

Key Points
IFNγ induces a highly oxidative metabolic phenotype with high rates of oxygen consumption by both mitochondria and NADPH oxidase.
IFNγ induces a NAMPT-dependent, NAD+ salvage pathway that is required for complete induction of the respiratory burst in human monocytes.

Abstract
Interferon γ (IFNγ) is an essential and pleiotropic activator of human monocytes, but little is known about the changes in cellular metabolism required for IFNγ-induced activation. We sought to elucidate the mechanisms by which IFNγ reprograms monocyte metabolism to support its immunologic activities. We found that IFNγ increased oxygen consumption rates (OCR) in monocytes, indicative of reactive oxygen species generation by both mitochondria and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase. Transcriptional profiling revealed that this oxidative phenotype was driven by IFNγ-induced reprogramming of NAD+ metabolism, which is dependent on nicotinamide phosphoribosyltransferase (NAMPT)-mediated NAD+ salvage to generate NADH and NADPH for oxidation by mitochondrial complex I and NADPH oxidase, respectively. Consistent with this pathway, monocytes from patients with gain-of-function mutations in STAT1 demonstrated higher-than-normal OCR, whereas chemical or genetic disruption of mitochondrial complex I (rotenone treatment or Leigh syndrome patient monocytes) or NADPH oxidase (diphenyleneiodonium treatment or chronic granulomatous disease [CGD] patient monocytes) reduced OCR. Interestingly, inhibition of NAMPT in healthy monocytes completely abrogated the IFNγ-induced oxygen consumption, comparable to levels observed in CGD monocytes. These data identify an IFNγ-induced, NAMPT-dependent, NAD+ salvage pathway that is critical for IFNγ activation of human monocytes.
Introduction
Interferon γ (IFNγ) is an essential activator of macrophages, inducing proinflammatory cytokines, augmenting intracellular microbial killing and stimulating the production of microbicidal and signal-propagating reactive oxygen/nitrogen species.1,2 IFNγ is dysregulated in myriad diseases. Functional IFNγ deficiency is caused by genetic mutations in IFNγ or its receptor or by acquired neutralizing anti-IFNγ autoantibodies, impairing control of intracellular bacteria and causing systemic immune dysregulation.3-11 Conversely, states of excessive IFNγ activity, such as signal transducer and activator of transcription 1 (STAT1) gain of function (GOF), have inflammatory complications.12-16 The effects of IFNγ on macrophages are pleiotropic and likely energetically expensive, requiring metabolic reprogramming to support this increased demand.
Much work has focused on the early metabolic changes in macrophages in response to lipopolysaccharide (LPS), known as the Warburg effect (aerobic glycolysis),17-21 but fewer data exist for metabolic responses to other stimuli. Cameron et al demonstrated that M1 (classically activated macrophages) depend on the nicotinamide adenine dinucleotide (NAD[H]) salvage pathway to sustain aerobic glycolysis and their M1 phenotype following stimulation with LPS or LPS plus IFNγ.22 However, the metabolic reprogramming in human monocytes and macrophages induced by IFNγ alone is not well characterized.
IFNγ is known to regulate phagocyte activation through metabolic adaptations that produce reactive oxygen species (ROS).23-27 Superoxide generation through oxidation of phosphorylated nicotinamide adenine dinucleotide phosphate (NADPH) by the NADPH oxidase complex is known as the respiratory burst.28 The same NAD+ salvage pathways that were identified as critical for maintaining aerobic glycolysis in LPS-stimulated macrophages also supply the intracellular NAD+ required to produce NADP(H) to support NADPH oxidase superoxide production.29 Therefore, we hypothesized that IFNγ-mediated activation of the respiratory burst might also depend on NAD+ salvage pathways.
We used primary human monocytes to characterize the metabolic reprograming that occurs in response to stimulation with IFNγ. We identified an IFNγ-induced, nicotinamide phosphoribosyltransferase (NAMPT)-dependent pathway of NAD+ salvage that is required for the highly oxidative metabolic phenotype induced by IFNγ. We used primary monocytes from healthy donors and patients with genetic defects in various steps of this pathway as well as chemical inhibitors to demonstrate that IFNγ augments NAD+ biosynthesis and reduction/oxidation (redox) metabolism to increase oxygen consumption. Our findings suggest that the IFNγ-induced respiratory burst in monocytes is dependent on NAMPT-mediated NAD+ salvage. This metabolic pathway may include novel targets for therapeutic modulation of monocyte activation.
Methods
Human monocyte collection, isolation, and stimulation
All patients and healthy volunteers signed onto approved National Institutes of Health protocols. The study was conducted in accordance with the Declaration of Helsinki. Peripheral blood was collected in sodium heparin tubes. Peripheral blood mononuclear cells were isolated by density gradient centrifugation using Lymphocyte Separation Medium (Corning, catalog #MT25072CI). CD14+ selection was performed using magnetic beads (Miltenyi Biotec CD14 MicroBeads, catalog #130-050-201) following the manufacturer’s protocol. CD14+ monocytes were plated at 105/50 μL per well in Seahorse XF96 V3 PS cell culture microplates (Agilent, 101085-004) in serum-free media. After 3 hours, 50 μL of complete media with 20% (2×) fetal bovine serum was added, and cells were rested overnight. Cells were then stimulated with media alone, IFNγ (1000 U/mL; Actimmune, NDC number 75987-111-10), and/or select inhibitors: rotenone (Sigma, #R8875), FK866 (Selleckchem, #S2799), diphenyleneiodonium (DPI) (Sigma, #D2926), Rotenone and Antimycin A, oligomycin, or trifluoromethoxy carbonylcyanide phenylhydrazone (FCCP) (Agilent Seahorse Mito Stress Test, #103015-100).
Seahorse metabolic rate assays
Oxygen consumption rates (OCR), indicative of mitochondrial respiration, and extracellular acidification rates (ECAR), indicative of glycolysis, were measured using the Seahorse Bioscience Extracellular Flux Analyzer (XFe96; Seahorse Bioscience Inc., North Billerica, MA). Prior to measurements, culture medium was removed and replaced with 180 μL pH-ready Seahorse Assay Media (Agilent, catalog #103575-100) and incubated in the absence of CO2 for 1 hour in the Biotek Cytation1 while preassay brightfield images were collected. For the Mito Stress Test, cells were sequentially treated with oligomycin (2 μM), FCCP (0.5 μM), and rotenone+ antimycin A (0.5 μM). OCR and ECAR were measured in a standard, 6-minute cycle. For the Glycolysis Stress Test, cells were sequentially treated with glucose (10 mM), oligomycin (1 μM), and 2-deoxyglucose (50 mM). For our modified assay, cells were treated with phorbol myristate acetate (PMA) (100 μM), then, in some cases, rotenone and antimycin A (0.5 μM).
All OCR and ECAR values were normalized by staining cells with 2 μg/mL Hoechst 33342 (ThermoFisher Scientific) for 30 minutes while performing postassay brightfield imaging. Cells were then imaged and counted using the Biotek Cytation1. Cell counts were calculated by cell imaging software (Agilent) and imported into Wave (Agilent) using the normalization function. OCR values were normalized to cell count data.
RNA-Seq
RNA sequencing (RNA-Seq) was performed on RNA isolated from primary CD14+ monocytes from 4 normal donors following differentiation with macrophage colony stimulating factor (30 ng/mL) for 5 days and subsequent stimulation with recombinant human IFNγ (100 ng/mL) for 24 hours.
Total RNA sample integrity was assessed using automated capillary electrophoresis on a Fragment Analyzer (Agilent) using the HS RNA Kit (15NT). For all samples RNA quality number > 8.0, total RNA amount of >75 ng was used as input for library preparation using the TruSeq Stranded mRNA Library Preparation Kit (Illumina). Sequencing libraries were quantified by real-time polymerase chain reaction using the KAPA Library Quantification Complete kit (Roche) and assessed for size distribution, absence of free adapters, and adapter dimers on a Fragment Analyzer. Sequencing libraries were pooled and sequenced on a HiSeq 3000 System (Illumina) using a HiSeq 3000/4000 PE Cluster Kit and SBS Kit (150 cycles) with run conditions of paired-end reads at 75 bp length.
Sequenced reads (100 bp, paired-end) were mapped to the human genome hg19 (GRCh38) using Bowtie 2.2.630 and Tophat 2.2.1.31 Uniquely mapped reads were retained, and raw counts that fell in coding regions were calculated and normalized using reads per kilobase per million mapped reads using given gene lengths from the University of California, Santa Cruz genome browser. Quantile normalization was applied to all samples,32 and data were log2-transformed. Nonexpressed and weakly expressed genes, defined as having <1 read per million in 4 of the samples,33 were removed prior to subsequent analyses, resulting in a count table of 13 873 genes. Limma (Bioconductor package) was used to conduct differential expression analyses.34 The voom module was used to transform data based on observational level weights derived from the mean-variance relationship prior to statistical modeling.35 Pairwise contrasts were done within limma to identify differentially expressed genes between conditions. Genes with a Benjamini-Hochberg multiple-testing adjusted P value of <.05 were defined as differentially expressed.
Volcano plot was generated using GraphPad Prism9 software. Metabolic genes involved in NAD(P)H metabolism and related pathways were extracted from the Kyoto Encyclopedia of Genes and Genomes pathway database. Metabolic genes that overlapped with RNA-Seq data are shown in heat maps generated using the web tool ClustVis.36 Unbiased assessment of the metabolic pathways transcriptionally regulated by IFNγ was performed using the ERGO™ 2. 0 analysis platform (Ingenbio, Chicago, IL).
Statistical analyses
Statistical analyses were performed using GraphPad Prism9 software. Data are expressed as mean ± standard error of the mean, and P values were calculated using 1-way analysis of variance (ANOVA) with Tukey’s multiple comparisons test or 2-way ANOVA with Sidak’s multiple comparisons test unless otherwise indicated. A confidence interval of 95% was used for all statistical tests. Sample sizes were determined based on the experiment type and standard practice in the field.
Results
IFNγ increases monocyte OCR
Although much is known about IFNγ-mediated transcriptional regulation, the metabolic processes that support IFNγ-mediated activation of primary human monocytes remain poorly understood. Therefore, we assessed cellular OCR and ECAR to quantify cellular oxygen metabolism and glycolysis, respectively.
Consistent with studies describing an immediate switch to glycolysis in murine bone marrow–derived macrophage’s response to LPS,37-39 we found that LPS and the combination of LPS plus IFNγ robustly increased ECAR (glycolysis) in primary human monocytes within 20 minutes of stimulation. IFNγ alone had no immediate effect on ECAR (Figure 1A) and none of the stimuli caused significant changes in OCR within 1 hour of stimulation (Figure 1B).
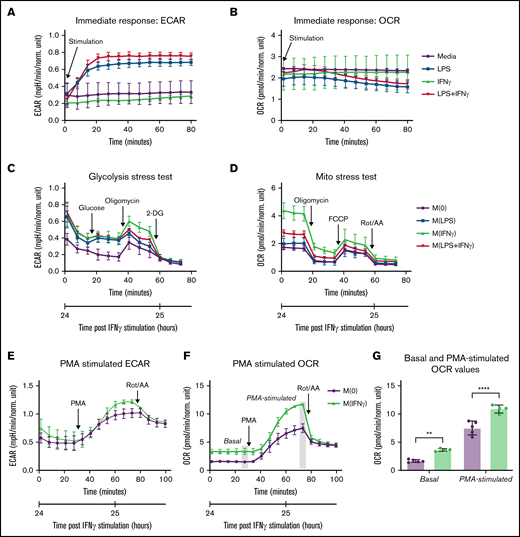
IFNγ Increases Monocyte OCR. (A-B) Real-time changes in ECAR (A) and OCR (B) measured using Seahorse extracellular flux analyzer in primary human monocytes immediately following injection of media alone, LPS, IFNγ, or LPS plus IFNγ (n = 5 technical replicates). (C-D) Primary human monocytes were stimulated with media alone, LPS, IFNγ, or LPS plus IFNγ for 24 hours prior to the start of the assay, then (C) ECAR was measured according to the Seahorse Glycolysis Stress Test or (D) OCR was measured according to the Seahorse Mito Stress Test. For the Glycolysis Stress Test, cells were sequentially treated with glucose (10 mM), oligomycin (1 µM), and 2-deoxyglucose (50 mM). For our modified assay, cells were treated with PMA (100 µM), then, in some cases, rotenone and antimycin A (0.5 µM). For the Mito Stress Test, cells were sequentially treated with oligomycin (2 µM), FCCP (0.5 µM), and rotenone+antimycin A (0.5 µM). OCR and ECAR were measured in a standard, 6-minute cycle. Data displayed in panels A-D are representative tracings of 3 independent experiments. (E-F) Modified Seahorse Assay: primary human monocytes treated with IFNγ or media alone for 24 hours prior to the start of the assay (n = 5 technical replicates). Basal ECAR (E) and OCR (F) measurements were collected, then PMA was injected to stimulate maximal activation, followed by rotenone/antimycin A injection to inhibit mitochondrial oxygen consumption. Real-time changes in ECAR (E) and OCR (F) were monitored. (G) Significant differences in OCR measurements from panel F are displayed, in which the last measurement before PMA injection is used to quantify “basal” OCR, and the last measurement before rotenone/antimycin A injection is used to quantify “PMA-stimulated” OCR throughout the study. Data in panel G were analyzed by 2-way ANOVA with Sidak’s multiple comparisons test. Error bars are mean ± standard error of the mean. **P < .01, ****P < .0001.
IFNγ Increases Monocyte OCR. (A-B) Real-time changes in ECAR (A) and OCR (B) measured using Seahorse extracellular flux analyzer in primary human monocytes immediately following injection of media alone, LPS, IFNγ, or LPS plus IFNγ (n = 5 technical replicates). (C-D) Primary human monocytes were stimulated with media alone, LPS, IFNγ, or LPS plus IFNγ for 24 hours prior to the start of the assay, then (C) ECAR was measured according to the Seahorse Glycolysis Stress Test or (D) OCR was measured according to the Seahorse Mito Stress Test. For the Glycolysis Stress Test, cells were sequentially treated with glucose (10 mM), oligomycin (1 µM), and 2-deoxyglucose (50 mM). For our modified assay, cells were treated with PMA (100 µM), then, in some cases, rotenone and antimycin A (0.5 µM). For the Mito Stress Test, cells were sequentially treated with oligomycin (2 µM), FCCP (0.5 µM), and rotenone+antimycin A (0.5 µM). OCR and ECAR were measured in a standard, 6-minute cycle. Data displayed in panels A-D are representative tracings of 3 independent experiments. (E-F) Modified Seahorse Assay: primary human monocytes treated with IFNγ or media alone for 24 hours prior to the start of the assay (n = 5 technical replicates). Basal ECAR (E) and OCR (F) measurements were collected, then PMA was injected to stimulate maximal activation, followed by rotenone/antimycin A injection to inhibit mitochondrial oxygen consumption. Real-time changes in ECAR (E) and OCR (F) were monitored. (G) Significant differences in OCR measurements from panel F are displayed, in which the last measurement before PMA injection is used to quantify “basal” OCR, and the last measurement before rotenone/antimycin A injection is used to quantify “PMA-stimulated” OCR throughout the study. Data in panel G were analyzed by 2-way ANOVA with Sidak’s multiple comparisons test. Error bars are mean ± standard error of the mean. **P < .01, ****P < .0001.
Wang et al reported that the metabolic program of murine bone marrow–derived macrophages stimulated with IFNγ for 24 hours was primarily characterized by increased glycolysis, without changes in OCR.40 To compare our findings in human monocytes to previously published data in murine macrophages, we used the same assays to measure OCR and ECAR in primary human monocytes cultured with media alone (M[0]), IFNγ (M[IFNγ]), LPS (M[LPS]), or LPS+IFNγ (M[LPS plus IFNγ]) for 24 hours. Although our data demonstrated an increase in ECAR in response to IFNγ (Figure 1C), there was also a significant increase in OCR in response to IFNγ (P < .0001) (Figure 1D), which may represent a metabolic phenotype unique to human monocytes. We also found that the effects of IFNγ on monocyte OCR were time dependent, with the greatest effects at 24 hours poststimulation (supplemental Figure 1A-B).
We then modified the standardized Seahorse assays to assess how IFNγ reprograms monocyte metabolism during a 24-hour stimulation period to prime cells metabolically to respond to the robust activating stimulus, PMA. Basal measurements of ECAR and OCR were collected, then PMA (100 ng/mL) was injected, and the real-time metabolic responses were monitored. M(IFNγ) monocytes demonstrated a trend toward increased PMA-stimulated ECAR (Figure 1E) and a significant increase in both basal (P = .003) and PMA-stimulated (P < .0001) OCR (Figure 1F-G). These findings demonstrate that primary human monocyte metabolism is reprogrammed by IFNγ, which has modest effects on ECAR and significantly increases OCR.
IFNγ-induced oxygen consumption is dependent on NAMPT-mediated NAD+ salvage
Wu et al reported increased OCR and fatty acid oxidation in response to type I interferons in plasmacytoid dendritic cells.41 Therefore, we first compared metabolic responses to IFNγ (type II interferon) to IFNα and IFNβ (type I interferons). We found that IFNα and IFNβ induced significantly less basal and PMA-stimulated OCR (∼1.5-fold increase) than IFNγ, (two- to threefold increase; Figure 2A). These data suggest a distinct metabolic effect of IFNγ that drives oxygen consumption.
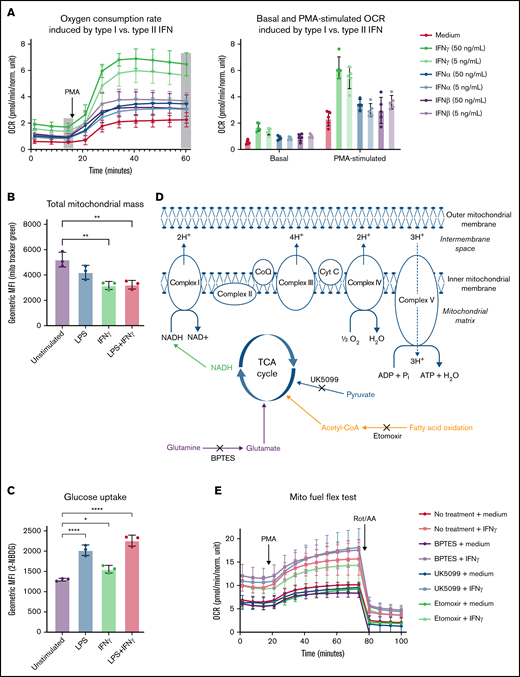
IFNγ-induced increases in OCR are unique to type II interferon and not dependent on total mitochondrial mass, glucose uptake, or substrate-specific catabolism. (A) Real-time changes OCR measured by Seahorse following treatment with media alone or high (50 ng/mL) or low (5 ng/mL) dose of IFNγ, IFNα, or IFNβ for 24 hours prior to the start of the assay. Basal OCR was measured, then PMA (100 ng/mL) was injected during the assay, and OCR was monitored (n = 5 technical replicates). Data in panel A are representative tracings, with bar graphs indicating basal and PMA-stimulated OCR of 3 independent experiments. (B-C) Primary human monocytes were stimulated with media alone, LPS, IFNγ, or LPS plus IFNγ prior to the start of the assay, then (B) total mitochondrial mass was measured using MitoTracker Green staining, and (C) glucose uptake was measured using 2-NBDG by flow cytometry, and the geometric mean fluorescent intensities are indicated in the bar graphs. Data in panels B-C were analyzed by 1-way ANOVA with Dunnett’s multiple comparisons test. Error bars are mean ± standard error of the mean. *P < .05, **P < .01, ***P < .001, ****P < .0001. (D) Schematic representation of the primary mitochondrial fuel sources and their respective inhibitors used in panel E. Primary human monocytes were stimulated with or without IFNγ and in the presence of absence of metabolic inhibitors according to the Mito Fuel Flex Test protocol; healthy control monocytes were left untreated or treated with etomoxir (4 µM), UK5099 (2 µM), or Bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethyl sulfide (3 µM) throughout the 24-hour IFNγ-stimulation period. Oxygen consumption was assessed by Seahorse assay according to a modified Mito Fuel Flex Test protocol (Agilent, 103260-100). Basal OCR measurements were captured after the 24-hour stimulation. PMA (100 ng/mL) was injected during the assay, and PMA-stimulated OCR measurement were collected. Rotenone and antimycin A were then injected, and OCR measurements were collected. For data in panels A and E, n = 5 technical replicates, and the tracings are representative of 3 independent experiments. TCA, tricarboxylic acid.
IFNγ-induced increases in OCR are unique to type II interferon and not dependent on total mitochondrial mass, glucose uptake, or substrate-specific catabolism. (A) Real-time changes OCR measured by Seahorse following treatment with media alone or high (50 ng/mL) or low (5 ng/mL) dose of IFNγ, IFNα, or IFNβ for 24 hours prior to the start of the assay. Basal OCR was measured, then PMA (100 ng/mL) was injected during the assay, and OCR was monitored (n = 5 technical replicates). Data in panel A are representative tracings, with bar graphs indicating basal and PMA-stimulated OCR of 3 independent experiments. (B-C) Primary human monocytes were stimulated with media alone, LPS, IFNγ, or LPS plus IFNγ prior to the start of the assay, then (B) total mitochondrial mass was measured using MitoTracker Green staining, and (C) glucose uptake was measured using 2-NBDG by flow cytometry, and the geometric mean fluorescent intensities are indicated in the bar graphs. Data in panels B-C were analyzed by 1-way ANOVA with Dunnett’s multiple comparisons test. Error bars are mean ± standard error of the mean. *P < .05, **P < .01, ***P < .001, ****P < .0001. (D) Schematic representation of the primary mitochondrial fuel sources and their respective inhibitors used in panel E. Primary human monocytes were stimulated with or without IFNγ and in the presence of absence of metabolic inhibitors according to the Mito Fuel Flex Test protocol; healthy control monocytes were left untreated or treated with etomoxir (4 µM), UK5099 (2 µM), or Bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethyl sulfide (3 µM) throughout the 24-hour IFNγ-stimulation period. Oxygen consumption was assessed by Seahorse assay according to a modified Mito Fuel Flex Test protocol (Agilent, 103260-100). Basal OCR measurements were captured after the 24-hour stimulation. PMA (100 ng/mL) was injected during the assay, and PMA-stimulated OCR measurement were collected. Rotenone and antimycin A were then injected, and OCR measurements were collected. For data in panels A and E, n = 5 technical replicates, and the tracings are representative of 3 independent experiments. TCA, tricarboxylic acid.
We determined that IFNγ-induced OCR was not a result of mitochondrial biogenesis leading to increased total mitochondrial mass (mitochondrial mass was significantly lower in M[IFNγ] monocytes [Figure 2B], and glucose uptake was only modestly increased [Figure 2C]). When we inhibited the 3 primary carbon sources fueling the TCA cycle and mitochondrial oxidative metabolism (Figure 2D), we found that none of the individual inhibitors tested significantly reduced IFNγ-induced increases in OCR (Figure 2E). Therefore, we concluded that IFNγ-induced oxygen consumption was not exclusively dependent on the oxidation of pyruvate, glutamine, or fatty acids.
Using RNA-Seq data from IFNγ-stimulated, human monocyte–derived macrophages, unbiased assessment of the metabolic pathways transcriptionally regulated by IFNγ42 identified “NAD and NADP metabolism” as the most significantly upregulated metabolic pathway (Figure 3A). We found that IFNγ both upregulated NAMPT, the rate-limiting enzyme in nicotinamide (NAM)-dependent NAD+ salvage,22,43 and simultaneously downregulated nicotinamide riboside kinase 2 (NRK2) (Figure 3B), which mediates the other arm of the NAD+ salvage pathway from nicotinamide ribose (Figure 3C).
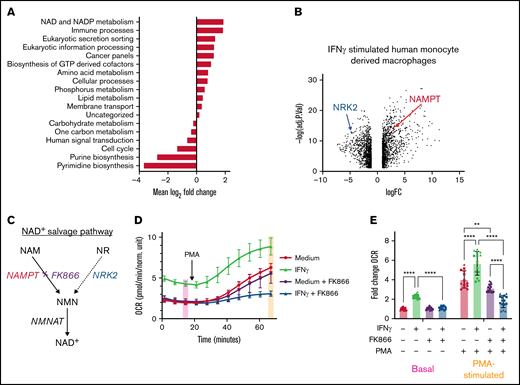
IFNγ induces NAMPT expression and IFNγ-induced increases in OCR are NAMPT-dependent. (A) Metabolic pathway analysis determined by the ERGO™ analysis platform based on differential gene expression from RNA-Seq data shown in panel B. (B) RNA sequencing of human monocyte–derived macrophages stimulated with IFNγ for 24 hours. (C) Diagram representing both NAM/NAMPT and nicotinamide ribose (NR)/NRK2-dependent NAD+ salvage pathways. (D) OCR measured by Seahorse in primary human monocytes treated with media alone or IFNγ with or without NAMPT inhibitor, FK866 (5 nM), for 24 hours prior to the start of the assay (n = 5 technical replicates). Data in panel D are from a representative tracing from 3 independent experiments. (E) Basal and PMA-stimulated OCR values from 3 independent experiments are summarized following normalization to the basal OCR values from samples without IFNγ, FK866, or PMA treatment. Data in panel E were analyzed by 2-way ANOVA with Sidak’s multiple comparisons test. Error bars are mean ± standard error of the mean. **P < .01, ***P < .001, ****P < .0001.
IFNγ induces NAMPT expression and IFNγ-induced increases in OCR are NAMPT-dependent. (A) Metabolic pathway analysis determined by the ERGO™ analysis platform based on differential gene expression from RNA-Seq data shown in panel B. (B) RNA sequencing of human monocyte–derived macrophages stimulated with IFNγ for 24 hours. (C) Diagram representing both NAM/NAMPT and nicotinamide ribose (NR)/NRK2-dependent NAD+ salvage pathways. (D) OCR measured by Seahorse in primary human monocytes treated with media alone or IFNγ with or without NAMPT inhibitor, FK866 (5 nM), for 24 hours prior to the start of the assay (n = 5 technical replicates). Data in panel D are from a representative tracing from 3 independent experiments. (E) Basal and PMA-stimulated OCR values from 3 independent experiments are summarized following normalization to the basal OCR values from samples without IFNγ, FK866, or PMA treatment. Data in panel E were analyzed by 2-way ANOVA with Sidak’s multiple comparisons test. Error bars are mean ± standard error of the mean. **P < .01, ***P < .001, ****P < .0001.
Addition of exogenous NAMPT substrate, NAM, did not augment IFNγ-induced increases in oxygen consumption (supplemental Figure 2A), consistent with the understanding that NAMPT activity is the rate-limiting step in NAM-dependent NAD+ salvage.44 We also assessed the dose-dependent inhibitory effects of the NAMPT inhibitor FK866 (APO866, Daporinad) and selected a dose of 5 nM FK866 to minimize off-target effects (supplemental Figure 2B).
FK866 completely blocked the IFNγ-induced increases in basal and PMA-stimulated OCR. Interestingly, NAMPT inhibition preferentially affected M(IFNγ) monocytes (Figure 3D), likely because IFNγ actively downregulates NRK2 expression, making M(IFNγ) monocytes even more dependent on NAMPT for NAD+ salvage (Figure 3B-C). Basal OCR levels in M(IFNγ) monocytes were 2.2-fold higher, and PMA-stimulated OCR levels were 1.4-fold higher, compared with M(0) monocytes. However, when FK866 was added throughout the 24-hour IFNγ stimulation period, the IFNγ-induced increases in basal and PMA-stimulated OCR were completely abrogated (Figure 3E).
Glycolysis is an NAD+-dependent pathway in which GAPDH activity depends on reduction of NAD+ to NADH.22 Consistent with this finding, we observed an inhibition of IFNγ-induced glycolysis with NAMPT inhibition (supplemental Figure 2C).22 Although other inhibitors of OCR (e.g., DPI or rotenone) cause a compensatory increase in basal ECAR when OCR is inhibited (supplemental Figure 2D), FK866 inhibits both ECAR and OCR. Therefore, we assessed the viability of FK866-treated monocytes. We first demonstrated that FK866 did not cause significant cytotoxicity at the doses used in these experiments (supplemental Figure 2E). We next rescued the diminished OCR and ECAR phenotypes with exogenous nicotinamide mononucleotide, the product of NAMPT. High doses of nicotinamide mononucleotide rescued both OCR and ECAR in M(IFNγ) monocytes treated with FK866, suggesting that these cells are both viable and metabolically active (supplemental Figure 2F-G). Taken together, these results suggest that IFNγ reprograms NAD+ salvage and renders monocytes dependent on NAMPT activity to maintain elevated levels of basal and PMA-stimulated OCR induced by IFNγ.
STAT1 is required for IFNγ-induced oxygen consumption and regulates oxygen consumption via mitochondrial complex I
To further elucidate the signaling pathways that mediate IFNγ-induced increases in OCR and the physiologic relevance of our assay, we assessed 2 patients with STAT1 GOF mutations (Table 1) and observed higher basal OCR (P = .192) and significantly higher PMA-stimulated OCR (P = .027) in M(IFNγ) monocytes. Though not significant, basal OCR levels in M(0) STAT1 GOF patient monocytes were higher than basal OCR levels in M(IFNγ) healthy control monocytes (Figure 4A). We also observed that expression of genes involved in NAD+ salvage (NAMPT, P2RX7, and CD38) were elevated in monocytes from a STAT1 GOF patient, and subsequently reduced to normal or below normal levels, after the patient was treated with the JAK inhibitor ruxolitinib (supplemental Figure 3A-C). These data show that STAT1 mediates IFNγ-induced increases in OCR and likely acts through modulation of NAD+ salvage gene expression.
Patients assayed for metabolic dysregulation
| Patient diagnosis | Patient age (y) | Patient sex (M/F) | Gene affected | Gain/loss of function |
|---|---|---|---|---|
| STAT1 GOF | 13 | F | STAT1 | GOF |
| STAT1 GOF | 34 | M | STAT1 | GOF |
| Leigh syndrome | 5 | F | MT-ND1 | LOF |
| STAT1 GOF (± ruxolitinib) | 34 | M | STAT1 | GOF |
| Leigh syndrome | 10 | M | MT-ND1 | LOF |
| CGD | 57 | F | p47phox | LOF |
| CGD | 24 | M | gp91 | LOF |
| CGD | 38 | M | gp91 | LOF |
| CGD | 14 | M | gp91 | LOF |
| Patient diagnosis | Patient age (y) | Patient sex (M/F) | Gene affected | Gain/loss of function |
|---|---|---|---|---|
| STAT1 GOF | 13 | F | STAT1 | GOF |
| STAT1 GOF | 34 | M | STAT1 | GOF |
| Leigh syndrome | 5 | F | MT-ND1 | LOF |
| STAT1 GOF (± ruxolitinib) | 34 | M | STAT1 | GOF |
| Leigh syndrome | 10 | M | MT-ND1 | LOF |
| CGD | 57 | F | p47phox | LOF |
| CGD | 24 | M | gp91 | LOF |
| CGD | 38 | M | gp91 | LOF |
| CGD | 14 | M | gp91 | LOF |
Patients’ clinical diagnosis, age, sex, affected gene, and functional consequence of their genetic mutation are listed as they appear in the corresponding figures.
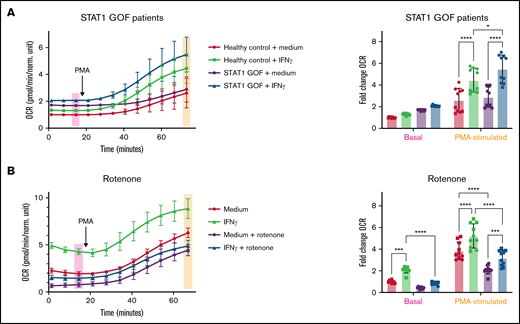
STAT1 is required for IFNγ-induced oxygen consumption and regulates oxygen consumption via mitochondrial complex I. (A) OCR measured by Seahorse in primary human monocytes treated with media alone or IFNγ for 24 hours prior to the start of the assay (n = 5 technical replicates). (A) Primary monocytes from 2 healthy controls and 2 STAT1 GOF patients are represented in the composite tracing from 2 independent experiments with values normalized to the unstimulated healthy control samples run in the same experiment. (B) Primary monocytes from a healthy control were stimulated with media alone or IFNγ with or without rotenone (10 μM) treatment throughout stimulation. Data are representative of 3 independent experiments and summarized in the bar graphs with basal and PMA-stimulated OCR values normalized to the healthy control unstimulated samples run in the same experiment. Basal and PMA-stimulated OCR values from respective experiments are summarized in bar graphs, and data were analyzed by 2-way ANOVA with Sidak’s multiple comparisons test. Error bars are mean ± standard error of the mean. *P < .05, **P < .01, ***P < .001, ****P < .0001.
STAT1 is required for IFNγ-induced oxygen consumption and regulates oxygen consumption via mitochondrial complex I. (A) OCR measured by Seahorse in primary human monocytes treated with media alone or IFNγ for 24 hours prior to the start of the assay (n = 5 technical replicates). (A) Primary monocytes from 2 healthy controls and 2 STAT1 GOF patients are represented in the composite tracing from 2 independent experiments with values normalized to the unstimulated healthy control samples run in the same experiment. (B) Primary monocytes from a healthy control were stimulated with media alone or IFNγ with or without rotenone (10 μM) treatment throughout stimulation. Data are representative of 3 independent experiments and summarized in the bar graphs with basal and PMA-stimulated OCR values normalized to the healthy control unstimulated samples run in the same experiment. Basal and PMA-stimulated OCR values from respective experiments are summarized in bar graphs, and data were analyzed by 2-way ANOVA with Sidak’s multiple comparisons test. Error bars are mean ± standard error of the mean. *P < .05, **P < .01, ***P < .001, ****P < .0001.
The primary connection between NAD+ metabolism and cellular oxygen consumption is mediated by mitochondrial complex I (NADH ubiquinone oxidoreductase), where NADH is oxidized, regenerating NAD+ for use in vital cellular reactions and initiating mitochondrial electron transport. Mitochondrial oxygen consumption occurs as a product of electron transport by 2 mechanisms: (1) production of water as molecular oxygen acts as the final electron acceptor and (2) electron leak, primarily from complexes I and III, which can generate superoxide.45-52 Treatment with the chemical inhibitor of mitochondrial complex I, rotenone, throughout the 24-hour IFNγ stimulation period abrogated IFNγ-induced increase in OCR (Figure 4B).
In addition to chemical inhibition of mitochondrial complex I, we also assessed primary monocytes from a patient with a clinical diagnosis of Leigh syndrome, caused by mitochondrial MT-ND1 (m.3697 G>A; Table 1), resulting in reduced function of mitochondrial complex I.53-56 Because this gene is maternally inherited, we assessed the proband’s father as an unaffected healthy control. One of the hallmarks of mutations in mitochondrial DNA is heteroplasmy, in which an individual may possess both wild-type and mutant mitochondrial DNA in the same cell,57 the balance of which determines mitochondrial function and cellular health. The proband was homoplasmic (100%) for m.3697 G>A, whereas her sibling was 80% heteroplasmic. As expected, the proband had significantly lower basal and PMA-stimulated OCR than her unaffected father, whereas her sibling had an intermediate level of OCR (supplemental Figure 3D-F). Therefore, both chemical and genetic disruptions of mitochondrial complex I inhibit IFNγ-induced OCR.
PMA-stimulated OCR measure NADPH oxidase activity
Besides oxidation by mitochondrial complex I, NAD(H) can also be converted into NADP(H) by cytosolic and mitochondrial NAD+ kinases43,58 or the mitochondrial enzyme nicotinamide nucleotide transhydrogenase, which transfers a hydride ion from NADH to NADP+.59,60 Oxidation of NADPH by the NADPH oxidase is the primary source of superoxide production during the respiratory burst, required for effective clearance of some pathogens by phagocytes. Patients with chronic granulomatous disease (CGD) have genetic defects in 1 of 6 NADPH oxidase components and characteristically present with recurrent infections and immune dysregulation including granulomatous inflammation.61-63 Therefore, we assessed 4 patients with loss-of-function mutations in various NADPH oxidase genes (Table 1). Interestingly, when compared with healthy controls, CGD patient cells demonstrated no increase in OCR in response to PMA stimulation. Although basal OCR levels (in M[0] and M[IFNγ] monocytes) were similar to those of healthy controls, there was no PMA-stimulated increase in OCR (Figure 5A), suggesting that the PMA-stimulated increases in OCR measured by our assay represented superoxide production by the NADPH oxidase complex.

PMA-stimulated OCR measures NADPH oxidase activity. (A-B) OCR was measured by Seahorse in primary human monocytes treated with media alone or IFNγ for 24 hours prior to the start of the assay (n = 5 technical replicates). (A) Monocytes from 4 healthy controls (black and green lines) and 4 CGD patients (red lines) were compared. (B) Monocytes were treated with (red lines) or without (black and green lines) DPI (100 uM) in addition to IFNγ or media alone for 24 hours. All OCR values were normalized by staining cells with 2 μg/mL Hoechst 33342 (ThermoFisher Scientific) for 30 minutes. Cells were then imaged and counted using the Biotek Cytation1. Cell counts were calculated by cell imaging software (Agilent) and imported into Wave (Agilent) using the normalization function. Raw OCR values were normalized by cell counts per well. (C) Bar graphs displaying PMA-stimulated OCR values normalized to the basal OCR levels in healthy control plus medium samples run in the same experiment (data not shown). Data are representative of at least 3 independent of experiments and were analyzed by 1-way ANOVA with Tukey’s multiple comparisons test. Error bars are mean ± standard error of the mean. ****P < .0001.
PMA-stimulated OCR measures NADPH oxidase activity. (A-B) OCR was measured by Seahorse in primary human monocytes treated with media alone or IFNγ for 24 hours prior to the start of the assay (n = 5 technical replicates). (A) Monocytes from 4 healthy controls (black and green lines) and 4 CGD patients (red lines) were compared. (B) Monocytes were treated with (red lines) or without (black and green lines) DPI (100 uM) in addition to IFNγ or media alone for 24 hours. All OCR values were normalized by staining cells with 2 μg/mL Hoechst 33342 (ThermoFisher Scientific) for 30 minutes. Cells were then imaged and counted using the Biotek Cytation1. Cell counts were calculated by cell imaging software (Agilent) and imported into Wave (Agilent) using the normalization function. Raw OCR values were normalized by cell counts per well. (C) Bar graphs displaying PMA-stimulated OCR values normalized to the basal OCR levels in healthy control plus medium samples run in the same experiment (data not shown). Data are representative of at least 3 independent of experiments and were analyzed by 1-way ANOVA with Tukey’s multiple comparisons test. Error bars are mean ± standard error of the mean. ****P < .0001.
We also tested a chemical inhibitor of NADPH oxidase, DPI.64 Like CGD patient monocytes, healthy control monocytes treated with DPI showed no increase in OCR in response to PMA. DPI treatment also reduced basal OCR levels (Figure 5B), which was consistent with reports of off-target effects of DPI, including inhibition of mitochondrial complex I, which has a structure similar to NADPH oxidase.65-67 Recognizing that the PMA-stimulated OCR in our assay represented NADPH oxidase-dependent oxygen consumption further clarified the mechanism by which IFNγ acts to augment the monocyte respiratory burst.23,24,26,68 -70 It also demonstrated that the reduction in PMA-stimulated OCR observed with NAMPT inhibition caused a diminished IFNγ-induced respiratory burst, comparable to that of CGD (Figure 5C). Our findings in CGD patient monocytes (and with chemical inhibition of NADPH oxidase) demonstrate that the measured OCR values are indicative of cellular ROS production. As opposed to conventional methods of ROS detection, which measure the reactive oxygen product, this method approximates ROS production based on the consumption of molecular oxygen in the context of ROS-promoting (or ROS-inhibiting) conditions.
IFNγ coordinately regulates transcription of multiple pathways to promote both NAM-dependent and -independent NAD salvage
In addition to NAMPT, we also found that IFNγ transcriptionally regulated several other genes that promote NAD(H) import and reduction/oxidation. IFNγ induced P2RX7, which forms an adenosine triphosphate (ATP)-gated pore that can import extracellular NADH, and CD38, an NAD+ consuming ectoenzyme that converts extracellular NAD+ into NAM, the membrane permeable substrate for NAMPT (Figure 6A-B). IFNγ also downregulated PDK4, an inhibitor of pyruvate dehydrogenase (PDH), which could allow for increased PDH activity, potentially increasing flux through the TCA cycle and NAD+ reduction (Figure 6B; supplemental Figure 5A). Furthermore, NADPH oxidase complex genes were upregulated by IFNγ, whereas nuclear-encoded mitochondrial complex I genes were mostly downregulated (supplemental Figure 4A-B). Minhas et al demonstrated a role for de novo NAD+ biosynthesis in the immunologic activity of resting human macrophages but did not assess metabolic changes in response to IFNγ.71 We found that genes involved in de novo and Preiss-Handler pathways of NAD+ biosynthesis were broadly downregulated in response to IFNγ (supplemental Figure 4C), as opposed to genes involved in NAD+ salvage.

IFNγ coordinately regulates transcription of multiple pathways to promote both NAM-dependent and -independent NAD salvage. (A) Diagram of pathways by which IFNγ regulates cellular NAD+ metabolism. IFNγ activates STAT1, inducing CD38, NAMPT, and P2RX7 and inhibiting PDK4 and NRK2. (B) Heat map generated from RNA-Seq analysis of human monocyte–derived macrophages stimulated with IFNγ for 24 hours displaying genes involved in NAD(H) salvage. ND, normal donor.
IFNγ coordinately regulates transcription of multiple pathways to promote both NAM-dependent and -independent NAD salvage. (A) Diagram of pathways by which IFNγ regulates cellular NAD+ metabolism. IFNγ activates STAT1, inducing CD38, NAMPT, and P2RX7 and inhibiting PDK4 and NRK2. (B) Heat map generated from RNA-Seq analysis of human monocyte–derived macrophages stimulated with IFNγ for 24 hours displaying genes involved in NAD(H) salvage. ND, normal donor.
We then used several inhibitors to determine which mitochondrial processes were required to maintain IFNγ-induced increases in OCR (supplemental Figure 5A). Compared with inhibitors that block both mitochondrial ATP synthesis and electron transport (rotenone, antimycin A, and oligomycin),48 -50,72,73 treatment with the mitochondrial ionophore FCCP, which uncouples ATP synthesis from electron transport, did not affect IFNγ-induced increases in basal or PMA-stimulated OCR (supplemental Figure 5D vs supplemental Figure 5B-C).74 Therefore, IFNγ-induced OCR is not dependent on mitochondrial ATP production but rather on the capacity to support electron transport. These findings suggest that IFNγ coordinately regulates a transcriptional program that increases cellular NAD(P)H available for oxidation. The result of this oxidation likely serves to both produce ROS and to recycle NAD(P)H between its oxidized and reduced states for activity as cofactors in vital cellular reactions.
Discussion
Metabolic reprogramming of immune cells in response to activation is essential for their immunologic activity,22,37,75 and disruption of these metabolic pathways has the potential to alter immune cell function. We investigated the metabolic reprogramming associated with IFNγ stimulation of human monocytes and identified a primary role for IFNγ in the regulation of NAD+ metabolism. This metabolic phenotype is not observed with type I interferons and is dependent on NAMPT. Inhibition of NAMPT with a specific chemical inhibitor, FK866, completely abrogated the IFNγ-induced increases in basal and PMA-stimulated OCR (Figure 2D-E).
We also demonstrated that IFNγ-induced increases in OCR are dependent on both STAT1 and mitochondrial complex I.76 Patients with STAT1 GOF mutations or MT-ND1 Leigh syndrome had altered OCR compared with healthy controls (Figure 3A-C). PMA-stimulated OCR levels measured in our assay represent oxygen consumption by the NADPH oxidase complex and were completely absent in CGD patient and DPI-treated monocytes (Figure 4A-B). Therefore, we have identified a metabolic pathway by which IFNγ-induced NAMPT augments NAD+ salvage, which is required for complete induction of the NADPH oxidase-mediated respiratory burst. Further investigation is required to directly assess the immunologic consequences of NAMPT inhibition. However, our data demonstrate the functional similarity between NAMPT inhibition and NADPH oxidase inhibition/dysfunction in monocytes, suggesting probable overlap in the immunologic effects of these pathways.
Our data provide a more detailed understanding of how IFNγ acts to activate monocytes in health and disease. Increasing microbicidal ROS production through an NAD(P)H-dependent mechanism may provide the added benefit of maintaining NAD(H) and NADP(H) availability intracellularly as cofactors for many cellular reactions, including the essential antioxidant processes involving glutathione and thioredoxin reductases.77-80 Therefore, the regulation of NAD+ metabolism by IFNγ may serve to augment ROS production and simultaneously enhance the activity of antioxidant processes required to protect the host cell from oxidative stress.81 Consistent with this hypothesis, we found that mitochondrial ATP synthesis was dispensable for maintaining the IFNγ-induced respiratory burst (supplemental Figure 6D), suggesting that IFNγ-induced NAD+ salvage does not primarily function to supply NADH for the purpose of ATP synthesis. Rather, increased NAD+ salvage likely supports mitochondrial ROS (mROS) production and synthesis/recycling of NAD(H) and NADP(H). These act as cofactors for critical oxidative and antioxidant processes.22,29,43,82
In contrast to superoxide production by the NADPH oxidase complex, less is known about the specific immunologic effects of mROS. Sander and Garaude recently reviewed the multifaceted relationship between innate immune responses and mitochondria,49,83 and Kiritsy et al provided evidence of crosstalk between IFNγ signaling and mitochondria, demonstrating that IFNγ-induced activation of antigen presenting cells is dependent on mitochondrial complex I activity.84 Other reports suggest that mitochondrial superoxide serves a similar microbicidal function as NADPH oxidase–derived superoxide.85 Interestingly, NADPH oxidase-produced ROS has been shown to induce mROS, and mROS can, in turn, activate NADPH oxidase.86
The NADPH oxidase complex and mitochondrial complex I (NADH ubiquinone oxidoreductase) are not only related in their ability to oxidize NAD(P)H to produce ROS but also in that their substrates are almost identical derivatives of NAD+. We now understand that oxygen consumption by both oxidases is regulated by IFNγ-induced NAMPT activity in human monocytes. This bidirectional relationship is likely important for both maximizing ROS production during the respiratory burst and for effectively inducing protective antioxidant programs through (1) recycling NAD(P)H between its oxidized and reduced states and (2) by activating redox sensitive mitochondrial antioxidants.86
Expression of NAMPT is precisely regulated and highly inducible in response to inflammatory stimuli, including IFNγ, compared with the resting state.22 Impaired upregulation of NAMPT, as in the setting of IFNγ signaling defects, could therefore affect bacterial killing during infection through its effects on superoxide production. Conversely, sustained or dysregulated expression of NAMPT could maintain/promote excessive inflammation, causing collateral tissue damage and chronic cellular stress. High levels of superoxide production have been associated with chronic inflammatory diseases such as atherosclerosis and are less prevalent in CGD patients who do not produce superoxide.87,88 For this reason, NADPH oxidase inhibitors have been trialed as immunomodulators to protect against oxidative stress.89-93 Our data suggest that modulation of NAMPT activity could have a similarly protective effect in states of chronic inflammation. By contrast, induction of NAMPT activity could be beneficial in states of IFNγ deficiency to augment the respiratory burst for microbicidal purposes.94
IFNγ was first trialed as a therapeutic in CGD with the aim of augmenting the respiratory burst. It was ultimately approved for clinical use to reduce the frequency and severity of infections associated with CGD but did not significantly increase superoxide production in CGD patients.95 Although the mechanism of action of IFNγ in CGD still remains elusive, our data provide a metabolic mechanism by which IFNγ enhances NADPH oxidase superoxide production in normal human monocytes. This may explain why this effect is not observed in CGD patients, namely that IFNγ acts to augment NADPH upstream of the defect in CGD.
In addition to NAMPT, we identified several other IFNγ-inducible genes involved in NAD(H) salvage, some of which have important clinical implications. P2RX7 has been associated with susceptibility to mycobacterial infection, an infection for which increased susceptibility is also associated with defects in IFNγ signaling. The mechanism of P2RX7-associated mycobacterial infection has not been completely elucidated, but our data suggest it could be related to its role in regulating the import of extracellular NADH.96,97 Similarly, drugs targeting CD38, such as daratumumab,98,99 may have additional immunomodulatory effects through altered monocyte NAD+ metabolism.100 Finally, the effects of IFNγ on PDH via downregulation of PDK4 suggest that metabolic diseases such as PDH deficiency likely have immunologic manifestations that are not yet completely recognized.53
Collectively, our data identify a novel immunometabolic phenotype of IFNγ-stimulated primary human monocytes, characterized by significantly increased oxygen consumption. We confirmed that these metabolic changes are physiologically relevant as they are dysregulated in specific patients and reproducible. Further investigation is required to determine the specific effects of NAMPT-mediated metabolism on IFNγ-induced immunologic functions and how flux through various NAD(H)-dependent metabolic pathways impacts cellular function more broadly. This understanding of the metabolic foundation of IFNγ-induced monocyte activation identifies several metabolic genes as potential causes or modifiers of immunologic and inflammatory diseases that should be evaluated as therapeutic targets for the modulation of IFNγ-mediated monocyte activation.
Acknowledgments
The authors thank the patients, their families, and the healthy volunteers who participated in this study. They also thank Peng Li for help with RNA-sequencing data.
This work was supported by the Intramural Research Program of NIAID and the NIH.
Authorship
Contribution: K.J.M., B.A.M., and S.M.H. conceived and designed experiments; K.J.M. performed experiments; C.L.D. and G.S. performed RNA sequencing and library preparation, respectively; S.M.C. and W.J.L. designed and analyzed RNA sequencing; K.J.M., S.M.C. and I.A.M. analyzed data; K.J.M., S.M.C., P.J.M., I.A.M., W.J..L, B.A.M., and S.M.H. interpreted data; P.J.M., C.S.Z., and S.M.H. provided clinical care; K.J.M. and S.M.H. wrote manuscript; and all authors read and approved the final manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Steven M. Holland, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Building 10/11N248 Mail Stop Code 1960, Bethesda, MD 20892; e-mail: smh@nih.gov.

