

TO THE EDITOR:
Langerhans cell histiocytosis (LCH) is a histiocytic neoplasm primarily driven by activating mutations of MAPK pathway genes in hematopoietic and myeloid cells.1-4 BRAFV600E mutation accounts for 50% to 60% of disease and is enriched in patients with multisystem LCH involving risk organs and LCH-associated neurodegeneration.5-9 Other mutations include small deletions of BRAF, mutations of MAP2K1 (MEK1), MAPK (ERK), ARAF, RAS, ERBB3, and CSF1R and fusions involving BRAF and other genes.4,10
The high prevalence of BRAFV600E and availability of sensitive allele-specific tests have facilitated several studies on the presence of mutation in the peripheral blood.5,11 ,-14 These have shown that BRAFV600E is rarely detectable at low levels in unsorted peripheral blood mononuclear cells (PBMCs) or cell-free DNA of patients with single-system disease (SS-LCH). In contrast, BRAFV600E is usually present at 1% to 10% in patients with multisystem LCH (MS-LCH), especially with involvement of the risk organs (bone marrow, liver, and spleen; [RO+]). At diagnosis, mutation is primarily found in classical and nonclassical monocytes and myeloid dendritic cells (DC).5,11,14 -16 Plasmacytoid DC and lymphocytes harbor mutation at lower levels in a minority of patients.15-17 Involvement of the hematopoietic stem cell and myeloid lineages correlates with high-risk status and has potential utility as a prognostic marker.5,6,9,11,15,17,18 Molecular analysis of the blood may also provide valuable diagnostic confirmation and assessment of response to treatment.
Interestingly, BRAFV600E has been associated in some pediatric series with more severe multisystem disease, treatment failure, and increased risk of LCH-associated neurodegeneration compared with other mutations.5,6,9,11,18 However, the relative contribution of disease genotype vs the extent of hematopoietic involvement in determining the severity of disease is not known for patients with non-BRAFV600E mutations. Polymerase chain reaction (PCR)–based methods exist for the detection of several other mutations causing LCH. Here we sought to define whether these mutations have similar associations as BRAFV600E and may also be used to inform risk stratification and treatment decisions.
Blood and surplus biopsy material were obtained from patients with LCH referred to local clinics, with institutional review board approval from Baylor College of Medicine or Newcastle and North Tyneside Research Ethics Committee 2.
PBMCs were isolated from whole blood by density centrifugation. Fluorescence-activated cell sorting was performed with the Becton Dickinson FACS Aria Fusion System using antibodies described in supplemental Table 1 and gating shown in supplemental Figure 1. Sorted cells were collected into microfuge tubes containing RPMI with 10% fetal calf serum. Data were analyzed with FlowJo (version 10; Treestar).
Genomic DNA from LCH lesions and PBMC samples was extracted using Qiagen QiAMP DNA Kits and used directly in droplet digital PCR (ddPCR) assays. DNA from sorted PBMC fractions was amplified using Qiagen Repli-G Kits to achieve sufficient quantities for testing.
ddPCR was performed with the BioRad QX200 ddPCR System using ddPCR Supermix for Probes (BioRad). Full details of assay codes are given in supplemental Table 2. The most sensitive threshold of detection was 0.01%, achievable with an input DNA of 200 ng. Graphs and statistics were performed with Prism (version 9; GraphPad).
Twenty patients with non-BRAFV600E mutations who had PBMCs stored at diagnosis were included in the study (Table 1). Eight patients with BRAFV600E mutations were analyzed using the same ddPCR platform for comparison. Probes were first validated on lesional DNA (supplemental Figure 2A). DNA from sorted PBMC subsets was amplified to obtain sufficient input for ddPCR, but this did not skew the reported allele frequency in lesional DNA using any of the probes tested (supplemental Figure 2B).
Summary of patient characteristics
| UPN | Stage | Risk | Age, y at diagnosis | Sex | Site | Gene | Amino acid |
|---|---|---|---|---|---|---|---|
| MB0375 | SS | — | 8.1 | M | Bone (femur) | BRAF | p.N486_P490del |
| MB0781 | SS | — | 3.2 | M | Bone (frontal) | BRAF | p.N486_P490del |
| A2570 | SS | — | 49.0 | M | Mucosae (oral and gingival) | BRAF | p.V600E |
| MB0347 | SS | — | 4.5 | F | Bone (pelvis) | MAP2K1 | p.E102_I103del |
| MB0064 | SS | — | 1.9 | M | Bone (orbit) | MAP2K1 | p.F53_Q58delinsL |
| MB0099 | SS | — | 3.0 | M | Bone (orbit) | MAP2K1 | p.F53_Q58delinsL |
| MB1072 | SS | — | 0.1 | M | Skin | MAP2K1 | p.K57T |
| MB0141 | SS | — | 0.9 | M | Bone (orbit) | MAP2K1 | p.Q58_E62del |
| MB0360 | SS | — | 4.5 | M | Bone (orbit) | MAP2K1 | p.Q58_E62del |
| A8035 | MS | RO− | 39.0 | M | Bone, DI | BRAF | p.V600E |
| MB0225 | MS | RO− | 16.4 | M | Lung, DI | BRAF | p.N486_P490del |
| MB0265 | MS | RO− | 3.5 | F | Skin, bone, lung | BRAF | p.N486_P490del |
| MB0276 | MS | RO− | 40.6 | M | Skin, DI | BRAF | p.N486_P490del |
| MB0352 | MS | RO− | 36.3 | M | Mulitfocal bone (mastoid, mandible, tibia, orbit), skin, kidney, nodes, DI | BRAF | p.V600E |
| MB0392 | MS | RO− | 4.7 | M | Bone, DI, ND-LCH | BRAF | p.V600E |
| MB0507 | MS | RO− | 0.5 | M | Multifocal bone (skull, tibia and pelvis), skin | BRAF | p.V600E |
| MB0165 | MS | RO− | 23.5 | M | Skin, bone, DI | ERBB3 | p.P921Q |
| MB0743 | MS | RO− | 1.4 | M | Multifocal bone (orbit, skull, mandible), skin, nodes | MAP2K1 | p.Q58_E62del |
| MB0381 | MS | RO− | 10.7 | F | Multifocal bone | MAP2K1 | p.Q58_E62del |
| MB0218 | MS | RO− | 2.9 | F | Multifocal bone, skin | MAP2K1 | p.E102_I103del |
| A2582 | MS | RO+ | 4.0 | M | Mucosae (gingival), BM, spleen | BRAF | p.N486_P490del |
| A7517 | MS | RO+ | 49.0 | F | Multifocal bone (scapula and ribs), skin, BM, spleen | BRAF | p.N486_P490del |
| A2951 | MS | RO+ | 73.0 | M | Skin, gut, nodes, BM, spleen | BRAF | p.V600E |
| A7495 | MS | RO+ | 18.0 | M | Bone (vertebrae), lungs, BM, spleen | BRAF | p.V600E |
| MB1035 | MS | RO+ | 0.8 | M | Skin, bone (skull), BM, spleen | BRAF | p.V600E |
| MB0124 | MS | RO+ | 0.5 | F | Bone, liver, nodes, BM, spleen | MAP2K1 | p.Q56P |
| MB0134 | MS | RO+ | 0.5 | F | Skin, bone (temporal skull), liver, nodes, BM, spleen | MAP2K1 | p.Q56P |
| MB0688 | MS | RO+ | 0.8 | M | Nodes, BM, spleen | MAP2K1 | p.Q58_E62del |
| UPN | Stage | Risk | Age, y at diagnosis | Sex | Site | Gene | Amino acid |
|---|---|---|---|---|---|---|---|
| MB0375 | SS | — | 8.1 | M | Bone (femur) | BRAF | p.N486_P490del |
| MB0781 | SS | — | 3.2 | M | Bone (frontal) | BRAF | p.N486_P490del |
| A2570 | SS | — | 49.0 | M | Mucosae (oral and gingival) | BRAF | p.V600E |
| MB0347 | SS | — | 4.5 | F | Bone (pelvis) | MAP2K1 | p.E102_I103del |
| MB0064 | SS | — | 1.9 | M | Bone (orbit) | MAP2K1 | p.F53_Q58delinsL |
| MB0099 | SS | — | 3.0 | M | Bone (orbit) | MAP2K1 | p.F53_Q58delinsL |
| MB1072 | SS | — | 0.1 | M | Skin | MAP2K1 | p.K57T |
| MB0141 | SS | — | 0.9 | M | Bone (orbit) | MAP2K1 | p.Q58_E62del |
| MB0360 | SS | — | 4.5 | M | Bone (orbit) | MAP2K1 | p.Q58_E62del |
| A8035 | MS | RO− | 39.0 | M | Bone, DI | BRAF | p.V600E |
| MB0225 | MS | RO− | 16.4 | M | Lung, DI | BRAF | p.N486_P490del |
| MB0265 | MS | RO− | 3.5 | F | Skin, bone, lung | BRAF | p.N486_P490del |
| MB0276 | MS | RO− | 40.6 | M | Skin, DI | BRAF | p.N486_P490del |
| MB0352 | MS | RO− | 36.3 | M | Mulitfocal bone (mastoid, mandible, tibia, orbit), skin, kidney, nodes, DI | BRAF | p.V600E |
| MB0392 | MS | RO− | 4.7 | M | Bone, DI, ND-LCH | BRAF | p.V600E |
| MB0507 | MS | RO− | 0.5 | M | Multifocal bone (skull, tibia and pelvis), skin | BRAF | p.V600E |
| MB0165 | MS | RO− | 23.5 | M | Skin, bone, DI | ERBB3 | p.P921Q |
| MB0743 | MS | RO− | 1.4 | M | Multifocal bone (orbit, skull, mandible), skin, nodes | MAP2K1 | p.Q58_E62del |
| MB0381 | MS | RO− | 10.7 | F | Multifocal bone | MAP2K1 | p.Q58_E62del |
| MB0218 | MS | RO− | 2.9 | F | Multifocal bone, skin | MAP2K1 | p.E102_I103del |
| A2582 | MS | RO+ | 4.0 | M | Mucosae (gingival), BM, spleen | BRAF | p.N486_P490del |
| A7517 | MS | RO+ | 49.0 | F | Multifocal bone (scapula and ribs), skin, BM, spleen | BRAF | p.N486_P490del |
| A2951 | MS | RO+ | 73.0 | M | Skin, gut, nodes, BM, spleen | BRAF | p.V600E |
| A7495 | MS | RO+ | 18.0 | M | Bone (vertebrae), lungs, BM, spleen | BRAF | p.V600E |
| MB1035 | MS | RO+ | 0.8 | M | Skin, bone (skull), BM, spleen | BRAF | p.V600E |
| MB0124 | MS | RO+ | 0.5 | F | Bone, liver, nodes, BM, spleen | MAP2K1 | p.Q56P |
| MB0134 | MS | RO+ | 0.5 | F | Skin, bone (temporal skull), liver, nodes, BM, spleen | MAP2K1 | p.Q56P |
| MB0688 | MS | RO+ | 0.8 | M | Nodes, BM, spleen | MAP2K1 | p.Q58_E62del |
BM, bone marrow; DI, diabetes insipidus; ND-LCH, neurodegenerative LCH.
An association between mutation burden and clinical risk was observed, similar to that previously reported for BRAFV600E (Figure 1A). All patients with RO+ MS-LCH had detectable mutation in PBMCs, ranging from 0.13% to 6.7%. Mutation <1% was also detectable in 4 of 7 patients with RO− MS-LCH but in none of 6 patients with SS-LCH. A similar distribution was observed for BRAFV600E mutations tested in parallel. This observation supports the model that self-renewing hematopoietic progenitors with MAPK mutations are present only in high-risk LCH and that transient mutation of committed progenitors results in low-risk disease.4 Although we cannot exclude that mutation is present but below the limit of detection in SS-LCH,16 the very different clinical courses of high- and low-risk LCH suggests the influence of biologic rather than simply quantitative differences.
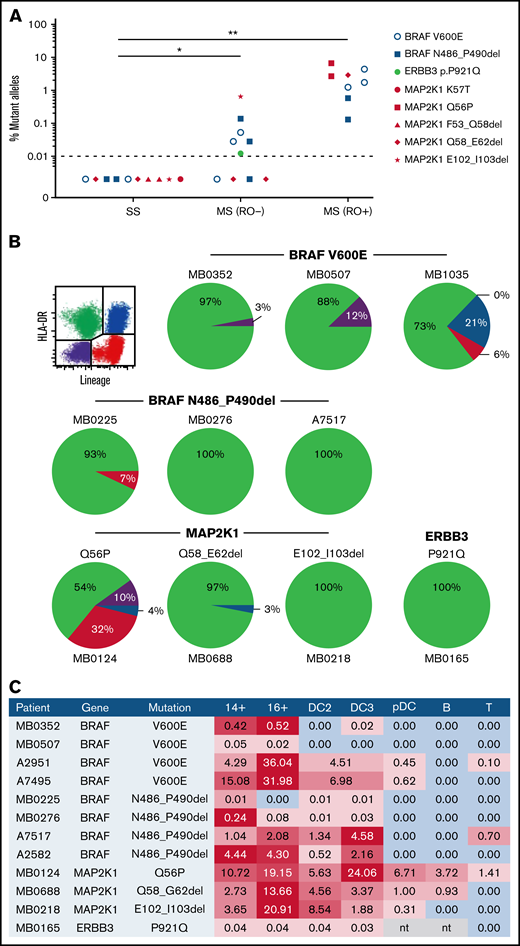
Distritubution of non-BRAFV600E mutations. (A) Allele burden of MAPK mutations at diagnosis according to clinical stage: SS, RO− MS-LCH, or RO+MS-LCH. (B) Distribution of MAPK mutations in PBMCs from patients with MS-LCH sorted into quadrants as shown according to expression of HLA-DR and lineage antigens (CD3, CD19, CD20, and CD56). Area of each quadrant in the pie is proportional to the total number of mutant alleles (mutated allele fraction multiplied by number of cells in the quadrant). (C) Distribution of MAPK mutations among PBMCs from patients with MS-LCH showing the percentage of mutated alleles detected. Red shading indicates positive fractions weighted by abundance within each sample. B, B cell; mono, monocyte; nt, not tested; pDC, plasmacytoid DC. *P < .05, **P < .001 by Mann-Whitney test (excluding patients with BRAFV600E).
Distritubution of non-BRAFV600E mutations. (A) Allele burden of MAPK mutations at diagnosis according to clinical stage: SS, RO− MS-LCH, or RO+MS-LCH. (B) Distribution of MAPK mutations in PBMCs from patients with MS-LCH sorted into quadrants as shown according to expression of HLA-DR and lineage antigens (CD3, CD19, CD20, and CD56). Area of each quadrant in the pie is proportional to the total number of mutant alleles (mutated allele fraction multiplied by number of cells in the quadrant). (C) Distribution of MAPK mutations among PBMCs from patients with MS-LCH showing the percentage of mutated alleles detected. Red shading indicates positive fractions weighted by abundance within each sample. B, B cell; mono, monocyte; nt, not tested; pDC, plasmacytoid DC. *P < .05, **P < .001 by Mann-Whitney test (excluding patients with BRAFV600E).
PBMCs with mutations were then segregated into apposing quadrant gates by HLA-DR and lineage antigen expression (CD3, CD19, CD20, and CD56) to determine the distribution of the total allele burden, derived from the product of mutation fraction and the relative number of cells in each quadrant (Figure 1B). In all patients, a majority of alleles were found in HLA-DR+ lineage− cells containing monocytes and DC. In 3 patients, lower mutation fractions were detectable in HLA-DR+ lineage+ B cells, HLA-DR− lineage+ T and NK cells, or the double-negative fraction, which contains some myeloid progenitors. In terms of the fraction of each lineage bearing mutation, monocytes and myeloid DCs were the highest, notably CD16+ nonclassical monocytes, as previously reported with BRAFV600E (Figure 1C). It was also evident that the recently described DC3 subset of CD1c+ myeloid DCs, which expresses low levels of monocyte antigens CD14 and CD163,19 contains mutation. In 2 patients, this reached a similar level as nonclassical monocytes.
The main implication of this study is that other MAPK pathway mutations are associated with the risk status of LCH in the same manner as BRAFV600E. Testing for non-BRAFV600E mutations requires an array of customized ddPCR reagents, some of which have been designed for this study and are now commercially available. Caveats of the study are that not all indels are amenable to analysis by ddPCR, and gene fusions were not represented, because these require patient-specific probes. However, the data suggest that all causative mutations of RO+ MS-LCH will be present in PBMC at diagnosis. The data show a similar enrichment of mutation in monocytes and myeloid DCs as observed in patients with BRAFV600E.5,11 -14 Without exception, these contain the highest allele burden and account for most of the alleles present in PBMC at diagnosis. This observation corroborates the idea that myeloid cells with mutation play a direct role in the pathogenesis of RO+ MS-LCH,20 a model that has so far been based exclusively on profiling patients with BRAFV600E. These findings support a personalized approach to LCH that may be further refined by defining the relative risks of specific mutations within the group of non-BRAFV600E genotypes. It would also be interesting to extend these observations using cell-free DNA, but stored plasma or serum was not available from this mostly infant population.
Acknowledgments: The authors thank patients and their families for their participation and the organizations providing funding.
This work was supported by Cancer Research UK grant C30484/A21025, the Histiocytosis Association, Histio UK, Bright Red, the HistioCure Foundation, Cookies for Kids Cancer, St Baldrick’s Foundation, the Leukemia and Lymphoma Society, and National Institutes of Health National Cancer Institute grants R01 CA154489, R01 CA154947, and P50 CA126752.
Contribution: P.M. designed research, performed experiments, analyzed data, and wrote the paper; H.A., B.S., and P.S. performed experiments; R.C. contributed patient material; and C.E.A. and M.C. supervised the study, designed research, and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Matthew Collin, Translational and Clinical Research Institute, Newcastle University, Newcastle upon Tyne, United Kingdom; e-mail: matthew.collin@newcastle.ac.uk.

