

TO THE EDITOR:
The proto-oncogene MYB encodes the transcription factor c-MYB (cellular MYB, hereafter called MYB), which is often upregulated or aberrantly activated in cancer, including hematological malignancies.1,2 High Myb levels were especially found in acute myeloid leukemia (AML).2-4 Myb was identified initially as a retroviral oncogene (v-Myb) of avian myeloblastosis virus and E26.5,6 These retroviruses are able to transform immature hematopoietic cells in vitro and induce AMLs in chickens7 and mice.8 In leukemia patients, MYB is highly expressed, and in a subset of patients, this is a consequence of translocations, genomic duplications, or somatic mutations that involve the MYB gene itself.9-12 Furthermore, compelling evidence is accumulating that MYB also acts as a dependency factor for the maintenance of most myeloid, T-, and B-cell leukemias.13-15 Overexpression of viral MYB, a truncated form of MYB that lacks its negative regulatory domain, results in the spontaneous formation of T-cell lymphomas in mice.16 However, the in vivo roles of cellular MYB in tumor initiation remain largely unexplored. Here, we show, for the first time, that hematopoietic-specific overexpression of Myb is sufficient to drive B-cell neoplasms and myeloid malignancies in mice.
To evaluate whether elevated MYB expression is sufficient to transform cells in vivo, we developed a conditional Myb overexpression (R26-Myb) mouse model (Figure 1A; supplemental Figure 1A,B) using an optimized pipeline for targeting the Rosa26 (R26) locus,17 which previously allowed us to model AML,17 mantle cell lymphoma,18 and immature T-cell leukemia19 in mice. Cre-mediated removal of the floxed stop cassette in mouse embryonic stem cells resulted in a 10-fold increase of Myb transcripts and a threefold upregulation of MYB protein (supplemental Figure 1C,D). R26-Myb mice were crossed with Vav-iCre mice20 to enable R26-driven overexpression of Myb and a Firefly luciferase-reporter in the entire hematopoietic system (supplemental Figure 1E). Homozygous R26-Myb mice have a 10- to 15-fold increase in Myb RNA levels in the thymus and bone marrow (BM) (supplemental Figure 1F,G). Of note, we were not able to show increased MYB protein level of 10-week-old thymocytes (data not shown), probably because T-cell progenitors already express high endogenous MYB levels. We monitored an aging cohort of R26-Mybtg/tg; Vav-iCretg/+ (hereafter named MybVav; n = 21 with 9 males and 12 females) mice (Figure 1A) and found that 7 out of 21 (33%) animals spontaneously developed hematological malignancies (Figure 1B). Detailed necropsy was performed on 4 animals. MybVav mice had mild anemia and showed increased white blood cell and lymphocyte counts in peripheral blood (Figure 1C). In addition, 3 of them displayed splenomegaly (Figure 1D). Detailed flow cytometric analysis revealed that Myb overexpression resulted in the spontaneous development of both B-cell neoplasms and myeloid malignancies (Figure 1E,F). A selection of 1 MybVav tumor per mouse is represented in Figure 1E, while an overview of the immunophenotype of all MybVav tumors per mouse is shown in Figure 1F. A comparison of these MybVav tumors with their Cre-negative littermate controls can be found in supplemental Figure 2. Although cumulative genetic evidence suggests a role for Myb in T-cell leukemia,9-11 no spontaneous T-cell malignancies were observed in MybVav mice. One explanation for this might be that Myb is already highly expressed in T-cell progenitors and that the additional R26-driven Myb does not have a major impact on the already high Myb levels in this lineage. One Myb-driven malignancy displayed increased percentages of conventional CD19+B220+ B cells in the liver and spleen compared with the corresponding organs of nondiseased Cre− littermate controls (supplemental Figure 2A). B-cell populations in this MybVav tumor (mouse #1) reached 44% and 58% in the spleen and lymph node (LN), respectively, while T-cell and myeloid populations were only underrepresented (supplemental Figure 2B,C). Two MybVav mice (mouse #2 and #4) had tumors that displayed dominant myeloid cell fractions in BM (62%) and splenomegaly (44%), respectively (Figure 1E,F). Here, the percentage of myeloid cells in the spleen and BM of mouse #2 was higher compared with nondiseased Cre− littermate control samples (supplemental Figure 2D). Remarkably, the lymphoma and spleen samples of MybVav mouse #2 and the spleen sample of MybVav mouse #3 presented with mixed immunophenotype malignancies, which were composed of both B cells and myeloid cells (Figure 1E,F). We wondered if these mixed MybVav malignancies were of ambiguous lineage, featuring both myeloid and B-cell markers on the same cell, or biphenotypic containing a mixture of both B- and malignant myeloid cells. By using flow cytometry, we confirmed that mixed MybVav malignancies were biphenotypic, containing both B- and malignant myeloid cells (supplemental Figure 2E). The lymphoid and myeloid features of the MybVav tumors were validated via histology, including liver infiltration of KI67- and MYB-positive tumor cells via the portal vein (Figure 1G; mouse #1 and right panel) or liver sinusoids (Figure 1G; mouse #4), which are typically observed in lymphoid and myeloid leukemias. All examined liver (n = 2) and LN (n = 3) infiltrations in MybVav mice were composed of MYB-positive cells (Figure 1G,H).

Myb overexpression enables the formation of B-cell and myeloid neoplasms in vivo. (A) Schematic representation of R26-Myb mice that allow Cre-dependent conditional expression of a bicistronic transgene transcript, encoding for Myb and the eGFP/Luciferase reporter, from the Rosa26 promoter. Breeding scheme to obtain R26-Mybtg/tg;VaviCretg/+ (MybVav) mice with hematopoietic-specific overexpression of Myb. R26, Rosa26; IRES, independent ribosomal entry site; eGFP, enhanced green fluorescent protein; PKG, phosphoglycerate kinase 1; NeoR, neomycin resistance gene. (B) Kaplan Meier survival curve of Cre-negative control (R26-Mybtg/tg) vs MybVav mice. A log-rank Mantel-Cox test showed that survival of MybVav mice was significantly lower. *P = .0302. (C) Peripheral blood values of Mybvav mice and nonrecombined controls. WBC, white blood cells; RBC, red blood cells. An unpaired t-test indicated that there was no significant difference between tumor-carrying Mybvav mice and nonrecombined controls. (D) Graph depicting the spleen-to-body weight ratio of Mybvav mice that developed neoplasm and age-matched Cre-negative littermate control mice. *P = .0450. (E) Flow cytometry analysis of 4 MybVav tumors. Single live cells were analyzed for the T-cell marker Thy1.1 (CD90), B-cell markers B220 and CD19, and myeloid markers Gr-1 and Cd11b. FSC-A, forward scatter area. (F) Left: heatmap summarizing flow data of tumor samples, including BM, LN, and spleen, from 4 Mybvav mice. Right: Graph depicting the percentage of T cells, B cells, or myeloid cells from panel E, which were pregated for single live cells. (G,H) Hematoxylin and eosin (H&E) staining or representative immunohistochemistry (IHC) for the proliferation marker KI67 or MYB on paraffin sections of Mybvav liver (G) and LN (H) tumors and of an aged-matched Cre-negative littermate control. Scale bar: 25 µm. Scale bar inset: 50 µm.
Myb overexpression enables the formation of B-cell and myeloid neoplasms in vivo. (A) Schematic representation of R26-Myb mice that allow Cre-dependent conditional expression of a bicistronic transgene transcript, encoding for Myb and the eGFP/Luciferase reporter, from the Rosa26 promoter. Breeding scheme to obtain R26-Mybtg/tg;VaviCretg/+ (MybVav) mice with hematopoietic-specific overexpression of Myb. R26, Rosa26; IRES, independent ribosomal entry site; eGFP, enhanced green fluorescent protein; PKG, phosphoglycerate kinase 1; NeoR, neomycin resistance gene. (B) Kaplan Meier survival curve of Cre-negative control (R26-Mybtg/tg) vs MybVav mice. A log-rank Mantel-Cox test showed that survival of MybVav mice was significantly lower. *P = .0302. (C) Peripheral blood values of Mybvav mice and nonrecombined controls. WBC, white blood cells; RBC, red blood cells. An unpaired t-test indicated that there was no significant difference between tumor-carrying Mybvav mice and nonrecombined controls. (D) Graph depicting the spleen-to-body weight ratio of Mybvav mice that developed neoplasm and age-matched Cre-negative littermate control mice. *P = .0450. (E) Flow cytometry analysis of 4 MybVav tumors. Single live cells were analyzed for the T-cell marker Thy1.1 (CD90), B-cell markers B220 and CD19, and myeloid markers Gr-1 and Cd11b. FSC-A, forward scatter area. (F) Left: heatmap summarizing flow data of tumor samples, including BM, LN, and spleen, from 4 Mybvav mice. Right: Graph depicting the percentage of T cells, B cells, or myeloid cells from panel E, which were pregated for single live cells. (G,H) Hematoxylin and eosin (H&E) staining or representative immunohistochemistry (IHC) for the proliferation marker KI67 or MYB on paraffin sections of Mybvav liver (G) and LN (H) tumors and of an aged-matched Cre-negative littermate control. Scale bar: 25 µm. Scale bar inset: 50 µm.
Next, we investigated whether these Myb-driven hematological malignancies could be transplanted into secondary recipients and could grow out as full-blown myeloid or B-cell malignancies (Figure 2A). The transgenic animals and derived malignant cells express a bicistronic transgene transcript that encodes both Myb as well as the Firefly luciferase-reporter (Figure 1A), which enables us to trace neoplastic cells upon transplantations in secondary hosts using in vivo bioluminescence imaging. In a first transplantation experiment, we transplanted 4 primary MybVav tumors from 3 mice (#1, #2, and #4), and engraftment in immunodeficient nonobese diabetic/severely compromised immunodeficiency γ (NSG) mice was observed using myeloid tumor material obtained from BM (MybVav mouse #2) (Figure 2B). In line with the bioluminescence data, a secondary neoplasm was identified in the BM of this NSG 21 weeks posttransplantation (Figure 2B), and the myeloid origin of the luciferase-positive transplanted material was confirmed by flow cytometry (Figure 2C-E; transplant #2). We monitored a second series of MybVav transplants (6 primary tumors from 4 MybVav mice) for 40 weeks, and although engraftment of a low number of luciferase-positive myeloid or B cells could be observed (Figure 2C-E), no secondary malignancies were formed within 40 weeks. Since no B-cell tumors formed secondary neoplasms upon transplantation, they should be called B-cell proliferative disease rather than B-cell malignancies. This also suggests that additional oncogenic hits and/or an immunocompetent microenvironment may be required in order to promote full-blown malignant transformation of MybVav tumors.
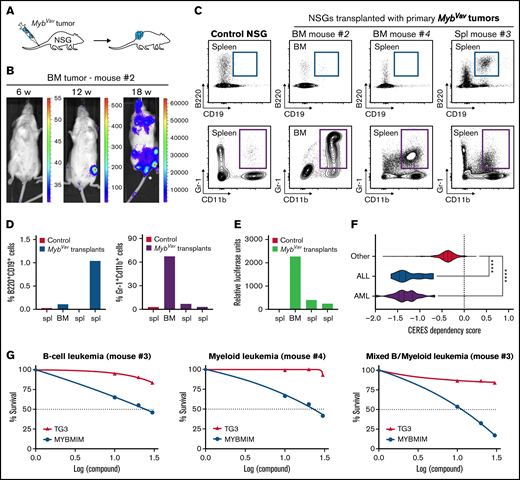
Transplantation and therapeutic targeting of the transcriptional activity of Myb. (A) Scheme of primary transplantation of MybVav tumor cells into immunocompromised nonobese diabetic/severely compromised immunodeficiency γ (NSG) mice. (B) Bioluminescence of a primary BM tumor sample from Mybvav mouse #2 that was transplanted in an NSG mouse. Bioluminescence was measured over time. (C) Flow cytometry analysis of myeloid (Gr-1+Cd11b+) and B cells (B220+CD19+) of a control NSG and NSGs that were transplanted primary Mybvav tumor cells. (D) Graphs showing the percentage of B cells or myeloid cells which were pregated for single live cells. (E) Luciferase assay on Mybvav transplants. (F) Violin plots showing CERES cell dependency scores for MYB from AML (n = 20), ALL (n = 10), and 772 other cancer cell lines which were taken from DepMap (https://depmap.org/portal/). CERES is a computational method that estimates gene dependency based on data from CRISPR-Cas9 screens. A CERES score of 0 indicates that MYB is not essential, while a lower score indicates a higher likelihood that MYB is essential in a given cell line. MYB, AML, and ALL cell lines had a significantly lower CERES score than other cell lines. ****P < .0001. (G) Graphs depicting the survival of myeloid, B-cell, and mixed MybVav hematopoietic malignancies, which were treated for 48 hours with increasing concentrations of either MYBMIM or TG3.
Transplantation and therapeutic targeting of the transcriptional activity of Myb. (A) Scheme of primary transplantation of MybVav tumor cells into immunocompromised nonobese diabetic/severely compromised immunodeficiency γ (NSG) mice. (B) Bioluminescence of a primary BM tumor sample from Mybvav mouse #2 that was transplanted in an NSG mouse. Bioluminescence was measured over time. (C) Flow cytometry analysis of myeloid (Gr-1+Cd11b+) and B cells (B220+CD19+) of a control NSG and NSGs that were transplanted primary Mybvav tumor cells. (D) Graphs showing the percentage of B cells or myeloid cells which were pregated for single live cells. (E) Luciferase assay on Mybvav transplants. (F) Violin plots showing CERES cell dependency scores for MYB from AML (n = 20), ALL (n = 10), and 772 other cancer cell lines which were taken from DepMap (https://depmap.org/portal/). CERES is a computational method that estimates gene dependency based on data from CRISPR-Cas9 screens. A CERES score of 0 indicates that MYB is not essential, while a lower score indicates a higher likelihood that MYB is essential in a given cell line. MYB, AML, and ALL cell lines had a significantly lower CERES score than other cell lines. ****P < .0001. (G) Graphs depicting the survival of myeloid, B-cell, and mixed MybVav hematopoietic malignancies, which were treated for 48 hours with increasing concentrations of either MYBMIM or TG3.
In conclusion, we modeled aberrant MYB overexpression, which is observed in many types of cancer, including breast and colon cancer, and hematological malignancies, using a newly established conditional Myb overexpression mouse model. We show that hematopoietic-specific overexpression of Myb is sufficient to induce B-cell neoplasms and myeloid malignancies in vivo, providing evidence for its function as an oncogene in the initiation of myeloid leukemia. Our data confirm the longstanding relationship between elevated Myb expression in myeloid leukemias1-4 and its high transformation potential in chicken and mice.7,8,16,21,22 In contrast, the newly identified role for Myb in the initiation of B-cell lymphoproliferative disease is rather unexpected as up to now, MYB was only associated with maintenance of B-cell leukemias.15 This conditional R26-Myb mouse model might also be of value to elucidate the role of MYB during the initiation of, for instance, breast or colon cancer.
Besides its role in tumor initiation, MYB also plays an important role in the maintenance of both AML and acute lymphoblastic leukemias,13,15 a notion that was confirmed in large scale functional screenings performed by the Broad Institute (data from the Cancer Dependency Map; https://depmap.org/portal/) (Figure 2F) using CRISPR/CAS9 knockouts and short hairpin RNA-mediated knockdown on 802 human cancer cell lines. The 20 AML and 10 ALL cell lines in this data set were significantly more dependent on MYB expression compared with nonleukemic cell lines (n = 772) (Figure 2F). Based on this observation, we wondered whether the murine Myb-driven B-cell neoplasms and myeloid malignancies relied on the continued transcriptional activity of Myb for their survival. MYB is a transcription factor and has a tripartite structure with an N-terminal DNA-binding domain, a central transactivation domain (TAD), and a C-terminal negative regulatory domain.1,23 The TAD of MYB allows the recruitment of its coactivators, such as CBP or its paralogue p300, through their KIX domain. It was shown that the interaction between MYB and CBP/p300 is required for leukemogenesis. Especially, MYB residue E308 is essential for this interaction, as was elegantly shown in the Booreana mouse strain, which has a naturally occurring Myb E308G mutation and is resistant to AML1-ETO- or MLL-AF9-induced leukemogenesis.24 Therefore, blocking the interaction between MYB and its transcriptional coactivators CBP/p300 is an attractive therapeutic strategy to treat AML. Indeed, human AML cells were effectively killed when the MYB:CBP/P300 binding was blocked with MYBMIM, a cell-penetrant peptidomimetic inhibitor that contains MYB residues 293-310.25 In contrast, human AML cell viability was unaffected when treated with an inactive version of MYBMIM, termed TG3, in which 3 MYB residues, which are important for the interaction with CBP/p300 (including E308), were replaced with glycines.25 Here, we confirmed that Myb-driven murine myeloid malignancies were also sensitive to MYBMIM (Figure 2G), using concentrations that were comparable to those that were used to effectively induce apoptosis and differentiation of human AMLs.25 At these concentrations, TG3 treatment had no significant effect on the viability of Myb-driven myeloid malignancies (Figure 2G). Finally, we also treated murine B-cell or mixed MybVav malignancies with either MYBMIM or TG3 and found that these malignancies were also selectively sensitive to MYBMIM treatment (Figure 2G). It remains to be determined if this reduction in cell viability is due to reduced cell proliferation or increased apoptosis. Our results confirm that MYB is not only sufficient to initiate myeloid malignancies but that MYB is also required for tumor maintenance.
In conclusion, and in line with the notion that MYB is highly expressed and required in both B-cell and myeloid malignancies2,13,15 and has in vitro transformation potential,21,22 we show that Myb drives spontaneous B-cell neoplasms and myeloid malignancies in mice.
Acknowledgments: The authors thank Jinke D’Hont and Frédérique Van Rockeghem for technical assistance.
This work was supported by the Baillet Latour Foundation, the Ghent University Research Fund (BOF-UGent), and the Research Foundation Flanders (FWO).
Contribution: T.P., A.A., S.T., K.L., and T.H. performed research; G.B. and A.K. provided research tools; and T.P., S.G., and P.V.V. designed research and wrote the paper.
Conflict-of-interest disclosure: A.K. is a consultant for Novartis and Rgenta. All other authors declare no competing financial interests.
Correspondence: Pieter Van Vlierberghe, Department of Biomolecular Medicine, Ghent University, Corneel Heymanslaan 10, 9000 Ghent, Belgium; e-mail: pieter.vanvlierberghe@ugent.be.

