

Key Points
CIC-39Na, a store-operated calcium entry inhibitor, reverts thrombocytopenia in KI-STIM1I115F mice and excess bleeding.
KI-STIM1I115F mice are characterized by increases in platelet clearance and in platelet cytosolic basal Ca2+.

Abstract
Store-operated Ca2+-entry is a cellular mechanism that governs the replenishment of intracellular stores of Ca2+ upon depletion caused by the opening of intracellular Ca2+-channels. Gain-of-function mutations of the 2 key proteins of store-operated Ca2+-entry, STIM1 and ORAI1, are associated with several ultra-rare diseases clustered as tubular aggregate myopathies. Our group has previously demonstrated that a mouse model bearing the STIM1 p.I115F mutation recapitulates the main features of the STIM1 gain-of-function disorders: muscle weakness and thrombocytopenia. Similar findings have been found in other mice bearing different mutations on STIM1. At present, no valid treatment is available for these patients. In the present contribution, we report that CIC-39Na, a store-operated Ca2+-entry inhibitor, restores platelet number and counteracts the abnormal bleeding that characterizes these mice. Subtle differences in thrombopoiesis were observed in STIM1 p.I115F mice, but the main difference between wild-type and STIM1 p.I115F mice was in platelet clearance and in the levels of platelet cytosolic basal Ca2+. Both were restored on treatment of animals with CIC-39Na. This finding paves the way to a pharmacological treatment strategy for thrombocytopenia in tubular aggregate myopathy patients.
Introduction
Store-operated Ca2+-Entry (SOCE; also known as capacitative-calcium entry) is a cellular mechanism that governs the replenishment of intracellular stores of Ca2+ on depletion caused by the opening of intracellular Ca2+ channels1,2 (eg, inositol-1,4,5-triphosphate receptors). This mechanism is orchestrated by 2 key proteins:1-3 (1) ORAI (of which there are 3 isoforms), a calcium channel located in the plasma membrane; and (2) STIM (of which there are 2 isoforms), a sensor of calcium located on the endo(sarco)plasmic reticulum membrane that triggers the mechanism when intraluminal Ca2+-concentrations drop.2
Gain-of-function mutations of STIM1 and ORAI1 lead to dominant ultra-rare genetic diseases that historically go under different names (eg, Stormorken syndrome, York syndrome) but that may be clustered as tubular aggregate myopathies4 (TAM). The characteristic histological findings of these disorders are tubular aggregates visible by electron microscopy in skeletal muscle,5 whereas the primary clinical features are muscle weakness and painful cramps.6 More than 20 dominant gain-of-function mutations have been reported on STIM1 and 6 dominant mutations have been reported on ORAI1.4,5 These mutations lead to a wide range of symptom severity, from asymptomatic patients to early-onset muscle weakness and multisystemic diseases.7,8 The full penetrance of the disorder is known as Stormorken syndrome and is characterized by the involvement of multiple districts alongside muscle.9 Recently, Morin et al have performed a systematic review of mutations and a genotype/phenotype correlation.10 Importantly, thrombocytopenia is present in the totality of patients bearing the classical Stormorken STIM1 mutation at position 304 (eg, R304W, R304Q) on the coiled-coiled domain thought to interact with ORAI1 and in about one-half of the patients that bear mutations in luminal EF-hand motifs of STIM1, responsible for sensing intraluminal calcium.4,11 Historically, STIM1 mutations have been described as causing at times solely thrombocytopenia (York syndrome),12,13 but further investigations have revealed that a muscle component is invariantly evident also in these patients.13 Bleeding episodes, likely correlated to thrombocytopenia, have also been described in patients that bear an ORAI1 mutation,14 but might not be as common as for STIM1 mutations because most patients are described as not having thrombocytopenia.10,15 Mutations of 1 of the intraluminal Ca2+-buffering proteins, calsequestrin, have been reported to lead to TAM, but thrombocytopenia has not been reported.16
SOCE has a crucial role in platelet aggregation and thrombus formation.17,18 Indeed, platelets from STIM-knockout animals display a reduced ability to switch to a procoagulant state.18 The presence of platelet dysfunction in TAM patients is therefore not surprising.
Four separate animal models have been developed that recapitulate TAM. In our previous work,19 we characterized a STIM1-mutated mouse model (KI-STIM1I115F) bearing a gain-of-function mutation (p.I115F; located in a EF-hand Ca2+-binding motif on the luminal side of the endoplasmic reticulum) associated in humans with muscle and platelet involvement.12,20 As expected, the p.I115F mutation in mice showed the muscular phenotype of TAM but also presented blood alterations. In particular, alongside an increase in LyC6high, a decrease in LyC6low myeloid cells, and a reduction in splenic NK cells, the KI-STIM1I115F mouse model presented severe thrombocytopenia associated with a pathologically prolonged bleeding time. Two separate mouse lines bearing the p.R304W mutation have been generated and described in the literature.21,22 In the mouse reported by Gamage et al,21 heterozygous R304W mice showed a normal number of platelets, which was associated with a significantly reduced expression of STIM1 and a consequent impairment of activation. Instead, in line with the KI-STIM1I115F mouse, the p.R304W mouse reported by Silva-Rojas et al22 manifested a marked reduction in platelet count. Importantly, Grosse et al23 reported a mouse model developed by random mutations in which an activating mutation on STIM1 (STIM1Sax/+), with no counterpart in human disorders, also resulted in thrombocytopenia and an associated bleeding disorder.
Several SOCE modulators have been postulated as of therapeutic interest in disorders characterized by SOCE overactivation. L-651582 (CAI) was the first inhibitor to enter clinical trials for cancer. Although its development was initially discontinued because of lack of promising efficacy in tumors, it is now gaining novel impulse in gliobastoma.24 The most recent phase 1b trial shows that the drug is tolerable with no dose-limiting toxicities. SOCE inhibitors are being tested also for other conditions, including acute pancreatitis, given the importance that Ca2+ has in triggering exocytosis of zymogen granules.25 In this respect, CM4620 (also known as Auxora26 ) has completed phase 1 trials and is now in phase 2 (NCT03401190) for acute pancreatitis as well as in Phase I/II trials for asparaginase-associated acute pancreatitis, a rare condition triggered by asparaginase treatment (NCT04195347). The same molecule is also in phase 2 for severe COVID-19 pneumonia (NCT04345614) for its potential to reduce pulmonary inflammation.27 A small, phase 2 study has been published on this compound showing that at a dose of 1.0 mg/kg to 1.4 mg/kg for 4 days it is well tolerated.26 A third compound, RP3128, developed for autoimmune disorders, has published data on its first-in-human trial again showing that the drug was well tolerated with no limiting toxicities.28 Importantly, it has been shown that 2 Food and Drug Administration- and European Medicines Agency-approved drugs, teriflunomide and leflunomide, bear at therapeutic doses inhibitory activity on SOCE.29 These emerging data therefore suggest that safety associated with SOCE inhibition is manageable.
Unfortunately, TAM is an orphan disorder with no treatment and no known drug development programs ongoing. In light of this, we decided to characterize the platelet dysfunction in the KI-STIM1I115F mouse model and to evaluate whether platelet number and function could be restored with chronic pharmacological SOCE inhibition. We now report that the thrombocytopenia and increased bleeding can be fully reverted by CIC-39Na, a recently described SOCE inhibitor.30 This provides for the first time encouraging evidence of a pharmacological strategy with small molecules targeting SOCE for TAM.
Methods
KI-STIM1I115F mice maintenance
Procedures were approved by the local animal health and ethical committee (Universita del Piemonte Orientale) and were authorized by the national authority (Istituto Superiore di Sanità; authorization N.98/2021-PR). Care, husbandry, and initial characterization have been described elsewhere.19
Study design of in vivo experiments
Thirty-two 12-week-old mice (16 wild-type [WT] and 16 KI-STIM1I115F) were randomly distributed in 4 groups following sex, weight (27-28 g for WT and 23-24 g for KI-STIM1I115F mice) and platelet number (6.0-6.2 × 108/mL for WT and 2.4-2.7 × 108/mL for KI-STIM1I115F mice) evaluation. Each strain, further divided in 2 subgroups, was anesthetized and Alzet osmotic minipumps (model 1002; volume: 100 μL; infusion rate: 0.25 μL/h; duration: 2 weeks) were filled with saline vehicle or CIC-39Na (60 mg/kg, dissolved in saline) and implanted intraperitoneally. Before implantation, the minipumps were incubated in saline solution at 37°C overnight. Because KI-STIM1I115F mice did not show sex differences in penetrance of platelet disorders,19 both sexes were considered for these in vivo treatments (supplemental Table 1). Each mouse was placed in a separate cage and its health impact was monitored twice a day for all the experimental period (30 days). All experimental procedures are summarized in Figure 1A. The hypothesis of the experiment was at least a 75% increase in platelet number at day 15 with an α error of 5% and a power of 80%. This led to groups of at least 5 animals. Not knowing the safety of the compound, it was decided to raise the group size to 8 animals.
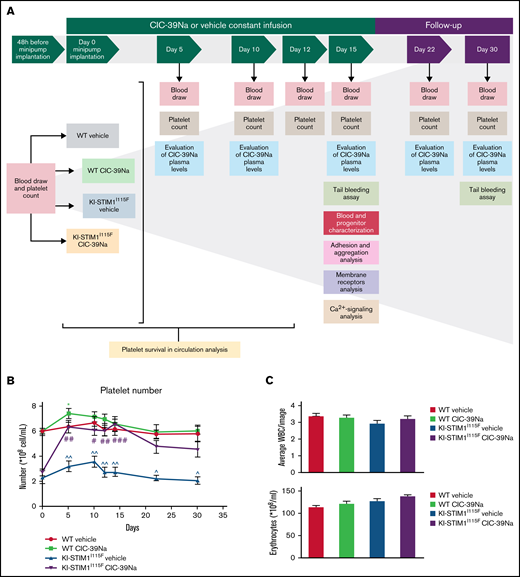
In vivo effect of CIC-39Na in WT and KI-STIM1I115F mice. (A) Study design of in vivo treatments in WT and KI-STIM1I115F mice. (B) Platelet count in the indicated groups. Mann-Whitney U test P value: *0.0334 for each experimental point vs WT vehicle. P value: #0.0154, ##0.016; ###0.0001 for KI-STIM1I115F vehicle vs KI-STIM1I115F CIC-39Na. (C) Circulating number of white blood cells and erythrocytes. (D) Absorbance values relative to blood lost in the indicated groups. Mann-Whitney U test. P value: ^^0.003, ^0.0208 for WT vehicle vs KI-STIM1I115F vehicle and #0.0208 for KI-STIM1I115F vehicle vs KI-STIM1I115F CIC-39Na. (E) Bleeding time in mice treated as indicated. P value: ^0.0208 for WT vehicle vs KI-STIM1I115F vehicle, #0.0304 for KI-STIM1I115F vehicle vs KI-STIM1I115F CIC-39Na. Data reported in panels B-E represent the mean ± SEM of 8 animals for each group from 2 independent experiments.
In vivo effect of CIC-39Na in WT and KI-STIM1I115F mice. (A) Study design of in vivo treatments in WT and KI-STIM1I115F mice. (B) Platelet count in the indicated groups. Mann-Whitney U test P value: *0.0334 for each experimental point vs WT vehicle. P value: #0.0154, ##0.016; ###0.0001 for KI-STIM1I115F vehicle vs KI-STIM1I115F CIC-39Na. (C) Circulating number of white blood cells and erythrocytes. (D) Absorbance values relative to blood lost in the indicated groups. Mann-Whitney U test. P value: ^^0.003, ^0.0208 for WT vehicle vs KI-STIM1I115F vehicle and #0.0208 for KI-STIM1I115F vehicle vs KI-STIM1I115F CIC-39Na. (E) Bleeding time in mice treated as indicated. P value: ^0.0208 for WT vehicle vs KI-STIM1I115F vehicle, #0.0304 for KI-STIM1I115F vehicle vs KI-STIM1I115F CIC-39Na. Data reported in panels B-E represent the mean ± SEM of 8 animals for each group from 2 independent experiments.
Evaluation of CIC-39Na plasma levels
Blood was drawn 24 hours before and 5, 10, 15, 22, and 30 days after minipump implantation. A total of 100 μL of blood extracted from the submandibular vein was mixed with 50 μL of EDTA solution (Sigma- Aldrich, Italy) and processed as follow. Aliquots of plasma samples (50 μL) were diluted by adding 50 μL of IS (BIP29Na final concentration 100 μg/L) and 100 μL of acetonitrile. Samples were homogenized and centrifuged at 13 000g for 10 minutes, and supernatants (5 μL) were injected into LC-ESI-HRMS system. Plasma calibration standards were prepared using the same procedure by spiking the appropriate amount of CIC-39Na DMSO working solutions into blank plasma aliquots. The calibration curves (y = ax + b) were constructed from the peak area vs plasma concentration using the weighted (1/x) linear least-squares regression method (calibration range, 1-10 000 μg/L).
Platelet isolation
Platelet-rich plasma (PRP) was obtained by collecting blood in acid citrate dextrose solution (citric acid 130 mM, Trisodic citrate 152 mM, and glucose 112 mM). Samples were then diluted in Tyrode solution (HEPES 10 mM, NaCl 137 mM, KCl 2.9 mM, NaHCO3 12 mM; pH 7.4) and centrifuged at 200g for 7 minutes with deceleration at 27°C. To isolate platelets, 0.02 U/mL apyrase (A6237), 1 μM of prostaglandin E1 (P5515), and 10 μM of indomethacin (I7378-5G) provided by Sigma Aldrich, were added to PRP before being recentrifuged at 850g for 6 minutes at 27°C and suspended in Tyrode. Platelets were counted as previously described.19
Platelet count in in vivo-treated mice
Blood was drawn 48 hours before and 5, 10, 12, 15, 22, and 30 days after minipump implantation. Platelet count was determined as previously described,19 with the exception that the executer of the platelet count was blinded of group assignment.
Tail bleeding assay and red blood detection
Fifteen and 30 days after minipump implantation, WT and KI-STIM1I115F mice were weighed, anesthetized, and placed in prone position. Two millimeters of the tail tip was amputated and the tail, positioned vertically, was immersed in a falcon containing 14 mL of saline solution at 37°C for 10 minutes. The precise bleeding time was calculated using a stop clock. If bleeding interruptions and resumes occurred, the sum of bleeding times over 10 minutes was calculated. At the end of the 10 minutes, the experiment was stopped and the tail was cauterized to stop bleeding. To determine blood loss, the falcon tube was centrifuged at 1000 RPM for 5 minutes at room temperature (RT), supernatant was removed, and erythrocytes were resuspended in 300 μL of double-distilled water. The optical density of the resulting solution was then determined using a spectrophotometer (Victor3 PerkinElmer) at 570 nm. At the end of experiment, animals were euthanized by cervical dislocation.
White blood cell count
Thin blood smears are prepared on precleaned glass slides from freshly drawn blood from a retro-orbital vein and anticoagulated with EDTA. The slides were allowed to air-dry for 10 to 15 minutes and then fixed with methanol for 1 minute. Blood smear was incubated in methylene blue solution and then in eosin solution for 5 minutes each step. The slides with the fixed blood smear were rinsed with distilled water 5 times and then air dry. The number of white blood cells was counted in 15 random fields using a microscope with a 40× objective.
Confocal microscopy analysis
Total bone marrow was flushed from the tibia of mice using a 1 mL syringe and embedded in OCT. Eight-micrometer sections were fixed in PFA (4%) and washed in phosphate-buffered saline (PBS). Permeabilization was performed in a PBS solution of Triton X-100 (0.1%). Sections were then incubated in bovine serum albumin (BSA) 2% and then with a primary polyclonal rabbit antibody to STIM1 (#4916 from Cell Signaling) for 1 hour. Sections were then incubated with an Alexa-488 conjugated goat anti-rabbit and a rat anti-mouse APC-conjugated CD41 monoclonal antibody. Sections were counterstained with DAPI and mounted in Prolong mounting medium (ThermoFisher). Slides were analyzed with the white light laser confocal microscope Leica TCS SP8 × equipped with a Leica DMI6000B-CS Microscope. Confocal images were obtained using a 40×/1.4 Oil CS2 or 63×/1.4 oil CS2 objectives and analyzed using the LasX software (3.5.5.19976 version).
Hematopoietic progenitors
Bone marrow cell suspensions were stained with a cocktail of PE-conjugated antibodies to Lin positive markers (CD3, CD11b, CD45R, Gr-1, F4/80, CD11c, Ter-119) and with antibodies to CD117 (c-Kit, PE-Cy7), SCA- 1 (APC), CD34 (FITC), and CD16/CD32 (PcP-Cy5). Myeloid progenitors (common myeloid progenitors [CMP], granulocyte-macrophage progenitors [GMP], megakaryocyte-erythroid progenitors [MEP]) were discriminated based on the expression of CD34 and CD16/CD32 within the gate of Lin–CD117+ cells. Common lymphoid progenitors were identified according to the expression of interleukin-7Ra+ (IL-7Ra+) within the Lin− gate. Reagents are shown in supplemental Table 2. Cells were detected using the BD FACSCanto II, BD LSRFortessa or BD Accuri C6 and analyzed with BD FACSDiva and FlowJo (9.3.2) software.
To evaluate LT-HSC (Long-Term Hematopoietic Stem Cells), ST-HSC (Short-Term Hematopoietic Stem Cells), and MMPs (Multipotent progenitors), we used the following Abs: CD34, CD16/32. CD41, Sca-1, CD150, CD48, CD105, and c-Kit all obtained from BD. The classification of the different MPPs was performed according to Pietras et al.31
Bone marrow colony assay
To evaluate colony-forming unit megakaryoctye (CFU Mk), we used a collagen-based assay (Stem Cell) bone marrow (BM) cells were resuspended at a concentration of 2.2 × 106/mL and in Iscove modified Dulbecco medium (without fetal bovine serum). A total of 0.1 mL of the cell suspension was added to 2 mL of MagaCultTM-C serum free media supplemented with human recombinant TPO, human recombinant IL-6 and mouse recombinant IL-3 (Peprotech). All the mixture was then added to 1.2 mL of collagen solution dispensed into a chamber slide (750 μL for each well) and transferred into a 37°C incubator for 8 days. To evaluate CFU-Mk, cultures were dehydrated, fixed in acetone, and stained in an acetylthiocholiniodide solution added with sodium citrate, copper sulfate, and potassium ferricyanide solution, according to manufacturer instructions. Slides were then fixed in ethanol and counterstained with the Harry’s hematoxylin (Sigma Aldrich). Megakaryocyte colonies (brown) were imaged and counted under an inverted microscope (Evos, Life Technologies).
Flow cytometry
Whole blood of WT and KI-STIM1I115F mice were collected from the abdominal vena cava in syringes containing 3.8% sodium citrate as anticoagulant. Blood was mixed with HEPES buffer, anti-CD41-BV510, anti-CD62P-BV421 (BD Biosciences), and JON/A-PE from Emfret Analytics. Samples of murine blood were stimulated with 50 ng/mL and 100 ng/mL convulxin, 0.5 µM, and 2 µM U46619 for 10 minutes at 37°C in the dark. The reaction was stopped adding 0.5% PFA and platelet activation was analyzed by flow cytometry using a BD FACS Lyric instrument equipped with BD FACSuite Flow Cytometry Software (BD Biosciences). Data analysis was performed using FlowJo v10 software (FlowJo LLC).
Adhesion
Washed platelets (2 × 108 cells/mL) were plated for 15 or 30 minutes on 13-mm glass coverslips coated with type I collagen or 0.5% BSA/PBS for 2 hours at 37°C. Platelets were washed twice with PBS and then fixed with 3% paraformaldehyde-4% sucrose in PBS for 5 minutes at RT, permeabilized by 0.2% Triton-X100 for 5 minutes, and saturated with 2% BSA in PBS for 15 minutes. Platelets were stained with Alexa488-phalloidin (#8878; Cell Signaling Technology) for 30 minutes in the dark. Coverslips were washed twice with 0.2% BSA in PBS and mounted with 20% MowiolR 4-88 (81381) and 2.5% Dabco (290734; Sigma Aldrich) in PBS. Platelets were viewed on a confocal microscope (Leica TCS SP2) and digital images (63×) were acquired. The number of adherent cells and the average cell area (index of platelet spreading) were determined using a purposely developed script suitable for Image-J 2.1.0/1.53c.
Thrombus formation under flow
Glass coverslips were coated with nonfibrillar type I collagen (100 μg/mL) and then blocked with 1% BSA in PBS, pH 7.4. The coverslips were mounted in RC-31 parallel-plate flow chamber (50 μm deep from Warner Instruments) and rinsed with a conditioning buffer (HEPES buffer supplemented with 2 mM CaCl2, 2 mM MgCl2, 5.5 mM glucose, 0,1% BSA, and 1 U/mL of heparin). The flow chamber was then connected to a pump system (Harvard Apparatus PHD 2000) and positioned under a fluorescence microscope. Before the perfusion, platelet count in WT and KI-STIM1I115F was determined, and the samples were brought to an identical platelet concentration. WT or KI-STIM1I115F murine blood anticoagulated with 40 μM PPACK and 5 U/mL heparin was then preincubated with 3 μg/mL of carboxyfluorescein diacetate succinimidyl ester for 15 minutes at 37°C and eventually flowed for 5 minutes at 1000 s-1. After the perfusion, the flow chamber was thoroughly rinsed with conditioning buffer, and at least 50 randomly taken fluorescence microscopic images were collected after 2 minutes and 10 minutes of rinse. Collected images were analyzed by ImageJ 1.51j8 software and the percentage of platelet covered area was determined. This value represents a cumulative index of a complex process involving platelet adhesion, spreading, and formation of aggregates on the collagen-coated surface; it is generally referred to as thrombus formation.
Aggregation
Murine platelets were prepared starting from blood collected from the abdominal vena cava in syringes containing ACD (152 mM sodium citrate, 130 mM citric acid, 112 mM glucose) and 3.8% sodium citrate (2:1) as anticoagulant. Briefly, anticoagulated blood was diluted with HEPES buffer (10 mM HEPES, 137 mM NaCl, 2.9 mM KCl, 12 mM NaHCO3, pH 7.4) and centrifuged for 10 minutes at 120g to obtain PRP. PRP was then transferred to new tubes and the remaining red blood cells were diluted with HEPES buffer and centrifuged again at 120g for 7 minutes. The upper phase was added to the previously collected PRP and 0.02 U/mL apyrase grade III (Sigma Aldrich), and 1 μM PGE1 (Santa Cruz Biotechnology) were added before centrifugation at 550g for 8 minutes. The supernatant platelet-poor plasma was removed and the platelet pellet was washed in PIPES buffer (20 mM PIPES, 136 mM NaCl, pH 6.5) and centrifuged at 550g for 8 minutes. Platelet pellet was finally gently resuspended in 300 μL of HEPES buffer, platelets were counted, and the platelet count was adjusted to 108/mL. Upon addition of 5.5 mM glucose, cells were allowed to rest for 30 minutes at 37°C. For aggregation studies, washed platelets from WT and KI-STIM1I115F mice (300 µL, 108 platelets/mL) were stimulated under constant stirring with 50 ng/mL and 100 ng/mL convulxin (provided by Dr. K. J. Clemetson, Theodore Kocher Institute, University of Berne, Switzerland), or 1 µM U46619 (Sigma Aldrich) in the presence of 1 mM calcium in a Chronolog Aggregometer (Mascia Brunelli). Platelet aggregation was monitored continuously over 5 minutes. The analysis was performed by Chronolog AGGRO/LINK Control Software version 4.75.
Western blot
Washed platelets were lysed in RIPA buffer. Lysates were clarified by centrifugation at 14 000g for 15 minutes at 4°C and 30 μg of protein from each sample were loaded on sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels. The antibodies against STIM1 (Cell signaling, 4916S), ORAI1 (Invitrogen, PA1-74181), and Actin (Millipore, MAB1501) were prepared in TRIS-buffered saline solution containing 0.1% Tween-20 (T-TBS) and supplemented with 3% to 5% nonfat dried milk or 3% to 5% BSA, according to manufacturer instructions.
Calcium imaging experiments
Washed platelets were loaded with 5 μM Fura-2 am (Life technologies, Italy) for at least 45 minutes and plated onto poly-l-lysine (P4832, Sigma-Aldrich)-coated glass coverslips for at least 30 minutes at RT. Store-operated-Ca2+-entry and cytosolic Ca2+ measurements following the stimulation with 100 ng/mL convulxin or 1 µM U46619 were performed as previously described.32
Clearance
Platelets were labeled in vivo with tail vein injection of 5 μg Alexa Fluor 488–conjugated anti-GPIX antibody in 100 μL of PBS. Diluted whole blood (50 μL) was incubated with 5 μL PE-Cy5–conjugated anti-GPIb antibody for 10 minutes at RT. The Alexa Fluor 488–PE positive platelet population was determined by flow cytometry.
Statistical analysis
The normality of data distributions was assessed using Shapiro-Wilk or Kolmogorov-Smirnov normality tests. Data are presented as mean ± standard error of the mean or median ± IQR. Parametric (unpaired t-test and One-way analysis of variance followed by Tukey post hoc) or nonparametric (Mann-Whitney U test and 1-way Kruskal-Wallis H test followed by Dunn post hoc) statistical analysis were used for comparisons of data. All statistical assessments were 2-sided and a value of P < .05 was considered statistically significant. Statistical analyses were performed using GraphPad Prism software (GraphPad Software, Inc., USA).
Results and discussion
SOCE inhibition leads to reversal of the platelet defects in KI-STIM1I115F mice
We have previously shown that KI-STIM1I115F mice are characterized by thrombocytopenia, which is also a feature of many patients with TAM.4,10 Given the availability of SOCE inhibitors, we speculated that their use could restore number and function of platelets in KI-STIM1I115F mice. To explore this, we made use of CIC-39Na, which we have previously shown to inhibit SOCE in HEK cells with an 50% inhibitory concentration (IC50) of around 800 nM and to possess a good aqueous solubility (in the manuscript, the compound was referenced as compound 3430 ). To evaluate whether KI-STIM1I115F was sensitive to this compound, we used myotubes from 6-month-old KI-STIM1I115F mice. Indeed, this was the case with an IC50 of around 1 µM (SOCE inhibition at 1 µM = 49.8% ± 3.6 and at 3 µM = 77.3% ± 4.8).
We next implanted intraperitoneally minipumps that allowed to have a constant infusion of CIC-39Na for about 2 weeks, thereby reducing animal manipulation. Preliminary experiments showed that a dose of 60 mg/kg/d intraperitoneally for 2 weeks was tolerated by mice and we therefore aimed to deliver a similar amount per day via infusion. To monitor pump efficiency, blood samples were taken from treated WT and KI-STIM1I115F mice at day 5, 10, 15, 22, and 30 (Figure 1A). The average plasma drug concentration found when pooling data from days 5, 10, and 15 (in which the drug is supposed to be released at similar levels) was 251 µg/L corresponding to 625 ± 89 nM, slightly below the estimated IC50. Although this concentration is below the IC50, we reckoned that to circumvent the gain-of-function mutation a modest inhibition was required because a large inhibition is likely to affect directly platelet function, as shown in studies in mice in which STIM1 was ablated17 and in patients with loss-of-function mutations.33 Animals were kept for a further 15 days and, during this time, drug concentrations dropped time dependently to 97 ± 48 nM at day 22 and 9.7 ± 2.1 nM at day 30, supporting that pumps deliver the drug for approximately 2 weeks. Treatments were well tolerated, and animals did not show any visible sign of suffering. To strengthen this, usually KI-STIM1I115F mice are characterized by small weight, whereas in these experiments treated animals gained weight compared with their controls (supplemental Figure 1).
As shown in Figure 1B, treatment of WT mice with CIC-39Na led to a small but significant increase in platelet count at day 5 that recovered in the following determinations. Strikingly, treatment of KI-STIM1I115F mice led to a threefold increase in platelet number at 5 days, which was maintained up to 15 days. The effect of CIC-39Na was specific, as no changes were observed in white blood cells or in erythrocytes (Figure 1C). The increase in platelets was paralleled by a significant decrease in blood loss (Figure 1D) and bleeding time (Figure 1E) after incision of the tail compared with control mice. After 30 days, in which drug release from the pump was significantly reduced, platelet number showed a significant drop from the peak (Figure 1B), and, similarly, bleeding time (Figure 1D) and blood loss (Figure 1E) were increased to levels comparable to untreated KI-STIM1I115F animals, suggesting a reversable drug effect.
These data therefore prove that inhibition of SOCE counteracts the effect on thrombocytopenia by gain-of-function mutations of STIM1, and in particular by the p.I115F mutation.
The bone marrow rearranges its megakaryocyte precursor pathways in KI-STIM1I115F mice
We next investigated whether an alteration of hematopoietic progenitor differentiation was at the origin of the observed thrombocytopenia and whether SOCE inhibition could alter hematopoiesis in WT and KI-STIM1I115F mice. To do so, we evaluated the frequency of the CMP, GMP, MEP, and megakaryocyte progenitors (MkP) in the bone marrow (see supplemental Figure 2A for the gating strategy). As represented in Figure 2A, the frequency of CMP, GMP, and MkP was not different between WT and KI-STIM1I115F mice, whereas we could observe a significant decrease in MEP. No significant differences could be observed in WT or KI-STIM1I115F mice treated with CIC-39Na compared with their respective controls.
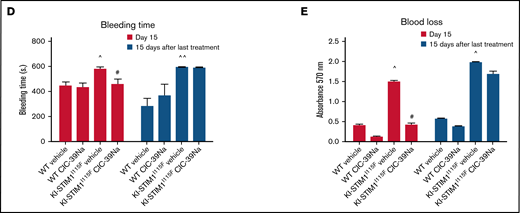
Progenitor characterization of WT and KI-STIM1I115F mice. (A-B) Frequency of the indicated progenitor cells; Mann-Whitney U test (n = 6 per group) P value: ^^≤0.0079 and ^≤0.00224 for WT vehicle vs KI-STIM1I115F vehicle. (C) Representative immunofluorescence staining for STIM1 (green), CD41 (blue), DAPI (cyano), and merged image on megakaryocytes of WT mice (see Supplemental Methods for the other groups). Top panel: Magnification 40 × 1.4 NA (numerical aperture). Bar = 25 μm. Bottom panel: Magnification 63 × 1.4 NA (numerical aperture). Bar = 10 μm. The data are representative of 5 experiments with similar results. (D) Box-and-whisker plots show median and IQR of the mean number of MK colonies/well in WT and KI-STIM1I115F mice. Mann-Whitney U test. P value: ^≤0.0111 WT vehicle vs KI-STIM1I115F vehicle.
Progenitor characterization of WT and KI-STIM1I115F mice. (A-B) Frequency of the indicated progenitor cells; Mann-Whitney U test (n = 6 per group) P value: ^^≤0.0079 and ^≤0.00224 for WT vehicle vs KI-STIM1I115F vehicle. (C) Representative immunofluorescence staining for STIM1 (green), CD41 (blue), DAPI (cyano), and merged image on megakaryocytes of WT mice (see Supplemental Methods for the other groups). Top panel: Magnification 40 × 1.4 NA (numerical aperture). Bar = 25 μm. Bottom panel: Magnification 63 × 1.4 NA (numerical aperture). Bar = 10 μm. The data are representative of 5 experiments with similar results. (D) Box-and-whisker plots show median and IQR of the mean number of MK colonies/well in WT and KI-STIM1I115F mice. Mann-Whitney U test. P value: ^≤0.0111 WT vehicle vs KI-STIM1I115F vehicle.
We next assessed whether noncanonical pathways leading to MkP differentiation could explain the inconsistency between MEP and MkP. In the noncanonical model of hematopoiesis, MkP differentiation occurs directly from MPP2, bypassing MEP, or from a subset of LT-HSCs, bypassing MMP2 and MEP differentiation.34 The overall frequency of MMP2 was similar between WT and KI-STIM1I115F; however, FACS analysis clearly showed an increase in the fraction of the HSC fractions in KI-STIM1I115F compared with WT (see supplemental Figure 2B for the gating strategy). In particular, we found statistically higher LK, LSK, and LT-HSCs (Figure 2B). Because we found no difference in MPP2, we conclude that the increase in LT-HSCs, which notably is also maintained when evaluated as total number of BM cells (supplemental Figure 2C), might be a compensatory mechanism that explains the normal number of MEPs despite a low number of MkP. Interestingly, we also found a decreased frequency in MMP4 that give rise to CLP in bone marrows from KI-STIM1I115F (Figure 2B). To some surprise, the changes in thrombopoiesis that occur in the KI-STIM1I115F bone marrows are not corrected by CIC-39Na.
Overall, despite the changes in thrombopoietic used pathways that lead to MkP, our data suggest that thrombocytopenia in TAM is not attributable to BM defects and that CIC-39Na does not act at this level. In support of this conclusion, the evaluation of STIM1 expression in normal BM by confocal microscopy analysis showed positivity for STIM1 expression in fully mature Mk (Figure 2C; supplemental Figure 2D), which prompts for an activity of STIM1 after differentiation. In line with this, the colony assay not only did not show a defect in CFU-Mk formation, on the contrary, it suggested an increase in KI-STIM1I115F mice (Figure 2D; supplemental Figure 2E). Again, no effect was observed in the BMs of KI-STIM1I115F mice treated with CIC-39Na compared with the relative control (supplemental Figure 2D,E).
KI-STIM1I115F platelets have a reduced half-life and an altered Ca2+-signaling
Given that the BM does not appear the site responsible for the thrombocytopenia, we next evaluated whether platelet turnover in the periphery could be responsible. To this end, we performed a chase-pulse experiment by labeling platelets with a nonsaturating dose of anti-GPIX antibody and drawing blood at sequential times to determine the number of bound platelets. As depicted in Figure 3A, no residual labeled platelets in KI-STIM1I115F mice were found after 48 hours, compared with around 35% in control animals. Control platelets also depicted a long plateau-phase and labeled platelets could still be detected after 4 days. Importantly, CIC-39Na was able to fully restore the clearance of platelets to the extent of WT, showing that this was most likely a SOCE-mediated event and at the basis of the increased number of platelets observed in Figure 1B.

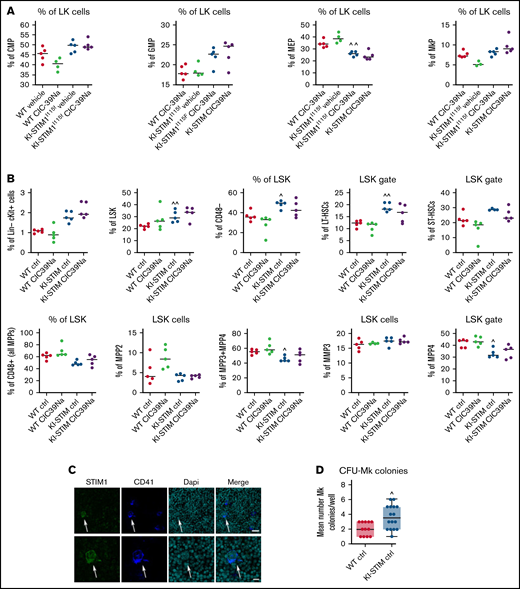
Clearance, STIM1 protein levels and Ca2+-signaling in WT and KI-STIM1I115F platelets. (A) Platelet lifespan in the indicated groups (n = 6). Mann-Whitney U test (n = 6 per group) P value: ^^^≤0.0015 and ^^≤0.0028 for WT vehicle vs KI-STIM1I115F vehicle; ##≤0.003 and #≤0.0014 for KI-STIM1I115F vehicle vs KI-STIM1I115F CIC-39Na. (B) Representative images and densitometry analysis of western blots for STIM1 and ORAI1 (n = 3); unpaired t test for STIM1 blot (P value: ****<0.0001 WT vehicle vs WT CIC-39Na; ###<0.0002 KI-STIM1I115F vehicle vs KI-STIM1I115F CIC-39Na and ^^^0.009 WT vehicle vs KI-STIM1I115F vehicle). ORAI1 blot (P value: ****<0.0001 WT vehicle vs WT CIC-39Na; ####<0.0001 KI-STIM1I115F vehicle vs KI-STIM1I115F CIC-39Na). (C) Basal platelet cytosolic Ca2+. Values are median ± SEM of at least 300 platelets from 2 experimental days. Mann-Whitney U test (n = 6 per group) P value: ^^^^<0.0001 WT vehicle vs KI-STIM1I115F vehicle and ####<0.0001 KI-STIM1I115F vehicle vs KI-STIM1I115F CIC-39Na. (D) Average Ca2+-traces depicting tBHQ release and SOCE. Statistical analysis of the AUC of the first 200 seconds after the respective addition are present in the insets. Arrows indicate addition of the SERCA poison in Ca2+-free conditions and the readdition of Ca2+. Traces are average of 300 cells from 2 different experimental days and color coding is as in panel C; to allow comparisons, F/Fo was offset to 0 in the graphs for all traces. Mann-Whitney U test. P value: ^^^^<0.0001 for WT vehicle vs KI-STIM1I115F vehicle; ###<0.001 for KI-STIM1I115F vehicle vs KI-STIM1I115F CIC-39Na. (E-F) Average Ca2+traces induced by the indicated agonists in Ca2+-containing media. Traces are average of 300 cells from 2 different experimental days; statistical analysis of the AUC of the first 200 seconds after the respective addition are present in the insets and color coding is as in panel C; Mann-Whitney U test. P value: ****<0.0001 and **<0.003 for KI-STIM1I115F vehicle vs KI-STIM1I115F CIC-39Na; ^^^<0.001 for WT vehicle vs KI-STIM1I115F vehicle. To allow visual comparisons, F/Fo was offset to 0 in the graphs for all traces.
Clearance, STIM1 protein levels and Ca2+-signaling in WT and KI-STIM1I115F platelets. (A) Platelet lifespan in the indicated groups (n = 6). Mann-Whitney U test (n = 6 per group) P value: ^^^≤0.0015 and ^^≤0.0028 for WT vehicle vs KI-STIM1I115F vehicle; ##≤0.003 and #≤0.0014 for KI-STIM1I115F vehicle vs KI-STIM1I115F CIC-39Na. (B) Representative images and densitometry analysis of western blots for STIM1 and ORAI1 (n = 3); unpaired t test for STIM1 blot (P value: ****<0.0001 WT vehicle vs WT CIC-39Na; ###<0.0002 KI-STIM1I115F vehicle vs KI-STIM1I115F CIC-39Na and ^^^0.009 WT vehicle vs KI-STIM1I115F vehicle). ORAI1 blot (P value: ****<0.0001 WT vehicle vs WT CIC-39Na; ####<0.0001 KI-STIM1I115F vehicle vs KI-STIM1I115F CIC-39Na). (C) Basal platelet cytosolic Ca2+. Values are median ± SEM of at least 300 platelets from 2 experimental days. Mann-Whitney U test (n = 6 per group) P value: ^^^^<0.0001 WT vehicle vs KI-STIM1I115F vehicle and ####<0.0001 KI-STIM1I115F vehicle vs KI-STIM1I115F CIC-39Na. (D) Average Ca2+-traces depicting tBHQ release and SOCE. Statistical analysis of the AUC of the first 200 seconds after the respective addition are present in the insets. Arrows indicate addition of the SERCA poison in Ca2+-free conditions and the readdition of Ca2+. Traces are average of 300 cells from 2 different experimental days and color coding is as in panel C; to allow comparisons, F/Fo was offset to 0 in the graphs for all traces. Mann-Whitney U test. P value: ^^^^<0.0001 for WT vehicle vs KI-STIM1I115F vehicle; ###<0.001 for KI-STIM1I115F vehicle vs KI-STIM1I115F CIC-39Na. (E-F) Average Ca2+traces induced by the indicated agonists in Ca2+-containing media. Traces are average of 300 cells from 2 different experimental days; statistical analysis of the AUC of the first 200 seconds after the respective addition are present in the insets and color coding is as in panel C; Mann-Whitney U test. P value: ****<0.0001 and **<0.003 for KI-STIM1I115F vehicle vs KI-STIM1I115F CIC-39Na; ^^^<0.001 for WT vehicle vs KI-STIM1I115F vehicle. To allow visual comparisons, F/Fo was offset to 0 in the graphs for all traces.
We next analyzed the effect of the mutation on the expression of ORAI1 and STIM1 in platelets. As it can be observed in Figure 3B, KI-STIM1I115F mice display a reduced level of STIM1 compared with WT and a normal level of ORAI1 channels. Platelets from treated KI-STIM1I115F animals showed an even lower level of STIM1 and a compensatory increase in ORAI1. Although these data might appear contradictory, a number of explanations can be postulated. First, it is possible that in KI-STIM1I115F mice there is an increased clustering of STIM1 (ie, the activation mechanism),35,36 and this would lead to a degradation of the protein. Alternatively, given that the platelets collected from KI-STIM1I115F mice are significantly reduced, these data might suggest that circulating platelets belong to those clones that express the least amount of STIM1. Last, it is possible that p.I115F is less stable compared with the WT protein, although in this instance the effect of the inhibitor would be difficult to explain. Our data tend to suggest that first hypothesis is more likely, given the observation in treated animals. Indeed, it could be speculated that in these animals clustering is even more increased because there is a reduced store replenishment given the blockage of ORAI1 channels by CIC-39Na. Although such observations would require to be more closely scrutinized to elucidate the mechanism, we did not investigate it further.
Given the strong effect on protein expression, we wondered whether Ca2+-signaling was still altered or whether protein expression was a sufficient compensatory mechanism. As in the case for myotubes with activating mutations,15,37 basal [Ca2+] levels were significantly increased in KI-STIM1I115F platelets compared with WT, resulting in a pre-activation state, and these ion concentrations were restored in platelets isolated from KI-STIM1I115F treated with CIC-39Na for 2 weeks (Figure 3C).
We next evaluated SOCE in platelets from animals that were either treated or not treated with CIC-39Na. As shown in Figure 3D, tBHQ elicited a statistically greater Ca2+-efflux from KI-STIM1I115F platelets compared with WT animals as determined by evaluating the area under the curve (AUC) of the first 200 seconds after stimulation. Similar results were obtained when determining maximum amplitude obtained by tBHQ (supplemental Figure 3A). Furthermore, SOCE activated by Ca2+ addition after tBHQ depletion was also statistically increased in KI-STIM1I115F platelets both in terms of AUC (Figure 3D) and maximum amplitude (supplemental Figure 3A). Importantly, platelets from KI-STIM1I115F animals treated with CIC-39Na displayed a restoration in the amount of stored Ca2+ and in the extent of SOCE.
We then explored the effect of Gq-coupled receptor or ITAM-mediated agonists on Ca2+-signaling in platelets. As shown in Figure 3E the response to convulxin (that acts via the ITAM-bearing GPVI receptor) was unchanged when evaluating the AUC of the first 200 seconds, although the shape of the response was significantly different. A steady increase over time was observable in KI-STIM1I115F platelets, which contrasted with the more canonical SOCE shape observed in WT. The inhibitor reduced significantly the AUC in WT and potentiated, albeit not significantly, SOCE in KI-STIM1I115F platelets, reverting the shape of the curve. The response to U46619 (that acts via the Gq-coupled thromboxane receptor) was significantly reduced in KI-STIM1I115F platelets compared with WT (Figure 3F). Animals treated with CIC-39Na displayed a significantly lower response and the inhibitor was unable to revert the effect observed in KI-STIM1I115F platelets (Figure 3F).
The data on Ca2+ signaling should be read in the context of an increased function of p.I115F STIM1 but also of a reduced expression of the protein, as shown in Figure 3B. The main findings, in agreement with the overall pathogenetic hypothesis, are that KI-STIM1I115F platelets have an increased basal Ca2+, an increased Ca2+ load in the ER, and trigger a more substantial SOCE (Figure 3D). All 3 of these key elements are reverted when evaluating platelets treated with CIC-39Na, with no significant effect of the inhibitor on WT animals.
Experiments in Ca2+-containing solutions with receptor agonists return a less interpretable picture. Indeed, when evaluating ITAM and thromboxane signaling we found a quantitative reduction in the response to U46619 but not to CVX, although in this latter case there was a substantial change in the kinetics of the Ca2+ rise. WT animals treated with CIC-39Na showed a significant reduction in the response to CVX and U46619, whereas platelets from KI-STIM1I115F treated animals reversed the kinetics on CVX treatment and showed no difference in response to U46619. Overall, therefore, the results on receptor triggered responses cannot be explained solely by increased entry and store loading and must involve other changes. We have not investigated receptor levels, and therefore are unable to determine whether it’s a change in expression that determines this effect. The ability to fully understand the changes that occur in receptor triggered responses is complicated, among other elements, by the design of the experiment, in which platelets are exposed chronically to the inhibitor in vivo and by the complexity of Ca2+ signaling, including the number of players involved in a single response.
We next evaluated whether the effects on degranulation and activation. A minimal increase of P-selectin exposure but not of integrin αIIbβ3 activation was detected in resting KI-STIM1I115F compared with WT platelets (Figure 4A-B). Stimulation of ITAM-bearing receptor GPVI by convulxin was unable to induce P-selectin exposure in KI-STIM1I115F platelets, and this inability was not restored by CIC-39Na. P-selectin exposure by stimulation of the thromboxane G-protein coupled receptor using U46619 was modest. Similarly, KI-STIM1I115F platelets showed a reduced integrin αIIbβ3 activation in response to convulxin, and to some extent also to U46619, but we could observe a modest recovery at high doses of agonists in KI-STIM1I115F platelets treated with CIC-39Na.
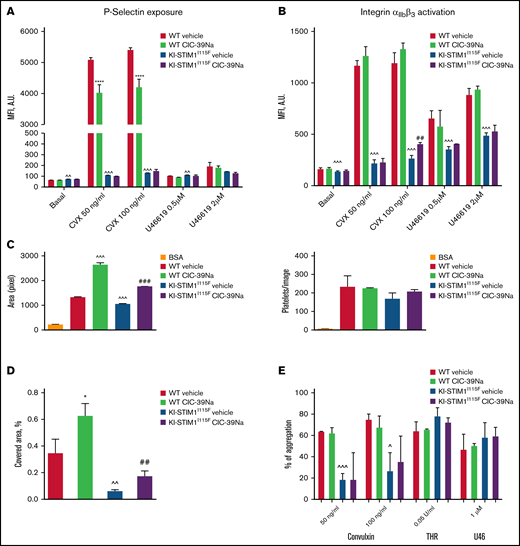
Platelet function WT and KI-STIM1I115F (A-B) P-selectin (A) and integrin αIIbβ3 exposure after 10-minute stimulation with the indicated stimuli. (C) platelet spreading to collagen in static conditions, indicated as are of adherent cells (left), and number of adhered platelets (right); BSA was used as negative control in place of collagen. (D) Thrombus formation under flow, indicated as percentage of platelet covered area; Mann-Whitney U test (n = 6 per group) P value: ^^^≤0.0002, ^^≤0.0039 WT vehicle vs KI-STIM1I115F vehicle; *****<0.00001 WT vehicle vs WT CIC-39Na and ##≤0.0036 KI-STIM1I115F vehicle vs KI-STIM1I115F CIC-39Na. Supplemental Figure 4A-B depicts fluorescent microscope images of the results; (E) % of aggregated platelets in the indicated conditions. To reach similar aggregations, WT platelets were diluted 1:3. Supplemental Figure 4C depicts slopes.
Platelet function WT and KI-STIM1I115F (A-B) P-selectin (A) and integrin αIIbβ3 exposure after 10-minute stimulation with the indicated stimuli. (C) platelet spreading to collagen in static conditions, indicated as are of adherent cells (left), and number of adhered platelets (right); BSA was used as negative control in place of collagen. (D) Thrombus formation under flow, indicated as percentage of platelet covered area; Mann-Whitney U test (n = 6 per group) P value: ^^^≤0.0002, ^^≤0.0039 WT vehicle vs KI-STIM1I115F vehicle; *****<0.00001 WT vehicle vs WT CIC-39Na and ##≤0.0036 KI-STIM1I115F vehicle vs KI-STIM1I115F CIC-39Na. Supplemental Figure 4A-B depicts fluorescent microscope images of the results; (E) % of aggregated platelets in the indicated conditions. To reach similar aggregations, WT platelets were diluted 1:3. Supplemental Figure 4C depicts slopes.
We next investigated adhesion and spreading of platelets to collagen in static conditions (Figure 4C; supplemental Figure 4A) as well as thrombus formation under flow (Figure 4D; supplemental Figure 4B). As shown in Figures 4C, the number of adherent platelets was comparable for control and KI-STIM1I115F platelets (right), whereas, spreading, measured as the surface area of adherent cells, was slightly but significantly hampered in KI-STIM1I115F platelets (left). Moreover, when flushed at arterial flow rate, the ability of KI-STIM1I115F platelets to arrest and form thrombi on a collagen matrix was reduced compared with that of WT platelets (Figure 4D). In both experimental settings, CIC-39Na was able to partially, but not completely, restore platelet spreading to collagen, as well as thrombus formation under flow. Alongside, we also observed an increased spreading of WT platelets from animals treated with CIC-39Na, as well as a more pronounced thrombus formation under flow, suggesting that chronic SOCE inhibition might potentiate this activity.
Last, we investigated platelet aggregation. Figure 4E and supplemental Figure 4C reveal a marked defect in aggregation of KI-STIM1I115F platelets in response to convulxin. Platelet aggregation induced by thrombin and U46619 were marginally, but not statistically, increased. CIC39-Na was unable to restore normal aggregation in convulxin-treated KI-STIM1I115F platelets, although a tendency for a slight increase of the response was observed.
Altogether, these results indicate that KI-STIM1I115F mice, in analogy to TAM patients, suffer from thrombocytopenia and increased bleeding. In the BM, the presence of p.I115F mutation leads to a shift from the canonical thrombopoiesis pathway to a noncanonical pathway but the resulting MkP cells are in normal number. Circulating platelets display a significantly faster turnover that accounts for the reduced platelet number and are also characterized by a modest preactivation state and a dramatic defect in response to GPVI stimulation. Silva-Rojas and Gamage have recently reported a R304W knock-in mouse that also recapitulates the features of tubular aggregate myopathy. Although Silva-Rojas et al reported a marked thrombocytopenia but did not characterize it further (other than finding megakaryocyte hyperplasia in the spleen22 ), Gamage et al reported a normal amount of platelets but a defect in activation and this was postulated to be the result of a significant reduction in STIM1 expression.21 On the contrary, Grosse et al reported a mouse generated by random mutations that presented a p.D84G mutation and fully characterized the thrombocytopenia. In line with our findings, they found thrombocytopenia, excessive bleeding, an increased platelet bleeding, reduced spreading, a preactivation because of elevated basal platelet cytosolic Ca2+, and a significantly affected ITAM-mediated signaling. Slight differences between these 2 models can also be observed: in the BM, the p.D84G presents similar CFU-MK colonies to control, whereas we found them increased, the p.D84G presents an increase in activated integrin αIIbβ3 while no noticeable differences were present in our model, and SOCE upon SERCA poisoning was decreased in the p.D84G model while we found it increased. In our manuscript, we also contribute a full characterization of thrombopoiesis that was not performed by Grosse et al. Overall, nonetheless, the similitudes between the 2 models significantly out way the differences.
An important limitation in our study is that we do not provide a direct mechanism by which the changes observed lead to premature clearance of platelets, and it will be necessary to investigate this in the future. Indeed, it would be expected that a higher basal Ca2+ would lead to an increased aggregation and this would be the mechanism by which the increased platelet clearance occurs, but this does not appear to be the case. Indeed, this finding is in line with the absence of thrombotic events in patients. Given that the p.I115F mutation is expressed in all cells, it could be speculated that other cell types, including hepatocytes or macrophages, participate in the clearance with higher efficacy or that KI-STIM1I115F platelets are more prone to such clearances.38
STIM1 is a single transmembrane protein that resides on the endoplasmic reticulum. Structurally, it is composed of a canonical and a noncanonical EF-hand Ca2+-binding motif located intraluminally, a sterile α motif located intraluminally, and a cytosolic portion of the protein that is mainly characterized by 3 coil-coil domains that interact with the ORAI1 protein. The p.I115F mutation is located in the noncanonical EF-hand motif and most likely confers the protein with a modified sensitivity to intraluminal Ca2+, thereby increasing the chance of activation. The p.D84G mutation is located in the canonical EF-hand motif and presumably confers similar properties. Numerous mutations have been reported on these 2 EF-hand domains and our prediction is that the thrombocytopenic phenotype would be similar. Mutations have also been reported for the sterile α motif domain and in the cytosolic portion of the protein, and it remains to be established whether our results can be automatically transferred to those mutations.
The most important advancement provided by the present manuscript is given by the demonstration that a SOCE inhibitor, CIC-39Na, is able to revert the key patient-centric features of the platelet dysfunction: thrombocytopenia and bleeding. These effects can probably be reconducted to the reversal of the fast platelet clearance observed in KI-STIM1I115F mice. Yet, when we evaluated platelets ex vivo, we found that CIC-39Na reverted fully the elevation in basal cytosolic Ca2+ and platelet spreading, but there was a blunted effect on other parameters, such as p-selectin exposure and integrin αIIbβ3 activation, spreading in flow conditions, and aggregation. A first possibility to explain the discrepancy is that STIM1 is a key player also in endothelium39,40 and that this cell type plays a key role in determining the platelet phenotype in KI-STIM1I115F mice. For the absence of effect on the BM after a 15-day treatment with CIC-39Na, this could be due to plastic changes that have occurred in the bone marrow during development triggered by the gain-of-function mutation or to the need for a longer treatment.
A second intriguing finding in our report is that chronic exposure to CIC-39Na leads to a significant decrease in platelet STIM1 levels, both in WT and in KI-STIM1I115F mice. This paradoxical effect on STIM1 levels is at present unexplained. A possibility is given by the fact that channel blockade by CIC-39Na might lead to exaggerated STIM1 clustering and subsequent degradation. Yet, it should be noticed that endoplasmic reticulum Ca2+-content appears normal in the presence of the inhibitor, somehow not providing fuel to the hypothesis of an increased clustering. Molecular glues that lead to protein degradation have been described,41 but given that CIC-39Na is thought to bind to ORAI1, a direct effect on STIM1 degradation by this drug appears unlikely. This effect by CIC-39Na is not observed in muscle (Pessolano, personal communication) and therefore it is likely that the absence of de novo mRNA synthesis in platelets unmasks or exaggerates this effect in these cells. This decrease in STIM1 upon CIC-39Na treatment occurred both in WT and in KI-STIM1I115F. In wild-type, though, we could not detect changes in basal Ca2+ and in SOCE, whereas differences between treated WT and untreated WT mice were observed in ITAM-mediated signaling, in thromboxane-mediated Ca2+-signaling, in p-selectin exposure and in spreading. It could be intriguing to evaluate whether other inhibitors lead to the same STIM1 downregulation.
Our results have important repercussions. First, we have paved the way for a treatment strategy for the platelet disorder that affects more than half of the sufferers of this ultra-rare disorder. The choice to test CIC-39Na was given by the availability of this compound over others that have already entered clinical trials, but we recognize that there are elements of this compound, namely whether it can be administered orally and a full toxicological characterization, which are still lacking. We believe that also other compounds that are already in clinical trials for other indications could be repurposed for thrombocytopenia in TAM with an appropriate clinical trial. Second, given the observation that a SOCE-mediated dysfunction is amenable of intervention with a SOCE inhibitor, this finding is the prelude to testing this or other compounds also for the muscular component of TAM. Were the effect of SOCE inhibition to have an impact on the muscle component in mice, these drugs could find even a stronger rationale in TAM patients. Importantly, given that TAM is a slowly progressive disease, clinical trials with hard muscular endpoints could be challenging as it would take a significant number of years to reach them. Were preclinical models to show a correlation between platelet number and muscle phenotype amelioration, improvement of thrombocytopenia could therefore also be used as a valid, relevant sensitive surrogate biomarker of efficacy in muscle strength in those patients that present both symptoms, thereby facilitating clinical trial design.
Acknowledgments
The authors thank ChemICare S.R.L. for providing CIC-39Na.
The work was supported by research funding from the Italian Telethon Foundation to A.A.G. (GGP19110). PoC instrument funding from LINKS Foundation and Compagnia di San Paolo, PoC UPO-Regione Piemonte art. 12 L.R. n. 4 05/04/2018 to B.R.
Authorship
Contribution: A.A.G. and A.B. contributed equally in supervising; A.A.G. provided funding; A.A.G., A.B., E.P., and C.C.-S. conceived the design of experiments; C.C.-S., E.P., B.R., M.V., S.M.G.T., M.T., N.C., F.A.R., P.P., N.F., I.Z., M.S., M.P.C., S.S., and T.P. performed experiments and analyzed the results; A.A.G., E.P., B.R., and C.C.-S. wrote the manuscript; and all authors have given approval to the final version of manuscripts.
Conflict-of-interest disclosure: B.R. is an employer of ChemICare, of which she also owns stocks. T.P. owns stocks of ChemICare, a company that owns the rights to CIC-39Na. All other authors declare no competing financial interests.
Correspondence: Armando A. Genazzani, Department of Pharmaceutical Sciences, University of Piemonte Orientale, Via Bovio 6, Novara (28100), Italy; e-mail: armando.genazzani@uniupo.it.

