

Key Points
Patient-derived SS cells show highly heterogeneous drug responses.
We have developed a joint in vitro/in vivo platform to predict SS therapy response.

Abstract
Current therapeutic approaches for Sézary syndrome (SS) do not achieve a significant improvement in long-term survival of patients, and they are mainly focused on reducing blood tumor burden to improve quality of life. Eradication of SS is hindered by its genetic and molecular heterogeneity. Determining effective and personalized treatments for SS is urgently needed. The present work compiles the current methods for SS patient–derived xenograft (PDX) generation and management to provide new perspectives on treatment for patients with SS. Mononuclear cells were recovered by Ficoll gradient separation from fresh peripheral blood of patients with SS (N = 11). A selected panel of 26 compounds that are inhibitors of the main signaling pathways driving SS pathogenesis, including NF-kB, MAPK, histone deacetylase, mammalian target of rapamycin, or JAK/STAT, was used for in vitro drug sensitivity testing. SS cell viability was evaluated by using the CellTiter-Glo_3D Cell Viability Assay and flow cytometry analysis. We validated one positive hit using SS patient–derived Sézary cells xenotransplanted (PDX) into NOD-SCID-γ mice. In vitro data indicated that primary malignant SS cells all display different sensitivities against specific pathway inhibitors. In vivo validation using SS PDX mostly reproduced the responses to the histone deacetylase inhibitor panobinostat that were observed in vitro. Our investigations revealed the possibility of using high-throughput in vitro testing followed by PDX in vivo validation for selective targeting of SS tumor cells in a patient-specific manner.
Introduction
Cutaneous T-cell lymphomas (CTCLs) are lymphoid malignant neoplasms that initially manifest in the skin. The most frequent CTCLs are mycosis fungoides (MF) and Sézary syndrome (SS). SS is clinically characterized by erythroderma associated with peripheral blood (PB) involvement manifested by circulating malignant lymphoid cells with cerebriform nuclei (Sézary cells). SS is considered an aggressive CTCL with no curative alternative. Interferon, oral retinoids (bexarotene), and nonspecific histone deacetylase (HDAC) inhibitors, alone or in combination with photopheresis, are currently prescribed as therapeutic options, but most cases achieve response rates of ∼30%. Multiple evidence shows that standard polychemotherapy is not effective in SS, showing very short-lived responses, associated in part with the high inter- and intra-tumoral heterogeneity of the disease. Genetic characterization of SS cells has been improved by next-generation sequencing studies.1-5 Because of the low incidence of SS,6 data from multiple small cohorts have been combined for identifying rare driving mutations and genomic copy number variations; this has led to multiple pathway alterations with impact on disease progression by modulating T-cell activation, inhibiting apoptosis, affecting chromatin remodeling and epigenetic regulators, DNA damage response, cell cycle, or immune surveillance impairment. Most relevant genomic alterations found include genes such as TP53, PLCG1, CARD11, TNFRSF1B, STAT5B, DNMT3A, ARID1, FASN, or ZEB1.7,8
Some of the described molecular alterations are currently being investigated in clinical trials for their therapeutic use in MF or SS. The effectiveness of using the phosphatidylinositol 3-kinase (PI3K) (eg, duvelisib) or NF-κB (eg, bortezomib) inhibitors has been discussed in translational research and tested in few patients in clinical trials, and both have shown good tolerance and durable responses. The HDAC inhibitors vorinostat and romidepsin are approved in the United States for use in MF, and resminostat is in a phase 3 trial in the European Union. Currently, alisertib, an oral inhibitor of the aurora A kinase, and cobomarsen, an oligonucleotide inhibitor of miR-155, are under investigation, among other interesting targeted drugs. Finally, JAK/STAT inhibitors are increasingly being used in different hematologic and skin conditions and represent promising drugs for further clinical development in CTCL. However, advances in the genetic knowledge of CTCL tumor cells have not yet been translated into the implementation of targeted therapies with curative response rates. Immunotherapy-related treatments such as mogamulizumab have changed the management of patients with SS, and brentuximab, as well as activators of the antitumor immune response (anti–programmed death-1 [PD1]/programmed cell death ligand 1), seem to be well positioned as alternative therapeutics.9-11
In this context, new pathway-based personalized treatments are clearly needed to combat SS. Currently, ex vivo culture of patient-derived cancer cells and xenograft (PDX) models represent a tool for expanding human tumors, which can then be banked and used as in vivo models for translational research. These models have recurrently been shown to closely resemble patient tumor features and retain molecular and histologic characteristics. Recently, a few PDX models of primary SS cells have been established,12 but most in vivo investigations have been performed by using SS cell lines.13,14 The predictive value of in vitro and in silico data are ultimately limited by the complexity of the whole-organism systems. Thus, combining in vitro and in vivo models will allow a better translation of in vitro drug testing to patient treatment.
Methods
Patients and study samples
Eight patients with SS were included in the study. Clinical characteristics, peripheral blood flow cytometry (FC) parameters, patient treatments, and clinical status are presented in supplemental Table 1. Primary Sézary cells for in vitro drug screening and in vivo generation of PDX were obtained during the apheresis procedure currently performed as adjuvant treatment for patients with SS. An enriched sample of mononuclear cells was obtained by Ficoll separation.
Monitoring and quantification of aberrant malignant Sézary cells in vitro and in vivo were determined by FC for the standardized SS markers CD3+/CD4low/CD26–/CD7–15-17 using fluorescein isothiocyanate/phycoerythrin (FITC/PE)–conjugated monoclonal antibodies (BD Biosciences, San Diego, CA).
In vitro drug screening platform
Inhibitors used in the study were obtained from Selleck Chemicals drug library (https://www.selleckchem.com/screening/fda-approved-drug-library.html) (Houston, TX). Selection of a single-agent multidrug testing panel was based on standard treatments used in the clinic (ie, bexarotene, steroids, methotrexate, HDAC inhibitors) and the signaling pathways previously identified as relevant for SS progression7 (ie, JAK/STAT, PI3K/protein kinase B/mammalian target of rapamycin, MAPK, or TCR/NF-kB pathway inhibitors). Compounds (supplemental Table 2) were prepared as dimethyl sulfoxide (DMSO) stocks and then diluted as indicated. Then, 4 × 105 cells per well were seeded in 96-well plates and treated with the therapeutic agents for 72 hours. Additional controls were included that were treated with the minimum and maximum doses of dimethyl sulfoxide used as vehicle (0.1% and 20%).
Quantification of the response to pharmacologic inhibition in the in vitro assay
The effect of pharmacologic inhibition on cell viability after 72 hours of incubation was determined by using CellTiter-Glo_3D Cell Viability Assay (Promega, Madison, WI). We initially tested all compounds at 20, 4, and 0.8 µM. Compounds showing a minimum of a 50% decrease in cell viability at a concentration of 1 µM were further tested in dose–response assays.
In vivo murine model
Transplantation of Sézary cells was performed by intradermal injection of 2 million mononuclear cells in 20 µL of phosphate-buffered saline (obtained from apheresis) in the ear of NOD-SCID-γ (NSG) mice (The Jackson Laboratory, Bar Harbor, ME) at 8 to 12 weeks of age. PDX assays were performed in accordance with the regulations of Parc de Salut Mar-Parc de Biomèdica de Barcelona-Universitat Autònoma de Barcelona (TG-09-1223P3-ABS) and the current regulations on Protection of Personal Data and Guarantees of Digital Rights (General Data Protection Regulation of the EU, Regulation EU-2016/679 and Spanish Organic Law 3/2018), and the Biomedical Research Law 14/2007 and Royal Decree RD1716/2011.
Mice treatment was initiated when a minimum of 1% human CD45+ cells were detected in circulation. Vehicle alone was used in the untreated controls at the same doses and protocol of administration as the therapeutic agents. Specific drug schedule treatment was determined according to pharmacokinetic, 50% inhibitory concentration dosage (obtained in the in vitro analysis), and/or maximum reported doses for the specific drug in the in vivo mouse models.
Quantification of the response to pharmacologic inhibition in the in vivo model
PB samples from mice were obtained at different time points after transplantation and treatment, and at the experimental end point. They were further analyzed by FC with antibodies for Sézary cells. First bleeding was performed at 4 weeks after transplantation and subsequently repeated every 2 weeks. We established detection of 1% of human CD45+ cells in circulation as the criterion to start the in vivo treatment. After starting treatment, mice were bled weekly to quantify the aberrant (and normal) human T-cell lymphoid populations by FC. Compounds used for in vivo testing were selected based on their therapeutic efficacy in the in vitro assay and the possibility of a rapid translation to the clinics (ie, we favored compounds already used in the clinical practice or in clinical trials). At euthanasia, either at the end point of the study or when mice showed symptoms of suffering, we analyzed tumor burden in PB by FC, and we also measured visceral infiltration by hematoxylin and eosin and immunohistochemical (IHC) analysis of paraffin-embedded tissue sections.
Histopathologic examination
Paraffin blocks of all tumor tissues in mice were fixed in 10% formalin solution, processed by standard methods, embedded in paraffin, sectioned at 2 μm, and stained with hematoxylin and eosin and with an IHC lymphoid panel.
Results
SS patients and sample characterization
We collected clinical samples from 8 patients with SS, newly diagnosed and under first-line treatments. Sézary cell count in PB ranged from 5.4% to 59% of total leukocytes, and up to 95% of the T lymphocytes (supplemental Table 1) measured by FC analysis (Figure 1). Of the 8 patients with SS analyzed, only 1 displayed peripheral lymph node involvement (stage IVA2) as determined by histopathologic examination of the biopsy specimens.

High efficiency purification of SS cells from patient photopheresis. Representative FC analysis of a blood sample obtained from the photopheresis of the indicated SS patient. Noteworthy is the high prevalence of malignant CD4+, CD26–, and CD7– lymphocytes.
High efficiency purification of SS cells from patient photopheresis. Representative FC analysis of a blood sample obtained from the photopheresis of the indicated SS patient. Noteworthy is the high prevalence of malignant CD4+, CD26–, and CD7– lymphocytes.
Primary Sézary cells display a highly heterogeneous sensitivity to specific pharmacologic inhibition
We first performed a single-agent titration (at 20, 4, and 0.8 µM) in primary patient-derived SS samples (schematic representation of the assay in Figure 2A) using 26 different therapeutic compounds that were selected based on their target selectivity and clinical applicability (supplemental Table 2). We detected a high heterogeneous response of individual primary SS samples against the different compounds tested (Figure 2B-C; supplemental Table 3). Likewise, single compounds produced very distinctive effects on the various patient samples, with the proteasome inhibitor bortezomib, bardoxolone methyl, and the HDAC inhibitor panobinostat showing the highest activity in most patient-derived samples (supplemental Figure 1A; supplemental Table 3).
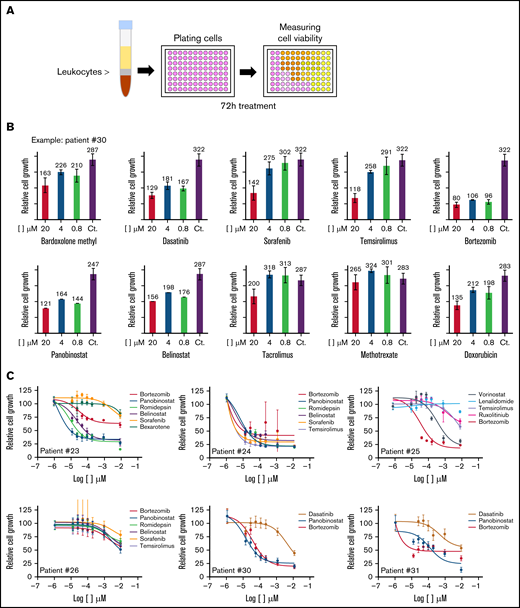
Variable drug sensitivity in leukemic cells from individual patients with SS. (A) Schematic representation of the protocol used for the in vitro drug screening. (B) Representative results obtained from patient #30. (C) Graphs representing the dose–response analysis of different SS patients treated as indicated. (D) Bar plots showing the high variation in the response of individual patients to specific compounds. (E) FC analysis of cells from patient #30 treated as indicated.
Variable drug sensitivity in leukemic cells from individual patients with SS. (A) Schematic representation of the protocol used for the in vitro drug screening. (B) Representative results obtained from patient #30. (C) Graphs representing the dose–response analysis of different SS patients treated as indicated. (D) Bar plots showing the high variation in the response of individual patients to specific compounds. (E) FC analysis of cells from patient #30 treated as indicated.
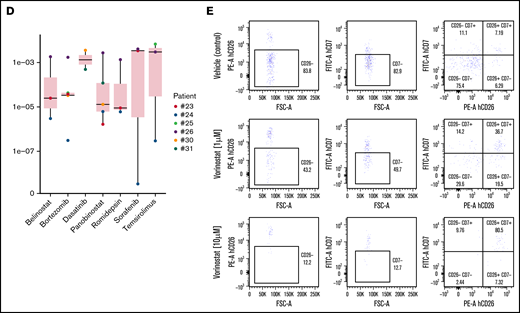
We next performed a more accurate dose–response assay for compounds that produced a minimum of a 50% decrease in cell viability (on specific SS samples) at concentrations of 1 µM (Figure 2C-D). Our results confirmed the high heterogeneous response of individual patient-derived SS cells against specific compounds, with some patients displaying high sensitivity against most drugs tested (ie, patient #24), whereas others were mostly insensitive to all treatments (ie, patient #26). Importantly, FC analysis of patient #25–derived SS cells with the HDAC inhibitor vorinostat confirmed selective killing of the malignant CD3+CD26– population after 72 hours of treatment at 1 µM and 10 µM (Figure 2E). In contrast, treatment with bortezomib imposed a nonspecific removal of the whole CD3 population (supplemental Figure 1B).
Generation of a robust model for primary SS PDX in mice
To establish an in vivo model for studying SS, we injected various numbers of fresh mononuclear cells (0.5, 1, and 2 × 106 cells) isolated from patient samples during apheresis procedures into the ear of NSG mice (n = 5 patients, 2 mice per condition). Schematic representation of the procedure used is shown in Figure 3A. Human CD45+ cells were consistently found in the circulation in the majority of recipient mice transplanted with 2 × 106 cells (Figure 3B), indicative of successful engraftment. In addition, a variable degree of human T lymphocytes was detected in the blood of mice that exhibited an aberrant phenotype (CD3+, CD4+, CD7–, and/or CD26–) consistent with Sézary cells (Figure 3C). As a control, NSG mice transplanted with mononuclear cells from healthy donors showed a consistent expansion of human T cells that were mainly CD7 and CD26 double positive (Figure 3D).
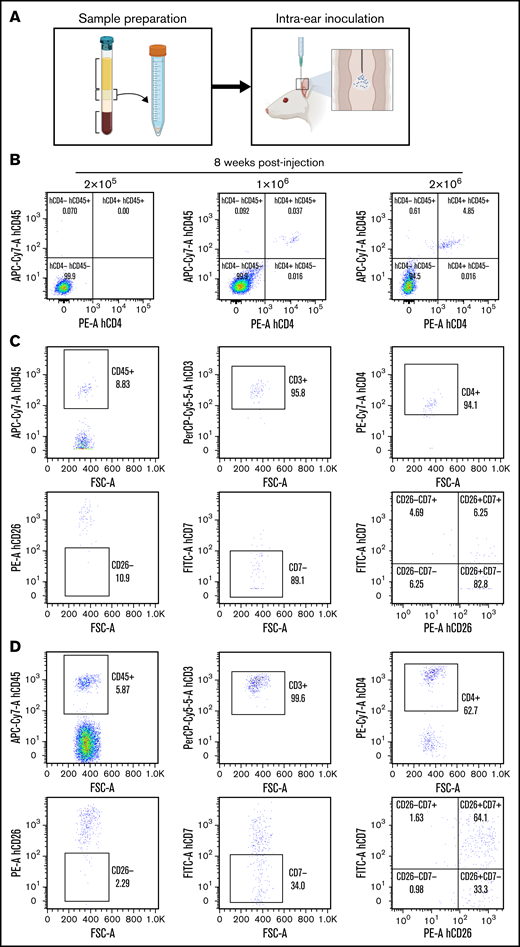
Robust in vivo xenograft model of patient-derived SS. (A) Schematic representation of the procedure used to generate the SS xenograft. (B-C) FC analysis of PB from mice at 8 weeks after transplantation with different numbers of SS cells from patient #23. (C) FC analysis with the indicated marker to confirm the outgrowth of malignant CD4+; CD7– SS cells in the recipient mice. (D) Same analysis as in panel C from the bone marrow (BM) of a mouse transplanted with mononucleated cells from a healthy donor.
Robust in vivo xenograft model of patient-derived SS. (A) Schematic representation of the procedure used to generate the SS xenograft. (B-C) FC analysis of PB from mice at 8 weeks after transplantation with different numbers of SS cells from patient #23. (C) FC analysis with the indicated marker to confirm the outgrowth of malignant CD4+; CD7– SS cells in the recipient mice. (D) Same analysis as in panel C from the bone marrow (BM) of a mouse transplanted with mononucleated cells from a healthy donor.
SS PDX cells reproduce the main characteristics of the original patient tumor
We next investigated whether this PDX model reproduced the clinicopathologic features of the original patient SS tumor. After 4 weeks of transplantation, mice injected with human SS cells rapidly developed symptoms of the disease; this included erythematous and scaly skin lesions and alopecia (Figures 4A) associated with the presence of a robust CD7– T-cell population in PB (supplemental Figure 2A-B) that was positive for the immune checkpoint PD1 (not depicted), as reported previously.18 However, we detected no obvious signs of SS infiltrates in the skin of mice (Figure 4B). From week 4 to week 6, the percentages of CD7– T cells were primarily maintained, but a rapid expansion of the human CD26– population was detected in most of the transplanted animals (supplemental Figure 3). Analysis of transplanted mice at euthanasia revealed the presence of human CD4+ cells in bone marrow, PB, spleen, lymph nodes, and liver (Figure 4C). IHC analysis of the spleens (Figure 4D) and livers (not depicted) showed extensive lymphomatous infiltrates that were CD3+, CD4+, and CD7– or low, further indicating that they correspond to malignant Sézary cells. Unexpectedly, by comparative analysis of the TCR repertoire in patient blood and PDX samples, we did not detect a predominant clonal amplification of the main patient-derived SS population in the transplanted mice but a heterogeneous expansion of different primary SS cell clones (Figure 4E).

Transplanted SS cells reproduce the human disease in mice. (A) Representative photograph of a mouse at 8 weeks after being transplanted with 2 million SS cells. (B) Hematoxylin and eosin (H&E) staining of a skin section of the same animal obtained at euthanasia. (C) FC analysis of the different organoids indicated the presence of SS infiltrates. (D) IHC analysis of liver sections to confirm FC data. (E) Clonality analysis by PCR analysis of SS from patient #23 and blood from 2 different mice transplanted with SS cells from the same patient. (F) IHC of the liver and spleen of a representative mouse transplanted with cells from patient #23. Note the high amounts of aberrant CD20+ B cells, already present in the patient, that infiltrate the organs in addition to the CD3+;CD4+;CD5+ SS population.
Transplanted SS cells reproduce the human disease in mice. (A) Representative photograph of a mouse at 8 weeks after being transplanted with 2 million SS cells. (B) Hematoxylin and eosin (H&E) staining of a skin section of the same animal obtained at euthanasia. (C) FC analysis of the different organoids indicated the presence of SS infiltrates. (D) IHC analysis of liver sections to confirm FC data. (E) Clonality analysis by PCR analysis of SS from patient #23 and blood from 2 different mice transplanted with SS cells from the same patient. (F) IHC of the liver and spleen of a representative mouse transplanted with cells from patient #23. Note the high amounts of aberrant CD20+ B cells, already present in the patient, that infiltrate the organs in addition to the CD3+;CD4+;CD5+ SS population.
Further supporting the concordance between patient samples and their corresponding PDX models, several of the mice transplanted with cells from SS patient #23 died as a result of massive B-cell lymphomatous infiltrates in the spleen and liver (Figure 4F); these were associated with the presence of an aberrant CD19++, CD20++. κ++ B-cell population (∼20% of lymphocytes) in the PB of this patient (supplemental Table 1).
These results indicate that the SS PDX model phenocopies the original SS patient populations.
SS PDX models provide a platform to screen targeted drug treatments
We investigated whether our in vitro drug screening was capable of predicting the therapeutic response of primary SS cells in the PDX model. Based on in vitro data, we selected bortezomib for in vivo treatment of samples from patient #23 (showing a medium response to the drug). We performed a first experiment with 10 mice (5 in the control group and 5 on treatment) and a second experiment with 12 mice (7 controls and 5 treated) (2 × 106 SS cells per mice transplanted). No significant differences were detected between the control and treatment groups in terms of survival or selective reduction of the pathologic cell population in the successive bleedings (data not shown) at the bortezomib doses reported in the literature.19 These results were in agreement with the nonselective activity of bortezomib observed in vitro (supplemental Figure 1B).
In a third experiment, we transplanted 12 NSG mice with 2 × 106 SS cells (per mice) from patient #30. Here, the HDAC inhibitor panobinostat was used as the therapeutic agent based on the high sensitivity of these cells in vitro (Figure 2C). Four weeks after transplantation, the presence of human SS cells was confirmed in the NSG mice circulation by FC. Because slight differences in the number of SS cells were detected in each recipient mouse, animals were homogeneously ascribed to the different experimental groups (control or panobinostat, 10 mg/kg oral administration for 5 consecutive days). After a first round of treatment (15 days), animals were left untreated for 15 days before starting a second round of treatment (15 days) to mimic the procedure used in patients (Figure 5A). FC analysis was performed every 2 weeks to monitor the presence of human T cells in the PB of mice. A significant increase was detected in the number of human CD45+CD3+ cells after the first round of treatment in the PB of control mice, which was not observed in 5 of 6 animals in the panobinostat-treated group (Figure 5B). The average percentage of CD45+CD3+ cells in the PB of control mice was 22.4 ± 20.8 compared with 2.0 ± 2.4 in the treated group (P = .06).
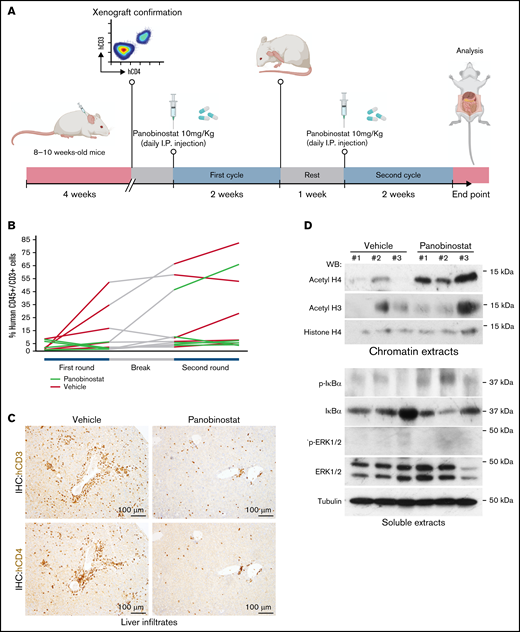
In vivo treatment reproduces the SS drug sensitivity determined in vitro. (A) Schematic representation of the treatment protocol in mice. (B) Representation of disease progression in the different groups of treatment as determined by FC analysis of PB. (C) Representative IHC analysis of the liver showing the presence of malignant SS cells specifically in vehicle-treated animals. (D) Western blot analysis of the indicated proteins in blood samples from 3 different vehicle-treated or panobinostat-treated mice. Note the increased levels of acetylated histone in the panobinostat-treated blood samples consistent with the HDAC inhibitory activity of this drug.
In vivo treatment reproduces the SS drug sensitivity determined in vitro. (A) Schematic representation of the treatment protocol in mice. (B) Representation of disease progression in the different groups of treatment as determined by FC analysis of PB. (C) Representative IHC analysis of the liver showing the presence of malignant SS cells specifically in vehicle-treated animals. (D) Western blot analysis of the indicated proteins in blood samples from 3 different vehicle-treated or panobinostat-treated mice. Note the increased levels of acetylated histone in the panobinostat-treated blood samples consistent with the HDAC inhibitory activity of this drug.
Further indicating the efficacy of panobinostat in vivo, we detected a transient increase in the human CD45+CD3+ population in both groups after concluding the first round of treatment, which was again reduced in the panobinostat group after restarting a second round of treatment. IHC analysis of the livers at euthanasia revealed the presence of a robust CD3+, CD4+ infiltrate, as previously found, in the vehicle-treated group that was absent in all but one of the animals treated with panobinostat (Figure 5C). These findings indicate that SS PDX retains the drug sensitivity observed in vitro and may represent a suitable preclinical model for human SS, as a previous step for personalized medicine.
Finally, we determined the effectiveness and specificity of panobinostat treatment by measuring the levels of acetylated histones in blood cells obtained at experimental end point. We performed western blot analysis of PB cell lysates with specific antibodies against acetylated histones H3 and H4. Increased histone acetylation was detected in samples from panobinostat-treated mice, indicative of effective HDAC inhibition even after long-term treatments (Figure 5D). In contrast, we did not detect off-target effects on IκBα, the classical readout of NF-κB activity, or p-ERK1/2, the downstream effector of the MAPK pathway.
Taken together, our data indicate that the SS PDX mouse model identified an efficacious therapy for patients with SS, strongly suggesting that the SS PDX model is a valid experimental platform that can guide clinical decision-making regarding therapeutic agents.
Discussion
CTCLs represent heterogeneous non-Hodgkin lymphomas, with SS being one of the most aggressive subtypes manifested by pruritic erythroderma, lymphadenopathy, and atypical circulating lymphocytes (Sézary cells). SS is a devastating disease with a high impact on the quality of life of patients. The pathogenic mechanisms are poorly understood, being molecularly complex and heterogeneous. Consequently, it is essential to obtain molecular-based personalized therapies (precision medicine).
There is enough evidence to state that standard polychemotherapy regimens are not effective in SS, showing very short-lived responses and short survivals. Thus, there is an urgent need for new effective treatments that have a safe profile, are well tolerated, and maintain long-term activity because standard therapeutic approaches in SS do not lead to a significant improvement in long-term survival. In recent years, knowledge regarding specific therapeutic targets has allowed the design of immunotherapies (both cytotoxic and immunoregulatory) such as brentuximab, mogamulizumab, or check-point inhibitors (PD1/programmed cell death ligand 1 inhibitors), which have modified the disease-free survival curves of CTCL patients in the short term. In younger patients, the therapeutic option of allogeneic hematopoietic stem cell transplantation can be proposed.
Integrated genomic data sets identified key elements and pathways that could be used to stratify patients with SS who would potentially benefit from future targeted therapies. Therefore, given the genetic and molecular heterogeneity of CTCL and particularly SS, it is essential to develop realistic and practical in vitro and in vivo models to test personalized targeted therapies in individual cases.
Many of the in vivo models use CTCL cell lines that do not represent an appropriate tool for personalized medicine. Among cell line–derived xenograft models, one the most notable was generated by injecting CTCL cells into mice in the context of an inflammatory environment, a scenario similar to the real pathologic situation.20,21 Also, intrahepatic injection of CTCL cell lines in NOD/SCID/IL2Rγ mice leads to successful engraftment and has been used to evaluate cell line tumorigenicity and therapeutic responses in preclinical studies.13 Using in vitro and in vivo models of human SS cell lines, we previously showed that canonical NF-kB is constitutively activated in CTCL cells, downstream of the TAK1 kinase. Inhibition of TAK1 prevented signaling through NF-κB and β-catenin, thus inducing apoptosis of CTCL cell lines and primary SS cells both in vitro and in vivo. In addition, the presence of activated TAK1, as determined by specific antibodies, correlated with activation of NF-κB and β-catenin, being indicative of CTCL, compared with different T lymphocyte–mediated inflammatory dermatoses. Thus, activated TAK1 was identified as a novel biomarker and therapeutic target for CTCLs,14 which functionally links NF-kB and β-catenin with CTCL initiation and/or progression. Recently, it was shown that transgenic mice with aberrant STAT3 activation in CD4 T cells develop clinical features of MF and SS, being microbiota required for acquisition of the complete clinical phenotype. This mouse model, which develops progressive disease, provides a new and useful tool for studying the cutaneous biology of T-cell lymphoma.22,23 Similarly, transgenic mice with constitutive overexpression of interleukin-15 develop an infiltration of the skin by mature T cells immunophenotypically mimicking the human disease.24 These models are an alternative tool for CTCL investigation.
PDX models currently represent a promising tool for translational research, as they closely resemble individual patient tumor features and retain molecular and histologic characteristics. PDX models have already been used in several solid tumors and validated as a robust model to evaluate drug response and therapeutic efficacy. However, their use in CTCL is still preliminary and scarce.21,25 The European consortium EuroPDX (www.europdx.eu) experience proposed PDX and orthotopic PDX as realistic and promising upcoming technologies to deep into: (1) the mechanisms of resistance to standard chemotherapy treatments; (2) the development of new therapeutic strategies; (3) the contribution of tumor microenvironment to cancer progression and its role in the development of resistance to targeted therapies; and (4) the improvement of models to evaluate personalized therapies in real time integrating the tumor genetics with the response to drugs.26,27
We have now established a robust PDX model for SS and showed its usefulness in predicting therapeutic response of tumor cells. Unexpectedly, we found that the main clone present in the patient was not predominant in the recipient mice, but several clones already present in the patient were randomly selected. Almost identical subclonal heterogeneity of the SS PDX system has been recently described by Poglio et al.12 Subclonal selection is likely associated with immunophenotypic plasticity and limited genomic evolution in response to environmental conditions. Unfortunately, we were not able to reproducibly expand human SS cells in secondary NSG recipients, suggesting that a low number of tumor-initiating cells recovered from the primary transplantation.
The present work compiles current methods for an SS PDX generation and its utility, providing new perspectives for the rapid translation of results to clinical practice. Our ongoing work is focused on studying the likely correlation between drug responses and the mutational landscape of individual SS patients to uncover biomarkers that could eventually inform future personalized therapies. This is particularly important in highly heterogeneous diseases such as SS in which specific therapies will fail in the absence of patient selection criteria. For example, we have now reported the preclinical efficacy of panobinostat on patient-derived SS cells previously characterized as sensitive in vitro; however, the same compound showed a modest but demonstrated efficacy in a phase 2 clinical trial but still higher in SS than in MF.28 In addition, we found that SS cells from patient #23 exhibited the highest response to inhibitors of the NF-kB pathway such as bardoxolone methyl and bortezomib in the in vitro assays (supplemental Table 3). Although we did not perform any in vivo experiment with these cells, patient #23 is now on mogamulizumab treatment, a blocking antibody against CCR4 that was found to induce NF-kB and PI3K pathways, and has achieved complete remission of both the circulating Sézary cell population and the aberrant B-cell population.
In case of treatments that are indicated for other tumor types, the possibility of recommending their use for treatments in selected patients could be considered. Our goal is to develop a realistic and practical preclinical strategy to identify new biomarkers of prognostic and pathway-based therapeutic strategies.
Acknowledgments
The authors thank all members of the Gallardo, Espinosa, and Bigas laboratories for their support and helpful comments and suggestions. They also thank Patricia Charoenrook for helping with English editing.
This work has been supported by grants PI19/00013, PI18/00021, and PI21/0390 from Instituto de Salud Carlos III/FEDER and grant 2017-SGR 135 from AGAUR from the Government of Catalonia and CIBERONC.
Authorship
Contribution: F.G., R.M.P., A.B., and L.E. designed the study, analyzed data, and wrote the manuscript; E.A., A.I., J.G., and L.S. performed experiments and analyzed data; Y.G., G.B., L.C., E.G., D.C., E.R., and I.B.-M. analyzed data; and M.I., B.B., and J.R.R. performed the phenotypic characterization of SS cells.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Fernando Gallardo, Dermatology Department, Hospital del Mar, Passeig Marítim 25-29, 08003 Barcelona, Spain; e-mail: fgallardo@parcdeslautmar.cat; Anna Bigas, Cancer and Stem Cells Research Group, Institut Hospital del Mar d'Investigacions Médiques, CIBERONC, Dr Aiguader 88, 08003 Barcelona, Spain; e-mail: abigas@imim.es; and Lluís Espinosa, Cancer and Stem Cells Research Group, Institut Hospital del Mar d'Investigacions Médiques, CIBERONC, Dr Aiguader 88, 08003 Barcelona, Spain; e-mail: lespinosa@imim.es.

