLL cells is not reliant on stimulation (Figure 2).
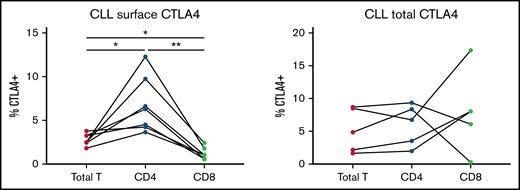
CLL CTLA4 expression is primarily stimulated by CD4 T cells. CLL cells were cocultured with activated total T cells, CD4+ T cells, or CD8+ T cells. CTLA4 expression on CLL cells was measured 48 hours later by surface and intracellular flow cytometry and compared by ANOVA (n = 7 surface, 5 total CTLA4). *P < .05 by ANOVA; **P < .01 by ANOVA.
CLL CTLA4 expression is primarily stimulated by CD4 T cells. CLL cells were cocultured with activated total T cells, CD4+ T cells, or CD8+ T cells. CTLA4 expression on CLL cells was measured 48 hours later by surface and intracellular flow cytometry and compared by ANOVA (n = 7 surface, 5 total CTLA4). *P < .05 by ANOVA; **P < .01 by ANOVA.
Soluble CTLA4 is an alternative isoform of CTLA4 that is secreted rather than bound to the cell membrane. It is produced by alternative splicing that removes the transmembrane domain (exon 3) and produces a frameshift in exon 4.41,42 Although this form of CTLA4 is less understood than membrane-bound CTLA4, it is elevated in various inflammatory conditions and malignancies, such as acute lymphoblastic leukemia, myelodysplastic syndrome, and AML, and has been implicated to be immunosuppressive.43-47 We decided to test if this alternative isoform is expressed by CLL cells in addition to the full-length version of CTLA4. Interestingly, the alternatively spliced soluble CTLA4 transcript is detectable in CLL cells at similar threshold cycle to the full-length transcript, indicating that this transcript is also expressed in CLL cells (Figure 3A). This transcript is suppressed by ibrutinib or acalabrutinib, similar to the total CTLA4 transcript (Figure 3B). However, soluble CTLA4 isoform protein was not detectable in CLL patient plasma by ELISA (Figure 3C). In this experiment, soluble CTLA4 was detectable (≥39 pg/mL) in 0 of 4 healthy donors, 1 of 16 CLL patients, and 3 of 4 AML patient samples. The lack of detectability in serum of this soluble isoform in the large majority of patients with CLL (94%) suggests it may not contribute significantly to CLL-induced T-cell dysfunction.
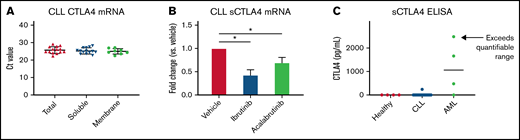
Soluble CTLA4 production by CLL. (A) mRNA of total, soluble, and membrane-bound CTLA4 was measured by qPCR using isoform-specific primers (n = 14 total CTLA4, 14 soluble, 8 membrane-bound). (B) CLL cells were treated with 1 μM ibrutinib or acalabrutinib for 1 hour before washing. sCTLA4 mRNA levels were measured 4 hours later by qPCR (n = 4 ibrutinib, 6 acalabrutinib). (C) Plasma was obtained from healthy donors, patients with CLL, and patients with AML by centrifugation of whole blood, and sCTLA4 levels were measured by ELISA (n = 4 healthy donors, 16 patients with CLL, 4 patients with AML). Graphs show mean ± standard deviation. *P < .05 by mixed effect modeling. Ct, threshold cycle.
Soluble CTLA4 production by CLL. (A) mRNA of total, soluble, and membrane-bound CTLA4 was measured by qPCR using isoform-specific primers (n = 14 total CTLA4, 14 soluble, 8 membrane-bound). (B) CLL cells were treated with 1 μM ibrutinib or acalabrutinib for 1 hour before washing. sCTLA4 mRNA levels were measured 4 hours later by qPCR (n = 4 ibrutinib, 6 acalabrutinib). (C) Plasma was obtained from healthy donors, patients with CLL, and patients with AML by centrifugation of whole blood, and sCTLA4 levels were measured by ELISA (n = 4 healthy donors, 16 patients with CLL, 4 patients with AML). Graphs show mean ± standard deviation. *P < .05 by mixed effect modeling. Ct, threshold cycle.
IFN-γ release from T cells has been shown to upregulate immune checkpoints such as PD-L1 on tumor cells.48 Therefore, we tested the effects of IFN-γ on CLL CTLA4 expression in vitro. We found that IFN-γ did significantly increase CTLA4 mRNA expression in CLL cells, which was prevented by ibrutinib or acalabrutinib treatment (Figure 4A). Although IFN-γ stimulation did somewhat increase CTLA4 surface expression and total CTLA4 protein, its effect was modest in comparison with coculture stimulation (Figure 4B). This indicates that IFN-γ contributes to the early stages of CTLA4 expression (transcription/translation), but that optimal translocation of CTLA4 protein to the leukemia cell surface is induced by other stimuli from T cells.
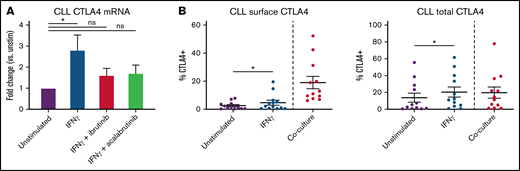
IFN-γ stimulates CTLA4 mRNA production. (A) CLL cells were treated with 1 μM ibrutinib or acalabrutinib for 1 hour before washing and stimulation with 100 IU/mL IFN-γ for 48 hours. Total CTLA4 mRNA was measured by qPCR (n = 10 unstim, 10 IFN-γ, 9 IFN-γ+ibru, 7 IFN-γ+acal). (B) CLL cells were stimulated with 100 IU/mL IFN-γ for 48 hours and then CTLA4 expression was quantified by flow cytometry. The left 2 columns represent paired unstimulated vs IFN-γ–stimulated samples (n = 13), and the right column represents unrelated coculture-stimulated CLL cells (n = 12). Graphs show mean ± standard deviation. *P < .05 by mixed effect modeling (A) or paired t test (B). ns, not significant.
IFN-γ stimulates CTLA4 mRNA production. (A) CLL cells were treated with 1 μM ibrutinib or acalabrutinib for 1 hour before washing and stimulation with 100 IU/mL IFN-γ for 48 hours. Total CTLA4 mRNA was measured by qPCR (n = 10 unstim, 10 IFN-γ, 9 IFN-γ+ibru, 7 IFN-γ+acal). (B) CLL cells were stimulated with 100 IU/mL IFN-γ for 48 hours and then CTLA4 expression was quantified by flow cytometry. The left 2 columns represent paired unstimulated vs IFN-γ–stimulated samples (n = 13), and the right column represents unrelated coculture-stimulated CLL cells (n = 12). Graphs show mean ± standard deviation. *P < .05 by mixed effect modeling (A) or paired t test (B). ns, not significant.
Finally, we conducted a preliminary analysis of the kinase responsible for CTLA4 regulation in CLL cells. CLL cells only express 4 kinases with a cysteine in the correct position for irreversible ibrutinib binding: BTK, TEC, BLK, and JAK3.40 Of these, our experiments with acalabrutinib demonstrate that BTK does not serve this role, and prior papers have demonstrated that TEC and JAK3 do not phosphorylate CTLA4.49,50 Therefore, we tested the effects of ibrutinib on B-lymphocyte kinase (BLK) to see if this may be a potential target. We found that 1-hour treatment with ibrutinib, but not acalabrutinib, did decrease BLK phosphorylation in CLL cells (Figure 5). This suggests that BLK may be the contributing kinase to CTLA4 membrane localization in CLL cells.
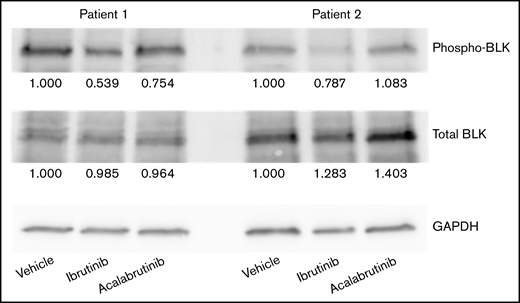
Ibrutinib inhibits BLK phosphorylation. CLL cells were treated with 1 μM ibrutinib or acalabrutinib for 1 hour before lysing, and BLK, phosphorylated BLK, and GAPDH levels were measured by western blotting. Numbers below each band indicate fold change vs the vehicle control, normalized by GAPDH. Figure shows 2 representative patient samples of 3 studied.
Ibrutinib inhibits BLK phosphorylation. CLL cells were treated with 1 μM ibrutinib or acalabrutinib for 1 hour before lysing, and BLK, phosphorylated BLK, and GAPDH levels were measured by western blotting. Numbers below each band indicate fold change vs the vehicle control, normalized by GAPDH. Figure shows 2 representative patient samples of 3 studied.
Discussion
Here we demonstrate a new mechanism of immunomodulation by ibrutinib: decreased CTLA4 on CLL leukemia cells. This effect is likely to be beneficial given this protein’s established roles in immunomodulation and CLL cell survival and proliferation. By showing that ibrutinib does not inhibit T-cell CTLA4 expression, we demonstrate a different regulatory mechanism driving CTLA4 expression in CLL cells vs T cells where we show that the decrease in T-cell CTLA4 expression seen during ibrutinib treatment occurs indirectly. CTLA4 is an established marker of T-cell exhaustion, so the decreased CTLA4 seen in CLL patients’ T cells may not be a marker of direct CTLA4 suppression but rather a marker of improved T-cell function and reversal of the exhausted phenotype seen in patients with CLL.10,37,51 The differential effect of ibrutinib vs acalabrutinib indicates that the suppression of CTLA4 protein expression occurs through a non–BTK-dependent mechanism. Although recent efforts have developed more selective BTK inhibitors in an effort to decrease medication side effects, our results indicate that off-target immunomodulatory effects may be an underappreciated benefit from ibrutinib treatment (although data suggest that more-selective BTK inhibitors do also have beneficial effects on immune function).30,37 Increased selectivity for BTK may instead have the unintended effect of removing the non-BTK benefit of CTLA4 suppression, a protein that both suppresses T cells and promotes malignant B-cell growth and survival.23,29
In T cells, CTLA4 trafficking is regulated by its phosphorylation, which inhibits binding to AP-2 and clathrin-mediated endocytosis.52 Our finding that the kinase inhibitor ibrutinib suppresses CTLA4 surface expression in CLL cells suggests that a similar mechanism may regulate CTLA4 in CLL cells, although the differences in ibrutinib response between CLL and T cells suggest a different kinase is involved. Our data demonstrate that ibrutinib treatment decreases BLK phosphorylation in CLL cells and, especially because the related Src-family kinases Fyn, Lyn, and Lck have been demonstrated to phosphorylate CTLA4 in T cells, it is plausible to speculate that Blk may serve a similar role in CLL, although more research will be needed to test this.49 In T cells, CTLA4 phosphorylation also regulates binding to AP-1 and subsequent lysosomal degradation, which may explain the decreased total CTLA4 protein seen after ibrutinib treatment of CLL cells.52
The soluble CTLA4 isoform is a little-understood aspect of CTLA4 biology, and we were surprised to see its expression also in leukemia cells. The discrepancy between active mRNA expression and absent CTLA4 protein may be related to a lack of translation, protein degradation, sCTLA4 release from the cell, or other factors, but overall, this isoform is unlikely to have a significant impact in CLL because of undetectable levels in patient plasma. IFN-γ has been tied to immune evasion in multiple studies of cancer-immune interactions.48 Although IFN-γ alone was not sufficient to induce significant CTLA4 surface expression in CLL cells, its ability to upregulate CTLA4 mRNA (which was also prevented by ibrutinib treatment) and total protein may contribute indirectly to CLL immune evasion and immunosuppression. The requirement of coculture for effective CTLA4 surface expression implicates membrane receptors, although it is difficult to speculate on a particular ligand given the numerous receptors that communicate between B and T cells; this may be a useful direction for future investigation. Overall, our data point to beneficial other-target effects of ibrutinib, and further development of treatments that target this mechanism may lead to beneficial immunomodulatory effects to trigger anti–leukemia immune responses in CLL.
Acknowledgments
The authors are grateful to the patients who contributed to these studies and OSU Comprehensive Cancer Center Leukemia Tissue Bank Shared Resource (P30CA016058).
This work is supported by National Institutes of Health, National Cancer Institute grant R35 CA197734 (J.C.B.) and the Pelotonia Graduate Student Fellowship (M.Y.).
Authorship
Contribution: M.Y. contributed to the design and conduct of experiments, data analysis, and manuscript writing; J.N. contributed to the design and conduct of experiments; X.M. performed statistical analysis and data interpretation; K.A.R. and J.A.W. contributed to providing patient samples and other materials and preparation of the manuscript; J.C.B. and N.M. designed the study, secured funding, directed the research, and provided overall guidance of the project and manuscript.
Conflict-of-interest disclosure: J.C.B. reports consulting, stock ownership with Vincerx and consulting with AstraZeneca, Syndax, Janssen, Pharmacyclics, Acerta, and Trillium that are outside of the submitted work. The remaining authors declare no competing financial interests.
Correspondence: Natarajan Muthusamy, The Ohio State University, 455H OSUCCC, 410 West 12th Ave, Columbus, OH 43210; e-mail: raj.muthusamy@osumc.edu; and John C. Byrd, Department of Internal Medicine, University of Cincinnati College of Medicine, 231 Albert Sabin Way, ML 0551, Room 6065, Cincinnati, OH 45267; e-mail: byrd2jc@ucmail.uc.edu.

