

TO THE EDITOR:
Myelodysplastic syndrome with ring sideroblasts (MDS-RS) constitutes a unique entity mainly driven by mutations in the SF3B1 splice factor gene. Studies of patients with lower-risk MDS show that distinct MDS stem cells propagate the disease, and we recently identified the hematopoietic stem cell (HSC) as the exclusive cell of origin in SF3B1-mutated MDS-RS.1-3
Although patients with MDS-RS rarely develop leukemia, a large proportion of patients develop progressive anemia, which frequently leads to a chronic need for transfusions.2,4,5 There is limited biological understanding as to why a particular patient shifts from stable to deteriorating erythropoiesis; consequently, specific preemptive strategies are not available. First-line treatment with erythropoietin-stimulating agents (ESAs) leads to clinical response in 45% to 75% of patients, and median duration of response is 1.5 to 3 years.6-8 A large prospective observational study found more durable effects if treatment was initiated before the onset of transfusion need.9 Current predictive models for response to ESAs encompass transfusion intensity (TI), serum erythropoietin (S-EPO), and International Prognostic Scoring System (IPSS) and Revised IPSS (IPSS-R) scores. However, patients with MDS-RS are now diagnosed earlier and rarely display high TI or elevated S-EPO, which limits the usefulness of these models.10,11
Earlier studies indicated that treatment with ESAs in MDS induces erythroid differentiation and reduces bone marrow (BM) apoptosis.12-14 However, our knowledge of how clonal involvement in hematopoietic stem and progenitor cells (HSPCs) correlates with progression of anemia and response to treatment remains limited. Furthermore, it is not known to what degree ESAs target mutant and wild-type (WT) BM cells.
Our study used diagnostic and longitudinal samples from patients with MDS-RS to address these questions. The primary aim was to analyze cellular and genetic parameters associated with treatment need (TN) status. Secondary aims encompassed the assessment of potential effects induced by successful ESA treatment on HSPCs and the status of ESA refractory patients. We selected SF3B1-mutated MDS-RS as a clinical model system because the MDS-RS phenotype is frequently driven by a mutation only in SF3B1, which facilitates clonal tracking.
Fifty-seven patients with SF3B1-mutated MDS-RS who were diagnosed and treated according to European guidelines6 (supplemental Table 1) were included in the study. The criteria for stable asymptomatic anemia (no treatment need [NTN]) and symptomatic anemia with or without transfusions (TN) were stable during the inclusion period (2000 to 2019) (supplemental Table 2). The study was approved by the Swedish Ethical Review Authority 2010/427-31/3 and 2017/4:6, and patients provided informed consent. Clinical and morphological information, including mutational status and SF3B1 variant allele frequency (VAF) in BM aspirates, was obtained at diagnosis using routine next-generation sequencing (NGS); 27 patients had NTN, whereas 30 had symptomatic anemia and received treatment with ESAs. SF3B1 VAF in clinical samples was assessed during response to ESAs in 16 patients (Table 1). HSPC sorting and analysis were performed for 21 patients, as previously described (supplemental Methods),1,3,15 8 of whom underwent paired HSPC analyses encompassing pre-treatment and response to ESAs stages.
Clinical characteristics of patients with MDS-RS and SF3B1 mutation with and without TN and during EPO response
| Variable | At diagnosis | At follow-up | ||||||
|---|---|---|---|---|---|---|---|---|
| NTN | TN (before EPO) | P* | Receiving EPO during response | P† | ||||
| No. (%) | Median (IQR) | No. (%) | Median (IQR) | No. (%) | Median (IQR) | |||
| Total no. of patients | 27 | 30 | 16 | |||||
| Age, y | 72 (67-77) | 76 (69-82) | .26 | |||||
| Sex | .79 | |||||||
| Male | 15 | 12 | ||||||
| Female | 12 | 18 | ||||||
| Blood | ||||||||
| Hemoglobin, g/dL‡ | 10.8 (10.2-11.6) | 9.0 (8.0-10.0) | <.001 | 10.7 (10.3-11.7) | <.001 | |||
| White blood cell count × 109/L | 4.7 (4.1-7.4) | 5.3 (4.8-6.7) | .53 | 4.9 (4.1-6.5) | .50 | |||
| Absolute neutrophil count × 109/L | 2.7 (2.0-5.2) | 3.4 (2.2-4.5) | .94 | 2.7 (1.6-4.1) | .61 | |||
| Platelet count × 109/L | 282 (205-312) | 277 (211-376) | .68 | 305 (238-375) | .79 | |||
| S-EPO, U/L | 32 (16-48) | 40 (29-62) | .13 | NA | NA | |||
| S-ferritin, µg/L | 358 (246-519) | 480 (281-756) | .43 | 472 (264-790) | .54 | |||
| BM | ||||||||
| Cellularity (%) | 60 (45-70) | 60 (50-70) | .58 | 70 (70-90) | .03 | |||
| Blasts (%) | 2.0 (1.0-3.5) | 2.5 (1.5-3.5) | .09 | 1.5 (0-2.5) | .04 | |||
| Erythropoiesis (%) | 34 (15-43) | 28 (18-40) | .30 | 32 (26-55) | .54 | |||
| Ring sideroblasts (%) | 40 (29-53) | 35 (25-52) | .64 | 40 (38-55) | .13 | |||
| IPSS-R (% of known) | ||||||||
| Very low | 17 (63) | 5 (18) | <.001 | NA | NA | |||
| Low | 10 (37) | 21 (75) | NA | |||||
| Intermediate | 0 (0) | 2 (7) | NA | |||||
| Not known/missing cytogenetics | 0 (0) | 2 (NA) | NA | |||||
| Mutations (% of known) | ||||||||
| SF3B1 only | 17 (63) | 16 (53) | .81 | 11 (69) | .47 | |||
| SF3B1 + 1 | 7 (26) | 9 (30) | 2 (12) | |||||
| SF3B1 + ≥2 | 3 (11) | 5 (17) | 3 (19) | |||||
| SF3B1 VAF | 36 (27-43) | 35 (30-39) | .85 | 36 (31-43) | .63 | |||
| Variable | At diagnosis | At follow-up | ||||||
|---|---|---|---|---|---|---|---|---|
| NTN | TN (before EPO) | P* | Receiving EPO during response | P† | ||||
| No. (%) | Median (IQR) | No. (%) | Median (IQR) | No. (%) | Median (IQR) | |||
| Total no. of patients | 27 | 30 | 16 | |||||
| Age, y | 72 (67-77) | 76 (69-82) | .26 | |||||
| Sex | .79 | |||||||
| Male | 15 | 12 | ||||||
| Female | 12 | 18 | ||||||
| Blood | ||||||||
| Hemoglobin, g/dL‡ | 10.8 (10.2-11.6) | 9.0 (8.0-10.0) | <.001 | 10.7 (10.3-11.7) | <.001 | |||
| White blood cell count × 109/L | 4.7 (4.1-7.4) | 5.3 (4.8-6.7) | .53 | 4.9 (4.1-6.5) | .50 | |||
| Absolute neutrophil count × 109/L | 2.7 (2.0-5.2) | 3.4 (2.2-4.5) | .94 | 2.7 (1.6-4.1) | .61 | |||
| Platelet count × 109/L | 282 (205-312) | 277 (211-376) | .68 | 305 (238-375) | .79 | |||
| S-EPO, U/L | 32 (16-48) | 40 (29-62) | .13 | NA | NA | |||
| S-ferritin, µg/L | 358 (246-519) | 480 (281-756) | .43 | 472 (264-790) | .54 | |||
| BM | ||||||||
| Cellularity (%) | 60 (45-70) | 60 (50-70) | .58 | 70 (70-90) | .03 | |||
| Blasts (%) | 2.0 (1.0-3.5) | 2.5 (1.5-3.5) | .09 | 1.5 (0-2.5) | .04 | |||
| Erythropoiesis (%) | 34 (15-43) | 28 (18-40) | .30 | 32 (26-55) | .54 | |||
| Ring sideroblasts (%) | 40 (29-53) | 35 (25-52) | .64 | 40 (38-55) | .13 | |||
| IPSS-R (% of known) | ||||||||
| Very low | 17 (63) | 5 (18) | <.001 | NA | NA | |||
| Low | 10 (37) | 21 (75) | NA | |||||
| Intermediate | 0 (0) | 2 (7) | NA | |||||
| Not known/missing cytogenetics | 0 (0) | 2 (NA) | NA | |||||
| Mutations (% of known) | ||||||||
| SF3B1 only | 17 (63) | 16 (53) | .81 | 11 (69) | .47 | |||
| SF3B1 + 1 | 7 (26) | 9 (30) | 2 (12) | |||||
| SF3B1 + ≥2 | 3 (11) | 5 (17) | 3 (19) | |||||
| SF3B1 VAF | 36 (27-43) | 35 (30-39) | .85 | 36 (31-43) | .63 | |||
Patients were diagnosed with (1) MDS-RS with single lineage dysplasia or multiple lineage dysplasia (MDS-RS-SLD/MLD) or (2) MDS/myeloproliferative neoplasm with ring sideroblasts and thrombocytosis (MDS/MPN-RS-T); all patients had SF3B1 mutation. Disease progression was rarely observed. Transformation to acute myeloid leukemia (AML) occurred in 1 of 57 patients; this patient had mutations in SF3B1, TET2, and RUNX1 at time of diagnosis, responded to EPO for more than 4 years, and developed AML after 7 years. A total of 20 patients were sequenced at 2 time points. In patients re-evaluated while receiving successful EPO treatment (at a median treatment period of 26 months [range, 10-80 months]), only 1 of 9 patients acquired additional mutations (ZRSF2 and TP53 in the same patient). In patients re-sequenced at time of loss of response to EPO, 1 of 11 gained an additional mutation (TET2).
IQR, interquartile range; NA, not applicable.
NTN vs TN (before EPO); Wilcoxon rank sum test for continuous variables, Fisher’s exact test for categorical variables.
Receiving EPO during response vs TN (before EPO).
Hemoglobin for transfused patients entered as 8.0 g/dL.
NGS was performed as described previously.3 Droplet digital polymerase chain reaction (ddPCR) analysis, PCR reaction mixture preparation, droplet generation procedures, and droplet analysis for emission in the HEX or FAM fluorescent signal channels using a QX200 Droplet Digital PCR System (Bio-Rad, Berkeley, CA) and QuantaSoft version 1.15 (Bio-Rad) are described in detail in the supplemental Methods.
Statistical analysis was performed by using GraphPad Prism v. 9. Unpaired testing was performed with the Mann-Whitney U test, and paired testing of values from the same donor at different time points was performed with the Wilcoxon matched-pairs signed-rank test in GraphPad Prism.
Figure 1A and supplemental Figure 1 summarize the experimental design and methods used to investigate HSPC subsets during stable anemia, at the onset of symptomatic anemia, during successful treatment with ESAs, and at ESA refractoriness and chronic transfusion need. Twenty-seven (47%) of 57 newly diagnosed patients with MDS-RS had asymptomatic stable anemia, and no patients had high TI or S-EPO >100 U/L at onset of TN; hence, no patients had a low probability of response according to published predictive models.6
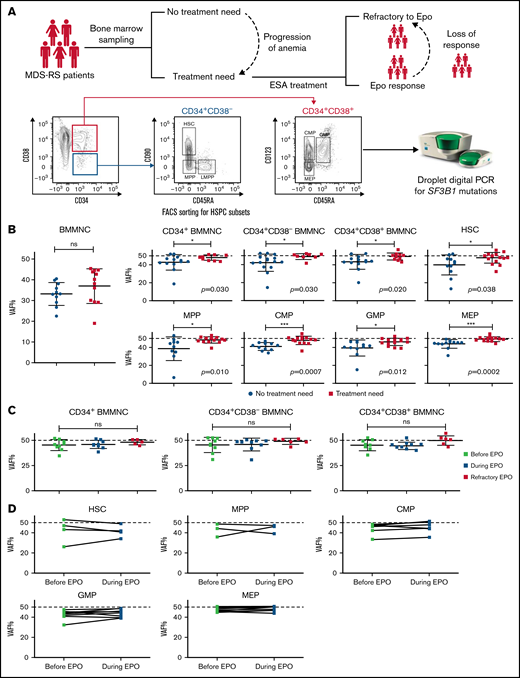
Mutation burden of CD34+ BM populations is significantly lower in patients with MDS-RS with asymptomatic anemia than in patients with TN. (A) Study overview. BM samples from patients with MDS-RS were taken at diagnosis, at onset of TN, during successful ESA treatment, and in a refractory state. Fluorescence-activated cell sorting (FACS) was used to isolate defined HSPC compartments. VAF was determined in all populations using ddPCR. (B) VAF of SF3B1 mutations measured in different HPSC subpopulations in patients with NTN (blue) and in patients with TN (yellow): bulk BM mononuclear cells (BMMNCs; n = 13), CD34+ BMMNCs (n = 12 NTN; n = 10 TN), CD34+CD38– BMMNCs (n = 15 NTN; n = 8 TN), CD34+CD38+ BMMNCs (n = 13 NTN; n = 10 TN), HSCs (n = 11 NTN; n = 14 TN), MPP cells (n = 10 NTN; n = 14 TN), CMP cells (n = 11 NTN; n = 14 TN), GMP cells (n = 11 NTN; n = 14 TN), and MEP cells (n = 12 NTN; n = 14 TN). The numbers of investigated subpopulations in each graph reflect subpopulations with enough cells to allow for statistical comparison. A dotted horizontal line is added at 50% VAF to indicate the theoretical maximum VAF of heterozygous SF3B1 mutation. Error bars indicate the standard deviation. (C) VAF of SF3B1 mutations measured in paired samples from patients with TN: before EPO treatment (n = 9; light green), during successful EPO treatment (n = 9; dark green), and in patients who had become refractory to EPO (n = 6; red). The response samples were taken at a median of 21 months (range, 6-71 months) from start of EPO treatment. (D) VAF analysis of SF3B1 mutations in paired sequential samples from patients (n = 8) taken before and during successful EPO treatment. Analysis was performed in HSCs, and MPP, CMP, GMP, and MEP cells. Significance was tested with paired two-tailed Wilcoxon tests. Mann-Whitney U test: *P < .05; ***P < .001. CMP, common myeloid progenitor; GMP, granulocyte-macrophage progenitor; LMPP, lymphoid-primed multipotent progenitor; MEP, megakaryocyte-erythroid progenitor; MPP, multipotent progenitor; ns, not significant.
Mutation burden of CD34+ BM populations is significantly lower in patients with MDS-RS with asymptomatic anemia than in patients with TN. (A) Study overview. BM samples from patients with MDS-RS were taken at diagnosis, at onset of TN, during successful ESA treatment, and in a refractory state. Fluorescence-activated cell sorting (FACS) was used to isolate defined HSPC compartments. VAF was determined in all populations using ddPCR. (B) VAF of SF3B1 mutations measured in different HPSC subpopulations in patients with NTN (blue) and in patients with TN (yellow): bulk BM mononuclear cells (BMMNCs; n = 13), CD34+ BMMNCs (n = 12 NTN; n = 10 TN), CD34+CD38– BMMNCs (n = 15 NTN; n = 8 TN), CD34+CD38+ BMMNCs (n = 13 NTN; n = 10 TN), HSCs (n = 11 NTN; n = 14 TN), MPP cells (n = 10 NTN; n = 14 TN), CMP cells (n = 11 NTN; n = 14 TN), GMP cells (n = 11 NTN; n = 14 TN), and MEP cells (n = 12 NTN; n = 14 TN). The numbers of investigated subpopulations in each graph reflect subpopulations with enough cells to allow for statistical comparison. A dotted horizontal line is added at 50% VAF to indicate the theoretical maximum VAF of heterozygous SF3B1 mutation. Error bars indicate the standard deviation. (C) VAF of SF3B1 mutations measured in paired samples from patients with TN: before EPO treatment (n = 9; light green), during successful EPO treatment (n = 9; dark green), and in patients who had become refractory to EPO (n = 6; red). The response samples were taken at a median of 21 months (range, 6-71 months) from start of EPO treatment. (D) VAF analysis of SF3B1 mutations in paired sequential samples from patients (n = 8) taken before and during successful EPO treatment. Analysis was performed in HSCs, and MPP, CMP, GMP, and MEP cells. Significance was tested with paired two-tailed Wilcoxon tests. Mann-Whitney U test: *P < .05; ***P < .001. CMP, common myeloid progenitor; GMP, granulocyte-macrophage progenitor; LMPP, lymphoid-primed multipotent progenitor; MEP, megakaryocyte-erythroid progenitor; MPP, multipotent progenitor; ns, not significant.
Except for hemoglobin levels, no clinical parameters, including S-EPO levels, number of co-mutations, or SF3B1 VAF in whole BM were significantly associated with TN (Table 1; supplemental Table 1). An IPSS-R status of “very low” was associated with stable asymptomatic anemia, likely reflecting the trend toward a lower percentage of BM blasts in this group (supplemental Table 2).
We then analyzed the HSPC compartment and measured the VAF in density gradient–separated BM mononuclear cells, total CD34+, and different HSPC subsets (Figure 1B). We previously reported that the frequencies of HSPC populations in MDS-RS are similar to those of normal age-matched controls,3 and here we show that HPSC frequencies do not differ statistically between patients with and without TN (supplemental Figure 1).
The SF3B1 VAF in BM mononuclear cells (Figure 1B) did not differ between patients with TN and those without TN, in line with SF3B1 VAF in BM aspirates (Table 1). However, patients without TN showed significantly lower clonal involvement in CD34+ and CD34+CD38+ sorted progenitors, as well as in immature CD34+CD38– HSCs. These differences were significant in all HSPC subsets (Figure 1B).
We next investigated whether a complete erythroid response to ESAs was associated with an increased fraction of residual SF3B1 WT HSPCs. We obtained paired samples from 8 patients with TN before the start of treatment with ESAs as well as in complete erythroid response (hemoglobin ≥11.5 g/dL without transfusions). Development of SF3B1 clonal involvement in HSPCs during response to ESAs showed an inconsistent pattern with no significant changes in any of the subfractions analyzed. Samples from transfusion-dependent patients obtained during transfusion need after failed treatment with ESAs showed fully clonal CD34+, CD34+CD38+, and CD34+CD38– subpopulations (Figure 1C-D). In addition, clinical and morphological parameters obtained during response did not change in comparison to pretreatment values (Table 1). These data suggest that treatment with ESAs mainly affects more mature cells than HSPCs or acts equally on WT and mutated cells.
Although patients with MDS-RS who have an SF3B1 driver mutation have a high risk of developing anemia and resistant transfusion dependency, this primary patient cohort showed that almost 50% of patients are diagnosed before the onset of symptomatic anemia. It has been demonstrated that the need for transfusions has a negative impact on overall survival and quality of life, and that initiating treatment with ESAs before the onset of a permanent TN is associated with significantly better response rates and duration.16 A clear goal for MDS-RS management is thus to prevent the development of symptomatic anemia and TN.
This is, to our knowledge, the first study assessing HSPC clonality in relation to the degree of anemia and treatment status. We demonstrate that conventional clinical predictors of response do not explain the degree of anemia in newly diagnosed patients. Instead, the prevalence of residual SF3B1-WT HSPCs, although a minority, is associated with freedom from TN. In agreement with this, deteriorating erythropoiesis and, in particular, refractoriness to ESAs is associated with a very high clonal involvement. Our studies indicate that treatment with ESAs does not change this balance within the investigated time frames; however, we suggest that it would be relevant to assess clonal involvement in HSPCs or more practically in total CD34+ BM cells when novel therapeutic approaches are evaluated.17 Our results also have general implications for studies of clonal hematopoiesis, and we propose that protection of residual WT HSPCs is an important goal for this patient cohort.
Acknowledgments: This work was supported by personal grants from the Swedish Cancer Society (I.J.F.H. and P.L.M.) and by project grants from the Wallenberg Foundation, the Swedish Cancer Society, and the Stockholm Cancer Society (E.H.-L.).
Contribution: I.J.F.H., M.D., P.S.W., S.-E.W.J., and E.H.-L. conceived the ideas for this study and designed the research; I.J.F.H. performed fluorescence-activated cell sorting (FACS) and ddPCR experiments; S.V.L. and E.M.E. contributed to FACS experiments; J.V., G.W., M.T., B.S., and M.J. provided samples and clinical information; I.J.F.H. and T.M.-B. analyzed all experimental data; I.J.F.H., T.M.-B., P.L.M., M.D., P.S.W., S.-E.W.J., and E.H.-L. analyzed and interpreted the experimental results; I.J.F.H., T.M.-B., and P.L.M. created the figures and wrote the article with the help of E.H.-L; and all authors commented on and approved the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Eva Hellström-Lindberg, Karolinska Institutet, Center for Hematology and Regenerative Medicine, Department of Medicine Huddinge, Karolinska Institutet, SE-141 83 Huddinge, Sweden; e-mail: eva.hellstrom-lindberg@ki.se.

