

Key Points
Inborn errors of immunity-associated lymphomas are characterized by distinct clinical features and genetic signatures.
Both germline and somatic alterations contribute to lymphomagenesis in patients with inborn errors of immunity.

Abstract
Patients with inborn errors of immunity (IEI) have a higher risk of developing cancer, especially lymphoma. However, the molecular basis for IEI-related lymphoma is complex and remains elusive. Here, we perform an in-depth analysis of lymphoma genomes derived from 23 IEI patients. We identified and validated disease-causing or -associated germline mutations in 14 of 23 patients involving ATM, BACH2, BLM, CD70, G6PD, NBN, PIK3CD, PTEN, and TNFRSF13B. Furthermore, we profiled somatic mutations in the lymphoma genome and identified 8 genes that were mutated at a significantly higher level in IEI-associated diffuse large B-cell lymphomas (DLBCLs) than in non-IEI DLBCLs, such as BRCA2, NCOR1, KLF2, FAS, CCND3, and BRWD3. The latter, BRWD3, is furthermore preferentially mutated in tumors of a subgroup of activated phosphoinositide 3-kinase δ syndrome patients. We also identified 5 genomic mutational signatures, including 2 DNA repair deficiency-related signatures, in IEI-associated lymphomas and a strikingly high number of inter- and intrachromosomal structural variants in the tumor genome of a Bloom syndrome patient. In summary, our comprehensive genomic characterization of lymphomas derived from patients with rare genetic disorders expands our understanding of lymphomagenesis and provides new insights for targeted therapy.
Introduction
Lymphomas are a heterogeneous group of malignancies derived from lymphocytes, accounting for ∼5% of cancers in Western countries.1 In the most recent World Health Organization classification, lymphomas are classified according to the cell of origin, with main categories including mature B-cell neoplasms, mature T and NK neoplasms, Hodgkin lymphoma (HL), and posttransplant lymphoproliferative disorders.2 Each type/subtype of lymphoma has distinct histological and immunological characteristics and genetic alterations.2-6 Immunodeficiencies, immune dysregulations, infections, lifestyle, medication, and occupational factors have all been implicated as risk factors for lymphoma.3,7-9 Although genome-wide association studies have identified loci linked to a heightened risk of non-Hodgkin lymphoma (NHL), much remains to be understood regarding how specific genes shape lymphoma development.10,11
Inborn errors of immunity (IEI), also referred to as primary immunodeficiencies, are groups of rare disorders characterized by an increased susceptibility to infections, autoimmunity, and autoinflammation due to genetic defects affecting the development and/or function of the immune system. To date, >450 monogenetic defects have been identified in IEI patients.12,13 Patients with IEI also show an increased risk for the development of cancer, especially lymphoma.14,15 Infection susceptibility may contribute to this increase. However, the mechanism of lymphomagenesis in IEI is more complex and may involve genome instability, mucosal defects permitting chronic antigen stimulation, dysregulated cellular functions, and defective tumor immunosurveillance.16-20
Studies of rare DNA repair disorders, including ataxia-telangiectasia (A-T), Blooms syndrome (BS), and Nijmegen breakage syndrome (NBS), in which the risk of lymphoma is dramatically increased, demonstrated an important role of chromosomal instability in the development of lymphoid malignancy in IEI patients.21,22 Moreover, genetic studies on primary antibody deficiency have provided some insight into the lymphoproliferative inclination of IEI patients. For example, gain-of-function mutations in the lymphocyte activator phosphoinositide 3-kinase δ (PI3Kδ)23 or loss-of-function mutations in T- and B-cell costimulators CD27/CD70 have been identified in primary antibody-deficient patients characterized by benign lymphoproliferation and predisposition to lymphoma.20,24-26 Overall, further genetic study of rare IEI disorders that predispose patients to lymphoma may advance our understanding of lymphomagenesis.
Lymphoid malignancies in patients with IEI are clinically and histologically heterogeneous and often difficult to diagnose.27 Due to their rarity, poor response to standard therapies, and increased risk for treatment-related toxicity, the clinical management of these patients remains highly challenging.21 In this study, using genomic sequencing approaches, we characterized both germline and somatic mutations in 23 IEI patients with rare disorders predisposing toward lymphoma. Further assessment of somatic translocations in the lymphoma genomes of the patients with chromosomal instability syndromes was also performed.
Methods
Detailed methods are available in supplemental Methods.
Patients and samples
Samples from patients were either described previously16,24,28 -30 or newly collected. IEI syndromes and lymphomas were diagnosed by local clinicians, as summarized in supplemental Tables 1 and 2. The Swedish national ethical review board approved the study, which was conducted in accordance with the Declaration of Helsinki.
Next-generation sequencing
Whole-genome sequencing (WGS) and whole-exome sequencing (WES) were performed using either the Illumina HiSeq or NovaSeq or the BGISEQ-500 platform. The depths and coverages of WES/WGS data were summarized in supplemental Table 3. Several samples have lower/nonoptimal coverage/depth, but considering the rarity of the disease (with both IEI and lymphoma), we kept data from all samples for further analysis.
High quality paired-end reads were aligned to the University of California Santa Cruz (UCSC) human reference genome (hg19) using BWA.31 Germline single-nucleotide polymorphisms (SNPs) and insertions and deletions (indels) were identified using the UnifiedGenotyper subtool in GATK32 and SOAPsnp.33 For the paired samples, somatic single nucleotide variants (SNVs) were identified using VarScan,33 and somatic indels were identified using the UnifiedGenotyper subtool in GATK32 and Platypus34 (supplemental Table 4). The copy number variations (CNVs) were identified using CNVkit35 (supplemental Table 5). The GISTIC algorithm was used to infer recurrently amplified or deleted genomic regions.36 Structural variations (SVs) from paired samples were identified by Manta algorithms.37 For tumor-only samples, SNVs and somatic indels were identified by filtering potential germline SNPs and indels. A panel containing the entire coding regions of 715 cancer-related genes (supplemental Table 6) was used to sequence one tumor-only sample.
Identification of disease-causing or -associated germline mutations
Germline variants were filtered according to a previously published strategy.38 Variants predicted to be rare, damaging, and in known IEI genes12,39 or known cancer susceptibility genes40,41 (supplemental Table 7) were reserved as candidates. If a mutation was furthermore indicated as “pathogenic” in ClinVar, the gene containing this mutation was classified as a disease-causing or disease-associated gene (supplemental Table 8).
Identification of somatic mutation targets in lymphoma
Potential lymphoma-associated genes/targets were selected among somatically mutated genes in our IEI lymphoma cohort if they were significantly mutated in previous lymphoma or pancancer studies42-45 or involved in DNA repair processes, which are required for normal B-cell development and genome stability (supplemental Table 9). Genes with “HIGH” gene damage index scores46 and not identified as cancer-associated genes in any previous studies in Integrative OncoGenomics47 were removed. The driver genes were also predicted in silico using OncoDriveFML48 (P < .005, q < 0.25, and mutated in at least 3 patients).
Mutational signature analysis
Identification of replication timing for SV breakpoints
The replication timing of all genomic loci was calculated by averaging wavelet-smoothed Repli-Seq signals across 6 B lymphocyte or leukemia cell lines. High and low values represent early and late replication in the synthesis (S) phase of the cell cycle, respectively.52-54
Statistical approach
Statistical analysis was performed using Fisher's exact test or the Mann-Whitney U test. A P value <.05 was considered statistically significant.
Results
Lymphoma subtypes in IEI patients
Twenty-three patients with previously diagnosed IEI who later developed lymphoma were recruited for this study. These included 18 primary antibody-deficient patients, 14 of whom were diagnosed with common variable immunodeficiency (CVID), 4 with activated PI3Kδ syndrome (APDS), and an additional 5 patients with DNA repair deficiency syndrome (supplemental Table 1).
Fourteen patients were diagnosed with diffuse large B-cell lymphoma (DLBCL). The remaining 9 patients developed marginal zone lymphoma (MZL, n = 3), Epstein-Barr virus (EBV)-associated B- cell lymphoma (n = 1), NHL with no subtype information (n = 1), T-cell lymphoma (n = 2), anaplastic large cell lymphoma (ALCL, n = 1), or HL (n = 1) (Figure 1A; supplemental Table 2). For the patient of PL13, samples were available for both primary (PL13P) and relapsed (PL13R) tumors, and for PL15, samples were collected from both lymph node (PL15L)- and stomach (PL15S)-derived tumors. In all patients, matched nonmalignant samples were analyzed in parallel, except for PL5 and PL21.
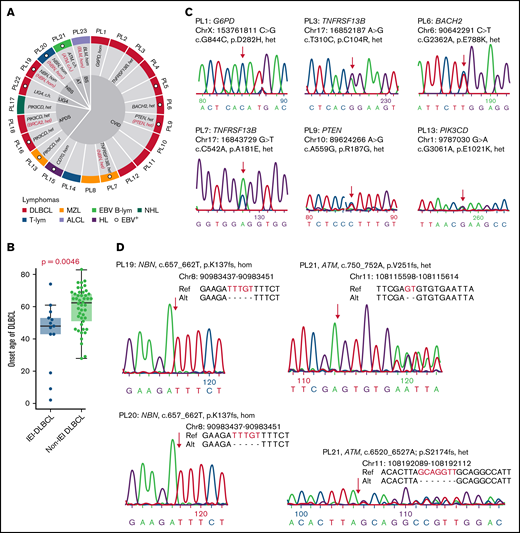
Germline mutations that underlie IEI and lymphoma. (A) Patient information and germline disease-causing or -associated genes are shown. IEI-causing or -associated genes are labeled in black. Cancer-causing or -associated genes are labeled in red. (B) A comparison of DLBCL onset ages between IEI and non-IEI patients. For the non-IEI cohort, HBsAg+ patients were excluded as these patients have previously been shown to have a younger age at diagnosis and distinct mutational patterns.44 (C-D) Newly identified disease-causing or -associated mutations were validated by Sanger sequencing. Alt, alteration; c.h., compound heterozygous; EBV B-lym, EBV-associated B-cell lymphoma; het, heterozygous; hom, homozygous; LIG4, LIG4 syndrome; Ref, reference; T-lym, T-cell lymphoma.
Germline mutations that underlie IEI and lymphoma. (A) Patient information and germline disease-causing or -associated genes are shown. IEI-causing or -associated genes are labeled in black. Cancer-causing or -associated genes are labeled in red. (B) A comparison of DLBCL onset ages between IEI and non-IEI patients. For the non-IEI cohort, HBsAg+ patients were excluded as these patients have previously been shown to have a younger age at diagnosis and distinct mutational patterns.44 (C-D) Newly identified disease-causing or -associated mutations were validated by Sanger sequencing. Alt, alteration; c.h., compound heterozygous; EBV B-lym, EBV-associated B-cell lymphoma; het, heterozygous; hom, homozygous; LIG4, LIG4 syndrome; Ref, reference; T-lym, T-cell lymphoma.
IEI and lymphoma diagnoses were made at average ages of 27 (range, 1-66) and 36 (range, 2-77) years old, respectively. Compared with a previously published non-IEI patient cohort with a representative age distribution,50 DLBCL was diagnosed in these IEI patients at a younger age (41 vs 55 years old, P = .0046, Mann-Whitney U Test, Figure 1B; supplemental Table 10) and at a later stage (10/10 vs 23/46 at stage III/IV, P = .0034, Fisher's exact test). Additionally, the onset age of IEI or lymphoma was significantly younger in patients with APDS or DNA repair deficiency syndromes than in those with CVID (Table 1). The onset age of lymphoma was also significantly younger in the patients with DNA repair deficiency syndromes than in those with APDS, although there was no significant difference in the onset age of IEI between these 2 groups. Additionally, the onset age of malignancies in our cohort was largely similar to that in previous studies (supplemental Table 11). Finally, no significant difference in sex or EBV infection status was observed among patients with CVID, APDS, and DNA repair deficiency syndromes.
Clinical characterization of IEI patients with lymphoma
| CVID | APDS | DNA repair deficiency | P value CVID vs APDS | P value CVID vs DNA repair deficiency | P value APDS vs DNA repair deficiency | |
|---|---|---|---|---|---|---|
| No. of patients | 14 | 4 | 5 | |||
| Sex | 0.2745† | 0.1409† | 1.0000† | |||
| Male (%) | 9 (64%) | 1 (25%) | 1 (20%) | |||
| Female (%) | 5 (36%) | 3 (75%) | 4 (80%) | |||
| EBV status | 1.0000† | 0.1409† | 0.5238† | |||
| Positive (%) | 5 (36%) | 2 (50%) | 4 (80%) | |||
| Negative (%) | 9 (64%) | 2 (50%) | 1 (20%) | |||
| Average IEI onset age (range), y | 42 (5-66)* | 1 (1-2) | 7 (1-14) | 0.0008‡ | 0.0016‡ | 0.4127‡ |
| Average lymphoma onset age (range), y | 50 (17-77) | 19 (16-20) | 11 (2-18) | 0.0046‡ | 0.0003‡ | 0.0317‡ |
| CVID | APDS | DNA repair deficiency | P value CVID vs APDS | P value CVID vs DNA repair deficiency | P value APDS vs DNA repair deficiency | |
|---|---|---|---|---|---|---|
| No. of patients | 14 | 4 | 5 | |||
| Sex | 0.2745† | 0.1409† | 1.0000† | |||
| Male (%) | 9 (64%) | 1 (25%) | 1 (20%) | |||
| Female (%) | 5 (36%) | 3 (75%) | 4 (80%) | |||
| EBV status | 1.0000† | 0.1409† | 0.5238† | |||
| Positive (%) | 5 (36%) | 2 (50%) | 4 (80%) | |||
| Negative (%) | 9 (64%) | 2 (50%) | 1 (20%) | |||
| Average IEI onset age (range), y | 42 (5-66)* | 1 (1-2) | 7 (1-14) | 0.0008‡ | 0.0016‡ | 0.4127‡ |
| Average lymphoma onset age (range), y | 50 (17-77) | 19 (16-20) | 11 (2-18) | 0.0046‡ | 0.0003‡ | 0.0317‡ |
Values are reported as the n (%) or age (minimal-maximal) of patients unless indicated otherwise.
Data are missing for 1 patient in this group.
Fisher’s exact test.
Mann-Whitney U test.
Identification of germline mutations that underlie IEI and lymphoma
Germline nonsilent SNPs and indels were detected for each nonmalignant sample (supplemental Table 3). Candidate disease-causing or -associated mutations for IEI and/or lymphoma were predicted for 21 patients (supplemental Figure 1).38 On average, 2 (range, 1-4) candidates were identified for each sample, and 14 of these were considered as disease-causing or -associated mutations (Figure 1A,C-D; supplemental Table 8).
Disease-causing or -associated mutations were identified in 6 CVID patients. PL1 was a male patient carrying a variant in G6PD (p.D282H) that has been reported in association with partial X-linked G6PD deficiency.55 PL3 and PL7 harbored 2 heterozygous variants in TNFRSF13B (p.C104R and p.A181E, respectively), which have previously been associated with CVID.56,57 In addition, a heterozygous NBN mutation (p.K137fs) was detected in PL7. PL6 harbored a heterozygous BACH2 (p.E788K) variant that leads to BACH2-related immunodeficiency and autoimmunity.58 A heterozygous variant in PTEN (p.R187G), which is known to cause PTEN hamartoma tumor syndrome, was identified in PL9.59 Recently, it was also reported that PTEN loss of function may cause APDS-like immunodeficiency.60 A homozygous frameshift indel in CD70 (p.S84fs) was found in PL15, validating our previous finding.24 No disease-causing/associated genes were identified in the remaining 8 CVID patients; however, several candidate genes were noted, including BACH2 for PL2 and NFKB1 for PL3 (supplemental Table 8).
Three APDS patients were initially diagnosed with hyper– immunoglobulin M syndrome with disease-causing heterozygous variants in PIK3CD (p.C416R for PL16 and PL17 and p.E1021K for PL18).28 The fourth APDS patient, PL13, had onset of IEI-related symptoms in her first year of life, and the diagnosis of APDS was based on the identification of a heterozygous variant in PIK3CD (p.E1021K, Figure 1C). Additionally, PL18 also carried a heterozygous deletion in BRCA2 (p.G602fs), a susceptibility gene for breast, ovarian, and prostate cancers,61,62 as well as lymphomas.63,64
Regarding patients with DNA repair deficiency syndrome, 2 cases were previously published: cases PL22 (ligase IV deficiency) and PL23 (BS).29,30 Three heterozygous variants in LIG4 (p.T9I; p.A3V; c.*3delC) were identified as previously reported for PL22, although none of them met our criteria for disease-causing variants. For PL23, a homozygous disease-causing mutation in BLM (p.Q548X) was identified. Among the other 3 unpublished cases, a homozygous frameshift 5-bp indel in NBN (p.K137fs) were identified in the 2 NBS patients (PL19 and PL20), and 2 heterozygous frameshift indels in ATM (p.V251fs; p.S2174fs) were detected in the A-T patient (PL21), confirming the clinical diagnosis (Figure 1D). BLM, NBN, and ATM genes are also known to be associated with high risks of developing lymphomas.65-67
Somatic mutation landscape of lymphomas in IEI patients
Tumor tissues were sequenced by WES (n = 19, CVID and APDS patients), WGS (n = 5, DNA repair-deficient patients), or targeted sequencing (n = 1, PL16, an APDS patient). A median of 66 (range, 1-848) nonsilent somatic SNVs and 8 (range, 0-90) somatic indels were detected for each lymphoma sample with paired control tissues. The greatest number of nonsilent mutations was observed in 2 samples, PL4 (DLBCL) and PL17 (NHL). PL4 carries a germline heterozygous mutation in ATR, which encodes an important DNA damage factor that has a partially overlapping role with ATM in regulating genome integrity.68 No germline mutations related to DNA repair processes or mutagenesis were identified in PL17. Two additional tumor samples, PL5 (DLBCL) and PL21 (EBV-associated B-cell lymphoma), showed a comparatively high number of mutations; this may be due to potential contamination by germline variants as paired control samples were not available for analysis of these 2 cases.
Potential lymphoma-associated somatic mutations and mutational targets/genes were first identified (supplemental Table 12). Among these, 15 genes were considered recurrently mutated in our cohort (ie, mutated in at least 3 patients) (Figure 2A). Among the recurrently mutated genes, BRWD3 and FAS (apoptosis) were predicted in silico as drivers by OncoDriveFML,48 and 7 additional genes have previously been reported in lymphoma genomic studies,42,44 including KMT2C and KMT2D (epigenetic regulation), CCND3 (cell cycle), BRCA2 (homologous recombination, HR), KLF2 (FoxO signaling), NCOR1 (transcriptional dysregulation in cancer), and TET2. BRWD3 and TET2 do not belong to any pathway in the Kyoto Encyclopedia of Genes and Genomes database69 ; however, biallelic germline loss-of-function TET2 mutations have recently been identified in 3 patients with childhood immunodeficiency and lymphoma.70 Of the remaining 6 recurrently genes that have not been reported in lymphoma studies, PLCG1 is a component of the NF-κB signaling pathway. MSH3 is involved in mismatch repair, and ABCA3 is an adenosine triphosphate–binding cassette transporter. In addition, 37 genes were mutated in at least 2 patients, including many well-known lymphoma-associated genes, such as B2M, CD70, NOTCH2, PIM1, and SGK1, as well as 6 that have been shown to be functional oncogenes in DLBCL by clustered regularly interspaced short palindromic repeats (CRISPR) screens,35,42 including ANKRD17, BCL6, CD22, CREBBP, HIST1H1E, and PIM2 (marked with # in Figure 2A).
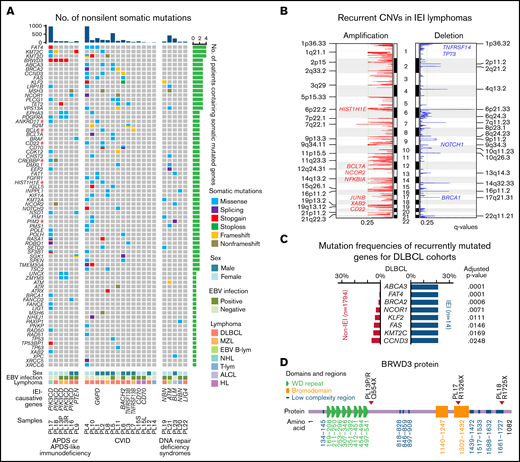
Somatically mutated genes in IEI-related lymphomas. (A) Genes affected by nonsilent, somatically occurring mutations in at least 2 IEI lymphomas or involved in DNA repair pathways are displayed as 3 groups, namely, patients with APDS (PIK3CD mutated) or APDS-like immunodeficiency (PTEN mutated), CVID, and DNA repair deficiency syndromes. Functional DLBCL oncogenes are marked with a red hash symbol. The non-IEI lymphoma data were downloaded from previous studies of DLBCL and MZL.42-44 (B) The recurrent CNVs in IEI lymphomas. (C) Significantly recurrently mutated genes in IEI DLBCL vs non-IEI DLBCLs are shown (adjusted P < .1). For the non-IEI cohort, which consisted of both HBsAg-positive and -negative patients, HBsAg+ patients were excluded. (D) The positions of the 3 BRWD3 stop-gain mutations are shown. #, previously described oncogenes for DLBCL; EBV B-lym, EBV-associated B-cell lymphoma; T-lym, T-cell lymphoma; WD, tryptophan-aspartic acid.
Somatically mutated genes in IEI-related lymphomas. (A) Genes affected by nonsilent, somatically occurring mutations in at least 2 IEI lymphomas or involved in DNA repair pathways are displayed as 3 groups, namely, patients with APDS (PIK3CD mutated) or APDS-like immunodeficiency (PTEN mutated), CVID, and DNA repair deficiency syndromes. Functional DLBCL oncogenes are marked with a red hash symbol. The non-IEI lymphoma data were downloaded from previous studies of DLBCL and MZL.42-44 (B) The recurrent CNVs in IEI lymphomas. (C) Significantly recurrently mutated genes in IEI DLBCL vs non-IEI DLBCLs are shown (adjusted P < .1). For the non-IEI cohort, which consisted of both HBsAg-positive and -negative patients, HBsAg+ patients were excluded. (D) The positions of the 3 BRWD3 stop-gain mutations are shown. #, previously described oncogenes for DLBCL; EBV B-lym, EBV-associated B-cell lymphoma; T-lym, T-cell lymphoma; WD, tryptophan-aspartic acid.
Somatic CNVs were also profiled for each tumor (supplemental Table 5), and significant recurrent CNVs in the IEI cohort were identified by GISTICS. Several regions were significantly amplified in IEI lymphomas, encompassing lymphoma-associated genes such as HIST1H1E, BCL7A, NCOR2, NFKB1A, and CD22. Recurrent focal deletion regions were further detected to affect tumor suppressor genes such as TNFRSF14, TP73, NOTCH1 and BRCA1 (Figure 2B). These changes may further contribute to lymphomagenesis in these IEI patients.
Mutational frequencies in recurrently mutated genes were further compared in DLBCLs from our IEI patients (n = 14) and previously published non-IEI DLBCL cases (n = 1794).42,44,45 Eight genes were found to be mutated more frequently in IEI DLBCLs, including ABCA3, CCND3, BRAC2, NCOR1, KLF2, and FAS (Figure 2C). No recurrent mutated genes were identified in the 3 IEI MZL cases, although KMT2D, the most frequently mutated gene described in non-IEI MZL (25%),71 was mutated in 1 MZL sample (PL17).
Coexistence of somatic BRWD3 mutations and germline PIK3CD mutations
BRWD3 was one of the frequently mutated genes in IEI lymphomas, and mutations in this gene were significantly enriched among APDS patients (3 of 4 patients, P = .0023, Fisher's exact test, Figure 2A). All identified mutations were stop-gain mutations and were located at distinct gene loci (Figure 2D). On the other hand, BRWD3 was not detected in the other IEI lymphoma patients and was rarely detected as being mutated in non-IEI lymphomas (12 in 1794 previously published data, 0.7%; Figure 2B). For PL16, the mutation status of BRWD3 was unclear as this gene was not covered by the targeted sequencing panel (supplemental Table 6).
Mutational signatures of lymphomas in IEI patients
In cancer genomes, somatic mutations may result from multiple mutational processes involving different types of DNA damage and repair.50,72 Mathematical methods have been developed to decipher mutational signatures based on mutational catalogs. To identify mutational processes associated with IEI lymphomas, somatic single-base substitutions (SBSs) from paired-sequenced lymphoma genomes were first cataloged into 96 classes. Five genomic signatures, referred to as Sig.1 to Sig.5, were subsequently identified using SigProfiler49 (Figure 3A; supplemental Figure 2).
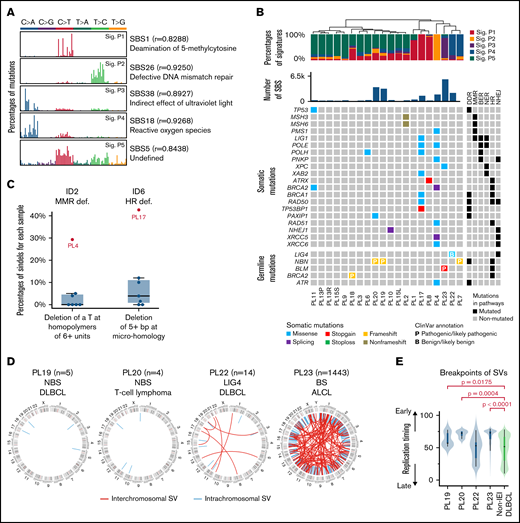
Mutational signatures and structural variants in lymphomas derived from IEI patients. (A) Mutational signatures of IEI-lymphoma genomes. Each signature is displayed according to the 96 substitution classifications defined by the substitution class (shown in different colors) and sequence context immediately 3′ and 5′ to the mutated base. The 96 possible mutated trinucleotides are located on the x-axis, and the frequency of the mutation type is shown on the y-axis. (B) Hierarchical clustering of IEI lymphomas based on the percentages of mutational signatures. DNA repair-related somatic and germline mutations identified in each tumor are shown below. (C) Percentages of specific types of somatic indels in PL4 and PL17. (D) SV of WGS-sequenced IEI lymphomas. (E) Comparison of the location of SV breakpoints at early and late replication sites between IEI and non-IEI patients. BER, base excision repair; DDR, DNA damage response; LIG4, LIG4 syndrome; MMR, mismatch repair; NER, nucleotide excision repair; NHEJ, nonhomologous end joining.
Mutational signatures and structural variants in lymphomas derived from IEI patients. (A) Mutational signatures of IEI-lymphoma genomes. Each signature is displayed according to the 96 substitution classifications defined by the substitution class (shown in different colors) and sequence context immediately 3′ and 5′ to the mutated base. The 96 possible mutated trinucleotides are located on the x-axis, and the frequency of the mutation type is shown on the y-axis. (B) Hierarchical clustering of IEI lymphomas based on the percentages of mutational signatures. DNA repair-related somatic and germline mutations identified in each tumor are shown below. (C) Percentages of specific types of somatic indels in PL4 and PL17. (D) SV of WGS-sequenced IEI lymphomas. (E) Comparison of the location of SV breakpoints at early and late replication sites between IEI and non-IEI patients. BER, base excision repair; DDR, DNA damage response; LIG4, LIG4 syndrome; MMR, mismatch repair; NER, nucleotide excision repair; NHEJ, nonhomologous end joining.
Sig.1 is characterized by dominant C to T transitions at NCG trinucleotides, resembling signature SBS1 in the COSMIC mutational signature database,51 which is a clock-like signature in most cancers and normal cells.73 Sig.2 is similar to the signature SBS26, which is associated with defective DNA MMR. Sig.3 showed high similarity with signature SBS38, which has been predominantly found in UV light-associated melanomas.51 Sig.4 is similar to signature SBS18, which may be associated with damage caused by reactive oxygen species.51 Finally, Sig.5 is similar to signature SBS5, for which the etiology is unclear.
The lymphoma samples were then clustered based on the proportion of different mutational signatures in each sample (Figure 3B). Sample PL4 was characterized with Sig.2, which is associated with a defective MMR and may be attributed to the somatic missense mutation of PMS1 (p.F701L). Sample PL23 (BS patient) showed a preference for Sig.3. This signature is associated with indirect damage due to UV light exposure, which is in accordance with the feature of sun sensitivity among BS patients.74 Finally, samples PL22 and PL7 featured Sig.4, which may be associated with damage by reactive oxygen species.
The somatic indels detected in lymphoma samples were also cataloged into 83 classes using a previously described method51 (supplemental Figure 3). Deletion of a T base at T homopolymers of at least 6 units, resembling the previously described signature ID2, a defective MMR indel signature,51 was observed more frequently in PL4, further supporting MMR deficiency for this tumor. Additionally, deletion of 5+ bp at microhomologies, similar to indel signature ID6, was more frequently observed in PL17, a PIK3CD-mutated APDS patient. As the ID6 signature was suggested to be associated with defective HR,51 the somatic BRCA1 mutation identified in this tumor may thus contribute to the enrichment of this signature. This tumor also carried several additional somatic mutations related to DNA repair, including LIG1, POLE, POLH, XAB2, RAD50, and TP53BP1. The latter encodes 53BP1, which plays a role in determining the balance between HR and nonhomologous end joining and, in its absence, leading to microhomology-based alternative end joining of the DNA double-stranded breaks.75 Thus, the specific somatic indel feature identified in PL17 can be explained by either the BRCA1 mutation and/or the TP53BP1 mutation identified in this tumor sample (Figure 3B).
Somatic structural variants identified in lymphoma genomes from IEI patients
The number of inter- and intrachromosomal SVs was investigated in lymphoma samples from 4 DNA repair-deficient patients with WGS data. A median of 10 (range, 4-1443) SVs were detected (supplemental Table 3). For PL19 and PL20, 2 NBS patients, 5 and 4 intrachromosomal SVs were detected in the tumor samples, respectively. For PL22, the LIG4-deficient patient, 7 inter- and 7 intrachromosomal SVs were detected in the tumors. Finally, for PL23, the BS patient, a strikingly high number of inter- (n = 150) and intra- (n = 1293) chromosomal SVs were observed in tumor cells compared with the other 3 IEI samples (Figure 3D). The higher number of SVs identified in PL23 was supported by the higher number of focal deletions and amplifications predicted by the CNV analysis (supplemental Table 5). Compared with the previously published WGS data from non-IEI DLBCLs,50 where the SVs were called using the same pipeline, the breakpoints in the 2 NBS patients and the BS patient were enriched at early-replication regions (Figure 3E), suggesting that the SVs in these 3 samples were likely generated in the early S phase of the cell cycle. Combined with the mutational signature data, the genome instability of the lymphoma sample from the BS patient is likely due to an inability to repair DNA damage induced during DNA replication, similar to those generated by UV light exposure, due to BLM deficiency. Additionally, the association between breakpoint loci and chromosome fragile sites was also investigated, and there was no significant difference observed between IEI lymphomas and non-IEI DLBCLs.
Discussion
In this study, a comprehensive analysis of both lymphoma genomes and matched germline sequences of 23 IEI patients was performed. Disease-causing or -associated germline mutations in ∼60% of patients were identified and validated, including in ATM, BACH2, BLM, CD70, G6PD, NBN, PIK3CD, PTEN, and TNFRSF13B. By focusing on somatic (tumor-specific) alterations, a unique set of genes was found to be more often mutated in DLBCLs from IEI patients, involving several signaling pathways important for lymphomagenesis, such as DNA repair (BRCA2), cell cycle control (CCDN3), apoptosis (FAS), and NF-κB/BCR signaling (KLF2) pathways.
Approximately half of the lymphomas in our cohort were EBV+ (11/23, 43%; supplemental Table 2), and germline disease-causing or -associated mutations were identified in a majority of these patients, including alterations in BACH2, PTEN, TNFRSF13B, CD70, PIK3CD, NBN, and ATM (Figure 1A). Thus, our result, in agreement with earlier studies, suggests that the inability to control EBV infection is one mechanism underlying lymphomagenesis in IEI patients, and multiple genetic defects may contribute to this etiology. Notably, 43% (6/14) of DLBCLs in our IEI cohort were EBV+ compared with ∼6% in non-IEI cohorts.44 EBV+ DLBCL is now defined as a new subtype of DLBCL in the 2016 World Health Organization lymphoma classification2 and usually develops in people >50 years.76 A previous study based on 47 samples identified frequently mutated genes in EBV+ DLBCLs, including ARID1A, KMT2A/KMT2D, ANKRD11, NOTCH2, CCR6, CCR7, DAPK1, TNFRSF21, and YY1.77 In our IEI patients, a different set of recurrent mutated genes was observed in EBV+ DLBCL genomes, including SGK1, KLF2, and NCOR1. This may suggest a different mutational profile in the EBV+ DLBCL genome from that in IEI patients, but due to the heterogeneity of EBV+ DLBCLs77 and the limited number of samples in our cohort, a larger number of samples will be required to confirm this finding.
A total of 5 mutational signatures were identified in this study, including 2 DNA repair deficiency-related signatures that are seldomly observed in non-IEI patients, suggesting that a defective DNA damage response/repair system is another key mechanism underlying lymphomagenesis in IEI patients. Sig.2, which was enriched in PL4, is associated with MMR deficiency; of all WES-sequenced lymphomas, PL4 showed the highest number of SBSs and a somatic indel pattern related to MMR deficiency. Only 1 mutation in MMR genes, a somatic PMS1 mutation, was detected in PL4. Thus, the genome instability of PL4 was likely due to this somatic DNA repair mutation. In contrast, Sig.3, which was enriched in PL23, a BS patient, is associated with indirect damage from UV light exposure. Extremely high numbers of somatic mutations and interchromosomal and intrachromosomal translocations were also identified in PL23, reflecting a predominant influence from the germline BLM mutation.78 Thus, our data demonstrate that both somatic and germline DNA repair gene mutations may lead to genome instability in IEI lymphomas. The specific DNA repair deficiency in IEI patients, on the other hand, presents an opportunity to enhance immunotherapy. Solid tumors with MMR deficiency and higher tumor mutational burden are associated with better prognosis for anti–PD-1/PD-L1 treatments.79,80 Thus, although most DLBCL patients do not respond well to immune checkpoint inhibitors,81 IEI DLBCL patients with selected DNA repair defects might benefit from such therapeutic strategies.
APDS patients are known to have an increased risk of B-cell lymphoma, and in the largest retrospective cohort, lymphoma was reported in 19 of 179 (10.6%) of these patients (PIK3CD mutated).82 Some of these lymphomas are unrelated to EBV.83 The mechanism underlying the progression from benign lymphoproliferation to malignant B-cell lymphoma remains elusive. In this respect, the coexistence of gain-of-function PIK3CD germline mutations and BRWD3 somatic mutations in these patients might provide new insights. BRWD3, which is located on chromosome X, can be disrupted in B-cell chronic lymphocytic leukemia.36 Germline mutations in BRWD3 have been found in patients with X-linked mental retardation and macrocephaly, and in female carriers, the chromosome carrying the mutation is preferentially inactivated.84 Somatic mutations in BRWD3 were observed more frequently in HPV− head and neck cancers from female vs male patients and were associated with worse 5-year overall survival.85 Of note, in our small cohort of samples, all BRWD3-mutated lymphomas were from female patients. dBRWD3, a homolog of BRWD3 in Drosophila, has been shown to positively regulate the JAK/STAT signaling pathway.86 Thus, the stop codon mutations identified in BRWD3 in APDS lymphomas are likely to dysregulate JAK/STAT signaling and provide a functional “second hit” needed for the malignant transformation of PIK3CD-mutated B cells. It is also of interest to note that we have identified an APDS-like patient with a loss-of-function germline mutation in PTEN, which encodes a protein that antagonizes the activity of the PI3K pathway. Specific PI3Kδ inhibitors that have been approved for the treatment of NHL,87 such as idelalisib, might provide a promising approach for the treatment of lymphoma in APDS and APDS-like patients.
In summary, IEI-associated lymphomas are characterized by distinct clinical and genetic features. Both germline and somatic mutations can contribute to genome instability and lymphomagenesis in IEI patients, and the specific genomic changes identified may provide treatment opportunities for selected patients. Although our study represents the most comprehensive genomic characterization of lymphomas derived from IEI patients, the cohort is small and heterogeneous, consisting of different types of IEIs as well as lymphomas. Our results will need to be validated by larger cohorts of patients, which may require extensive international collaborations.21 Furthermore, in most of the studied IEI-lymphoma cases, only formalin-fixed paraffin-embedded samples are available, which may result in some sequencing artifacts and preclude the possibility of performing transcriptome analysis. Such analysis, especially at the single-cell level,88 may help to further study the malignant cell states and the composition of the tumor microenvironment and may provide new direction for the development of therapies, especially immunotherapies, for this group of rare patients.
Acknowledgments
This work was supported by the Swedish Cancer Society (Cancerfonden), the Swedish Research Council, the Swedish Childhood Cancer fund, the National Natural Science Foundation of China (81670184), the Radiumhemmet research fund, the Center for Innovative Medicine, the Knut and Alice Wallenberg Foundation, the Guangdong Enterprise Key Laboratory of Human Disease Genomics (2020B1212070028), the Shenzhen Engineering Laboratory for Innovative Molecular Diagnostics (DRC-SZ [2016] 884), and the National Institute of Allergy and Infectious Diseases, National Institutes of Health grant AI137183.
Authorship
Contribution: X.Y. designed the study, performed the data analysis, and wrote the manuscript; P.J.M. collected the clinical data and wrote the manuscript; P.J.M., C.W., E.D., S.B., A.P., H.A., H.-B.K., J.K.G., B.G., A.S., A.F., S.D.R., K. Warnatz, and C.C.-R. provided patient samples and clinical data; B.S. confirmed the clinical data; D.L., X.L., H.W., Y.H., S.Z., and K. Wu performed the sequencing data analysis; L.D., Y.G., and Y.W. performed the validation experiments; H.A. and L.H. supervised the data analysis; and Q.P.-H. designed and supervised the study and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Qiang Pan-Hammarström, Department of Biosciences and Nutrition, Karolinska Institutet, Blickagången 16, 14183 Huddinge, Sweden; e-mail: qiang.pan-hammarstrom@ki.se.

