

Key Points
Loss of miR-31 in allogeneic T cells alleviates progression of scleroderma and lung dysfunction in cGVHD but not graft-versus-leukemia response.
MiR-31 inhibition attenuates CD4+ T-cell activity in hypoxic environments.

Abstract
Chronic graft-versus-host disease (cGVHD) remains a major obstacle impeding successful allogeneic hematopoietic cell transplantation (HCT). MicroRNAs (miRs) play key roles in immune regulation during acute GVHD development. Preclinical studies to identify miRs that affect cGVHD pathogenesis are required to develop these as potential lifesaving interventions. Using oligonucleotide array, we identified miR-31, which was significantly elevated in allogeneic T cells after HCT in mice. Using genetic and pharmacologic approaches, we demonstrated a key role for miR-31 in mediating donor T-cell pathogenicity in cGVHD. Recipients of miR-31–deficient T cells displayed improved cutaneous and pulmonary cGVHD. Deficiency of miR-31 reduced T-cell expansion and T helper 17 (Th17) cell differentiation but increased generation and function of regulatory T cells (Tregs). MiR-31 facilitated neuropilin-1 downregulation, Foxp3 loss, and interferon-γ production in alloantigen-induced Tregs. Mechanistically, miR-31 was required for hypoxia-inducible factor 1α (HIF1α) upregulation in allogeneic T cells. Therefore, miR-31–deficient CD4 T cells displayed impaired activation, survival, Th17 cell differentiation, and glycolytic metabolism under hypoxia. Upregulation of factor-inhibiting HIF1, a direct target of miR-31, in miR-31–deficient T cells was essential for attenuating T-cell pathogenicity. However, miR-31–deficient CD8 T cells maintained intact glucose metabolism, cytolytic activity, and graft-versus-leukemia response. Importantly, systemic administration of a specific inhibitor of miR-31 effectively reduced donor T-cell expansion, improved Treg generation, and attenuated cGVHD. Taken together, miR-31 is a key driver for T-cell pathogenicity in cGVHD but not for antileukemia activity. MiR-31 is essential in driving cGVHD pathogenesis and represents a novel potential therapeutic target for controlling cGVHD.
Introduction
Chronic graft-versus-host disease (cGVHD) remains a major cause of mortality and morbidity after allogeneic hematopoietic cell transplantation (allo-HCT).1 cGVHD is characterized by systemic inflammation, multiorgan fibrosis, and increased risk of infection.2 Uncontrolled donor T-cell activation and expansion in target organs contribute to the initiation of cGVHD. By differentiating into effector T helper (Th) subsets, including Th1, Th17, extrafollicular T cells, and follicular Th (Tfh) cells, CD4 T cells are key mediators of cGVHD. Loss of immune tolerance caused by impaired differentiation and homeostasis of regulatory T cells (Tregs) perpetuates cGVHD.3 Eventually, aberrant production of cytokines and autoantibodies leads to fibroblast activation and tissue fibrosis.4
MicroRNAs (miRs) are short, noncoding RNAs that repress gene expression at the posttranscriptional level via binding to target messenger RNAs, subsequently promoting degradation or impeding translation.5 MiRs play critical roles in regulating T-cell response6-11 and dendritic cell function12-14 in GVHD development. We previously reported that miR-17-92 enhances T- and B-cell pathogenicity in GVHD but is dispensable in T cell–mediated graft-versus-leukemia (GVL) effect.6,7 Recently, miR-31 emerged as an important regulator of T-cell response. Downregulation of miR-31 in human CD4 T cells contributed to immunosuppression during sepsis.15 MiR-31 expression was downregulated by Bcl6, which caused stabilized Tfh programming in tonsils.16 In mice, miR-31 enhanced CD4 T-cell pathogenicity in experimental autoimmune encephalomyelitis (EAE)17 while reducing CD8 T-cell potency in controlling lymphocytic choriomeningitis virus infection.18 However, whether or how miR-31 affects allogeneic T-cell responses is essentially unknown. Using genetic and pharmacologic approaches, we uncovered a key role for miR-31 in promoting T-cell expansion and Treg/Th17 imbalance and augmenting donor T-cell pathogenicity in cGVHD; however, it remains dispensable for CD8 T-cell function and GVL response. At a molecular level, by inhibiting factor inhibiting hypoxia-inducible factor 1 (HIF1; FIH1), miR-31 enhances HIF1α expression and promotes glycolytic metabolism and T-cell function under hypoxic conditions.
Materials and methods
Mice
CD45.1+ B6, BALB/c, and B6D2F1 mice were purchased from the National Cancer Institute (Frederick, MD). B10.BR and B10.D2 mice were purchased from The Jackson Laboratory (Bar Harbor, ME). MiR-31flox/flox mice on a B6 background were provided by Zhang et al17 and were bred with CD4Cre mice to generate miR-31 knockout (KO; miR-31flox/flox CD4Cre+) and wild-type (WT; miR-31flox/floxCD4Cre−) mice. All mice were housed in a pathogen-free facility at the American Association for Laboratory Animal Care–accredited Animal Resource Center at the Medical University of South Carolina. All animal studies were carried out under protocols approved by the Institutional Animal Care and Use Committee at the Medical University of South Carolina.
Experimental procedures and materials
Bone marrow transplantation (BMT), GVHD scoring, lung function measurement, miR detection, locked nucleic acid antagomir treatment, T-cell differentiation in vitro, flow cytometry, western blot, seahorse assay and histopathology, and trichrome staining are described in previously published work7,19,20 and in the data supplement.
Statistics
GraphPad Prism was used to perform statistical analysis. The log-rank test was used to determine statistical significance in recipient survival. Normality of data was assessed and statistical significance was determined with 2-tailed unpaired Student t test.
Results
MiR-31 expression in donor T cells is critical for the induction of cutaneous and pulmonary cGVHD in mice
To search for candidate miRs that regulate allogeneic T-cell responses, we used microarray to profile miR expression of donor T cells in allogeneic or syngeneic recipients after BMT in mice. We found many miRs that were differentially expressed in T cells during allogeneic (alloantigen-driven) vs syngeneic (homeostasis-driven) response, including miR-17-92, -155, -146, -181, and -142, which were closely associated with GVHD pathogenesis, as we6,7 and others8,9,11,21 have previously demonstrated. We also identified novel miR candidates that potentially affect allogeneic T-cell response, among which miR-31 was significantly upregulated in allogeneic T cells (Figure 1A). Increased expression of miR-31 in allogeneic T cells compared with either syngeneic or naïve T cells was confirmed by quantitative polymerase chain reaction (Figure 1B).
![MiR-31 is upregulated in allogeneic T cells and enhances their pathogenicity in cGVHD induction in mice. (A-B) T cells isolated from Ly5.1+ B6 mice were injected together with T cell–depleted (TCD; Ly5.2+) BM into lethally irradiated BALB/c (allogeneic [Allo]; 700 cGy) or Ly5.2+ B6 (syngeneic [Syn]; 1200 cGy) mice. (A) MiR expression was evaluated in H2Kb+Ly5.1+ donor T cells by oligonucleotide arrays on day 14. The heat map represents the log2 value of the relative amount of each miR. (B) The messenger RNA expression of miR-31 in naïve T cells and activated donor T cells isolated from Allo or Syn recipients was evaluated by real-time polymerase chain reaction. (C-I) Splenocytes (0.35 × 106) and TCD BM (5 × 106) from WT or miR-31 KO B6 mice were injected into lethally irradiated BALB/c mice. Recipients of WT BM cells only without splenocyte injections served as the no-GVHD control (white circles). Recipient clinical scores and body weight loss were recorded weekly and displayed along the time after BMT (C). Cutaneous pathologic scores (D) and collagen deposition in skin evaluated with Masson’s trichrome stain (E, original magnificaiton ×200) are shown on day 50. Pathologic scores of salivary glands in recipients were recorded on day 60 after BMT (I). (F-H) WT or miR-31 KO T cells (2.75 × 106) plus WT TCD BM were transferred into lethally irradiated B6D2F1 mice. Clinical scores (F), cutaneous pathologic scores (G), and collagen deposition (H, original magnificaiton ×200) are shown on day 50. Data shown are from 1 representative of 3 individual experiments (n = 5-6 mice per group for each experiment). (J-K) B10.BR mice were conditioned with cytoxan (120 mg/kg) and irradiation and then underwent transplantation with WT or miR-31 KO T cells (5 × 104) plus WT TCD BM. Body weight loss (J) and pulmonary compliance, elastance, and constriction analyzed with a flexiVent system (K) are shown on day 50 after BMT. *P < .05, **P < .01.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/6/10/10.1182_bloodadvances.2021005103/3/m_advancesadv2021005103f1.png?Expires=1763484038&Signature=oVzo4sdLG~nrGt1gVXZad0w8-jzEOPQCcOpYa2nk-PeyQb1wVP-joP7Qk2hi0jW9nN4-FGv7pFq9BN~OMUlfdGHKWcbelcsub6cXoNOJmcTSAeC2pVz4EGu8BGBAVvQfoN4~nI6buamCvJ4gRsdAL14F6vITDpMoAAiN6KKvoN3pSilwP01rLhu77f6kP6w6j4v-TeouE6A1aKW4pmUcLt8dvVlqBh7h7Syn~1Pv3BjnWTPO2u8Dm2zTAwpxqpCtl~yg4~4wNhRFiL9yMFFEUFvNg4eykwyfv0vknV9rLBiLrkmmeKwbJQO7sJu-xfzSN29XX0a0Jrnal67~ntCdUQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
MiR-31 is upregulated in allogeneic T cells and enhances their pathogenicity in cGVHD induction in mice. (A-B) T cells isolated from Ly5.1+ B6 mice were injected together with T cell–depleted (TCD; Ly5.2+) BM into lethally irradiated BALB/c (allogeneic [Allo]; 700 cGy) or Ly5.2+ B6 (syngeneic [Syn]; 1200 cGy) mice. (A) MiR expression was evaluated in H2Kb+Ly5.1+ donor T cells by oligonucleotide arrays on day 14. The heat map represents the log2 value of the relative amount of each miR. (B) The messenger RNA expression of miR-31 in naïve T cells and activated donor T cells isolated from Allo or Syn recipients was evaluated by real-time polymerase chain reaction. (C-I) Splenocytes (0.35 × 106) and TCD BM (5 × 106) from WT or miR-31 KO B6 mice were injected into lethally irradiated BALB/c mice. Recipients of WT BM cells only without splenocyte injections served as the no-GVHD control (white circles). Recipient clinical scores and body weight loss were recorded weekly and displayed along the time after BMT (C). Cutaneous pathologic scores (D) and collagen deposition in skin evaluated with Masson’s trichrome stain (E, original magnificaiton ×200) are shown on day 50. Pathologic scores of salivary glands in recipients were recorded on day 60 after BMT (I). (F-H) WT or miR-31 KO T cells (2.75 × 106) plus WT TCD BM were transferred into lethally irradiated B6D2F1 mice. Clinical scores (F), cutaneous pathologic scores (G), and collagen deposition (H, original magnificaiton ×200) are shown on day 50. Data shown are from 1 representative of 3 individual experiments (n = 5-6 mice per group for each experiment). (J-K) B10.BR mice were conditioned with cytoxan (120 mg/kg) and irradiation and then underwent transplantation with WT or miR-31 KO T cells (5 × 104) plus WT TCD BM. Body weight loss (J) and pulmonary compliance, elastance, and constriction analyzed with a flexiVent system (K) are shown on day 50 after BMT. *P < .05, **P < .01.
MiR-31 is upregulated in allogeneic T cells and enhances their pathogenicity in cGVHD induction in mice. (A-B) T cells isolated from Ly5.1+ B6 mice were injected together with T cell–depleted (TCD; Ly5.2+) BM into lethally irradiated BALB/c (allogeneic [Allo]; 700 cGy) or Ly5.2+ B6 (syngeneic [Syn]; 1200 cGy) mice. (A) MiR expression was evaluated in H2Kb+Ly5.1+ donor T cells by oligonucleotide arrays on day 14. The heat map represents the log2 value of the relative amount of each miR. (B) The messenger RNA expression of miR-31 in naïve T cells and activated donor T cells isolated from Allo or Syn recipients was evaluated by real-time polymerase chain reaction. (C-I) Splenocytes (0.35 × 106) and TCD BM (5 × 106) from WT or miR-31 KO B6 mice were injected into lethally irradiated BALB/c mice. Recipients of WT BM cells only without splenocyte injections served as the no-GVHD control (white circles). Recipient clinical scores and body weight loss were recorded weekly and displayed along the time after BMT (C). Cutaneous pathologic scores (D) and collagen deposition in skin evaluated with Masson’s trichrome stain (E, original magnificaiton ×200) are shown on day 50. Pathologic scores of salivary glands in recipients were recorded on day 60 after BMT (I). (F-H) WT or miR-31 KO T cells (2.75 × 106) plus WT TCD BM were transferred into lethally irradiated B6D2F1 mice. Clinical scores (F), cutaneous pathologic scores (G), and collagen deposition (H, original magnificaiton ×200) are shown on day 50. Data shown are from 1 representative of 3 individual experiments (n = 5-6 mice per group for each experiment). (J-K) B10.BR mice were conditioned with cytoxan (120 mg/kg) and irradiation and then underwent transplantation with WT or miR-31 KO T cells (5 × 104) plus WT TCD BM. Body weight loss (J) and pulmonary compliance, elastance, and constriction analyzed with a flexiVent system (K) are shown on day 50 after BMT. *P < .05, **P < .01.
To investigate how miR-31 contributes to T-cell pathogenicity in cGVHD, we used B6 mice that had miR-31 conditionally knocked out in T cells (miR-31flox/flox CD4Cre+).17 No significant differences were observed in the baseline composition of splenocytes of miR-31–deficient mice and WT controls (miR-31flox/flox CD4Cre−) with respect to the frequencies of CD4+, CD8+, B220+, CD25+, Foxp3+, CD44+, and CD62L+ cells (supplemental Figure 1). To study this, we used allogeneic BMT (allo-BMT) models that have cutaneous fibrosis as a feature because it is also a common clinical manifestation of cGVHD (B6 → BALB/c and B6 → B6D2F1). Consistent with a low intensity of acute inflammation, most recipients with cGVHD survived long term, whereas the recipients of miR-31 KO grafts had improved survival (supplemental Figure 2). In the acute GVHD to cGVHD transition model (B6 → BALB/c),22 the recipients of BM and splenocytes from miR-31 KO donors showed significantly attenuated clinical scores and body weight loss compared with those receiving WT grafts (Figure 1C). In addition, cutaneous manifestations, including pathologic scores and collagen deposition, were alleviated in KO graft recipients (Figure 1D-E). Using another clinically relevant, haploidentical model of cutaneous cGVHD (B6 → B6D2F1),23 we consistently found improved clinical scores, body weight maintenance, and attenuated cutaneous manifestations in the recipients of miR-31–deficient T cells compared with those of WT controls (Figure 1F-H). In addition, salivary glands, prototypical targets of cGVHD,24 showed milder pathologic damage in the BALB/c recipients of miR-31 grafts (Figure 1I). GVHD delays the reconstitution of donor lymphocytes and increases the risk of life-threatening infections in patients.25 Compromised donor T-cell reconstitution is partially attributed to impaired T-cell development in the recipient thymus, a direct target organ of alloreactive T cells.26 Consistent with attenuated cGVHD severity, reconstitution of donor CD4+ and CD8+ T and B220+ B lymphocytes was significantly improved in the recipients of miR-31 KO grafts (supplemental Figure 3A-D). These mice also displayed improved thymic regeneration and positive selection, reflected by significantly increased cell numbers of total and CD4+CD8+ thymocytes (supplemental Figure 3E-G).
To extend our findings, we used an additional preclinical cGVHD model (B6 → B10.BR) characterized by lung cGVHD to further evaluate how miR-31 regulates T-cell pathogenicity in pulmonary cGVHD.27 Recipients of miR-31 KO T cells showed significantly better body weight maintenance and lung function preservation, indicated by reduced alveolar constriction and elastance and increased compliance (Figure 1J-K). Therefore, in 3 preclinical cGVHD models, our data consistently demonstrate that lack of miR-31 reduces T-cell pathogenicity in the skin and lung damage.
Loss of miR-31 attenuates the expansion of pathogenic donor T cells after allo-BMT
T lymphocytes in the recipients are derived from 2 sources: expansion of mature alloreactive T cells in donor grafts, and generation from donor hematopoietic stem cells. Thymic negative selection during de novo T-cell development largely eliminates alloreactive T cells, and therefore, mature T cells in donor grafts are considered the key mediators of GVHD. To examine how miR-31 affects the expansion of mature alloreactive T cells, we performed BMT using congenic donor mice to distinguish mature alloreactive (CD45.2+) vs BM-derived (CD45.1+) donor T cells. We first examined frequencies of donor-type (H2Kb+) T cells and confirmed successful engraftment of donor cells in recipient splenic and single positive thymic T cells (supplemental Figure 4). Across 3 models, recipients of KO T cells displayed significantly attenuated expansion of CD45.2+ T cells in spleens (Figure 2A; supplemental Figures 5A and 6A). Consistent with improved thymic regeneration, thymic infiltration of CD45.2+ donor T cells was significantly reduced in the recipients of KO grafts (Figure 2B). Furthermore, these recipients had fewer CD45.2+ T cells in skin-draining lymph nodes (LNs) and lungs (supplemental Figures 5B and 6B). Mechanistically, miR-31 KO T, especially CD4+, cells displayed reduced proliferation and cell survival after being transferred into haploidentical recipients (supplemental Figure 5C-D). Consistently, in a model of pulmonary cGVHD, miR-31 KO T cells showed increased cell death in recipient spleens and lungs (supplemental Figure 6C-D), which may be attributed to increased expression of the death receptor Fas (supplemental Figure 6E). Reduced Ki67 expression in KO CD4 T cells in recipient spleens and increased Lag3 expression in KO T cells in spleens and lungs were observed (supplemental Figure 6F-H), indicating less activation and more exhaustion in these cells after allo-BMT.


Loss of miR-31 attenuates alloreactive T-cell expansion and differentiation of effector T (Teff) cells during cGVHD development. (A-B) Splenocytes (CD45.2+) from WT or miR-31 KO B6 donors were injected together with WT T cell–depleted (TCD) BM (CD45.1+) into lethally irradiated BALB/c recipients. Frequencies of CD45.2+ cells in gated live donor H2Kb+ CD4 or CD8 T cells and their absolute numbers are shown in recipient spleens (A) and thymuses (B) on day 50. (C-F) In separate experiments, splenocytes and TCD BM cells from WT or miR-31 KO B6 mice were injected into lethally irradiated BALB/c mice. On day 50 to 60, representative flow figures of CXCR5+PD-1hi cells gated on live donor H2Kb+CD4+ cells (upper), germinal center (GC) B cells (Fas+GL7+) gated on H2Kb+B220+ cells, and frequency of CXCR5+PD-1hi cells in donor H2Kb+CD4+ cells and their numbers are shown (C). Percentages of follicular Treg (Tfr) (Foxp3+) and Tfh (Foxp3−) cells in gated H2Kb+ CD4+ CXCR5+ PD-1hi cells (D) and percentages of GC B cells in live H2Kb+ B cells and CD138+B220− plasma cells in live H2Kb+ cells (E) and their absolute numbers are displayed. Representative flow figures and bar graphs showing the frequencies and numbers of interferon-γ–positive (IFN-γ+), interleukin-17A–positive (IL-17A+), and tumor necrosis factor α (TNFα+) cells in gated live donor H2Kb+ CD4 T cells in recipient spleens are displayed (F). Data shown are from 1 representative of 3 individual experiments (n = 4-5 mice per group for each experiment). *P < .05, **P < .01, ***P < .001.
Loss of miR-31 attenuates alloreactive T-cell expansion and differentiation of effector T (Teff) cells during cGVHD development. (A-B) Splenocytes (CD45.2+) from WT or miR-31 KO B6 donors were injected together with WT T cell–depleted (TCD) BM (CD45.1+) into lethally irradiated BALB/c recipients. Frequencies of CD45.2+ cells in gated live donor H2Kb+ CD4 or CD8 T cells and their absolute numbers are shown in recipient spleens (A) and thymuses (B) on day 50. (C-F) In separate experiments, splenocytes and TCD BM cells from WT or miR-31 KO B6 mice were injected into lethally irradiated BALB/c mice. On day 50 to 60, representative flow figures of CXCR5+PD-1hi cells gated on live donor H2Kb+CD4+ cells (upper), germinal center (GC) B cells (Fas+GL7+) gated on H2Kb+B220+ cells, and frequency of CXCR5+PD-1hi cells in donor H2Kb+CD4+ cells and their numbers are shown (C). Percentages of follicular Treg (Tfr) (Foxp3+) and Tfh (Foxp3−) cells in gated H2Kb+ CD4+ CXCR5+ PD-1hi cells (D) and percentages of GC B cells in live H2Kb+ B cells and CD138+B220− plasma cells in live H2Kb+ cells (E) and their absolute numbers are displayed. Representative flow figures and bar graphs showing the frequencies and numbers of interferon-γ–positive (IFN-γ+), interleukin-17A–positive (IL-17A+), and tumor necrosis factor α (TNFα+) cells in gated live donor H2Kb+ CD4 T cells in recipient spleens are displayed (F). Data shown are from 1 representative of 3 individual experiments (n = 4-5 mice per group for each experiment). *P < .05, **P < .01, ***P < .001.
Recent studies showed a critical role for B cells and autoantibodies in cGVHD pathogenesis in mice4 as well as in humans.28 Tfh cells instruct GC B cells to differentiate into antibody-secreting plasma cells,29 a process inhibited by follicular Tregs.30 In contrast, recent studies have revealed extrafollicular CD4+ T-B interactions as a key mechanism for cGVHD induction, in that GC formation is dispensable.31,32 To further study whether miR-31 expression affects Tfh differentiation in cGVHD, we examined Tfh cells by flow cytometry and found significantly reduced frequencies but increased absolute numbers of splenic follicular T cells (CXCR5+PD-1hi) among total donor T cells in the recipients of miR-31 KO grafts (Figure 2C; supplemental Figure 7). Among these cells, a greater proportion of follicular Tregs (Foxp3+) and a smaller proportion of Tfh (Foxp3−) cells were observed in KO cells compared with WT counterparts (Figure 2D). Furthermore, the frequencies of GC B cells and plasma cells, but not their absolute numbers, were reduced in the recipients of KO grafts (Figure 2C,E). The structure of B-cell follicles in spleens was similar between WT and KO graft recipients (supplemental Figure 8). In addition, fewer proinflammatory cytokines, including IFN-γ, IL-17A, and tumor necrosis factor α, were produced by total donor T cells in the recipients of KO grafts (Figure 2F; supplemental Figure 9A-B). Although the frequencies of naïve/memory populations were comparable between pretransplantation WT and KO cells (supplemental Figure 1), fewer effector memory, but more naïve and central memory, populations were observed among KO donor T cells after allo-BMT (supplemental Figure 9C-D). In addition, a majority of CD45.2+ donor T cells were CD44+ memory cells after allo-BMT (supplemental Figure 10C). These data indicate that miR-31 is required for the proliferation and survival of alloreactive T cells, and deficiency of miR-31 attenuates the expansion of pathogenic T cells after allo-BMT.
MiR-31 contributes to Th17/Treg imbalance, extrafollicular CD4 T-cell differentiation, and induced Treg instability post-BMT
Because mature alloreactive T cells are the major source of proinflammatory cytokines, we evaluated how miR-31 affects the differentiation of mature donor T cells (CD45.2+CD45.1−). A significant reduction of IL-17A expression in CD45.2+ KO CD4 T cells compared with WT counterparts was observed in recipient skin-draining LNs (Figure 3A). Furthermore, increased Treg differentiation from CD45.2+CD4+ KO T cells were found in recipient spleens and skin-draining LNs (Figure 3A-B). No difference regarding Tfh differentiation was observed within CD45.2+CD4+ WT vs KO T cells (data not shown). However, there were significantly lower frequencies of extrafollicular cells (CD4+CD44hiCD62LloPSGL-1hi)31,32 within splenocyte-derived KO T cells compared with WT counterparts (Figure 3B). The IFN-γ expression was comparable in CD45.2+ CD4 T cells but was increased in CD45.2+ KO CD8 T cells compared with WT counterparts in recipient spleens (supplemental Figure 10A-B). Consistently with attenuated GVHD and alleviated lymphopenia in the recipients of miR-31 KO grafts, we found the absolute numbers of total donor CD4 or CD8 T cells and Foxp3+ Tregs in spleens were significantly increased in these recipients (supplemental Figure 11A-B). The numbers of CD45.2+ Tregs and IFN-γ–expressing CD8 T cells were significantly elevated in these recipients (supplemental Figure 11C-D). Our data suggest that miR-31 regulates differentiation of mature donor T cells by influencing Th17/Treg imbalance after allo-BMT.

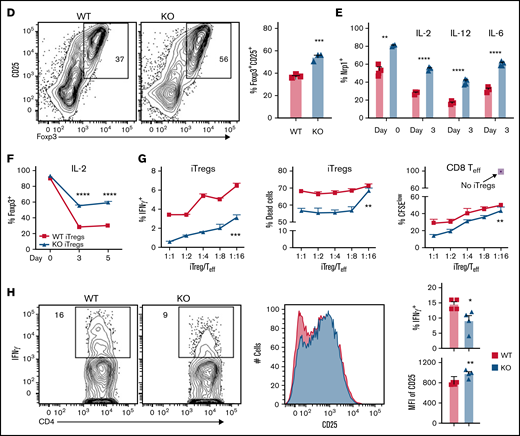
MiR-31 facilitates Th17/Treg imbalance and increases extrafollicular CD4 T-cell differentiation while inhibiting Treg stability. (A-C) Splenocytes (CD45.2+) from WT or miR-31 KO B6 mice were injected together with WT T cell–depleted (TCD) BM (CD45.1+) into lethally irradiated BALB/c mice. Frequencies of IL-17+ or Foxp3+ cells in gated live donor H2Kb+CD45.2+ CD4 T cells are shown in recipient skin-draining LNs (A) and spleens (B) on day 50 to 60 after BMT. Frequencies of CD62LloPSGL-1lo cells in gated live H2Kb+CD45.2+CD44+CD4 T cells are shown in recipient spleens on day 60 after BMT (top WT, bottom KO). Histograms for neuropilin 1 (Nrp1), PD-L1, and Bcl2 expression on gated H2Kb+CD45.2+Foxp3+ cells are shown (C). Data shown are from 1 representative of 3 individual experiments (n = 4-5 mice per group in each experiment). (D) CD4+CD25− T cells isolated from WT or miR-31 KO mice were stimulated with CD11c+ dendritic cells from BALB/c together with IL-2 (5 ng/mL), transforming growth factor β (5 μg/mL), and retinoic acid (40 nM). Frequency of Foxp3+ cells in gated live H2Kb+CD4+ cells is shown on day 5. (E-F) CD25hi-enriched induced Tregs (iTregs) were cultured in IL-2 (5 ng/mL), IL-12 (5 ng/mL), or IL-6 (10 ng/mL) in the presence of BALB/c APCs. Frequencies of Nrp1+ cells in gated live H2Kb+CD4+Foxp3+ cells (E) and Foxp3+ cells in gated live H2Kb+CD4+ cells (F) are shown. (G) Enriched iTregs (CD45.2+) were cocultured with B6 T cells (CD45.1+) at different ratios as indicated on the x-axis in the presence of BALB/c APCs. Frequencies of IFN-γ+ cells and live/dead dye+ cells in gated CD45.2+CD4+ cells and CFSElow cells in gated live CD45.1+ CD8 T cells are shown on day 4. (H) Enriched iTregs were injected together with BM cells (Rag1−/−) into lethally irradiated BALB/c mice followed by Teff cell (CD45.1+) transfer 3 days later. Expression of IFN-γ and CD25 on gated H2Kb+CD45.2+Foxp3+ cells is shown 4 days after Teff transfer. Data shown are from 1 representative of 2 individual experiments. *P < .05, **P < .01, ***P < .001, ****P < .0001. MFI, mean fluorescence intensity.
MiR-31 facilitates Th17/Treg imbalance and increases extrafollicular CD4 T-cell differentiation while inhibiting Treg stability. (A-C) Splenocytes (CD45.2+) from WT or miR-31 KO B6 mice were injected together with WT T cell–depleted (TCD) BM (CD45.1+) into lethally irradiated BALB/c mice. Frequencies of IL-17+ or Foxp3+ cells in gated live donor H2Kb+CD45.2+ CD4 T cells are shown in recipient skin-draining LNs (A) and spleens (B) on day 50 to 60 after BMT. Frequencies of CD62LloPSGL-1lo cells in gated live H2Kb+CD45.2+CD44+CD4 T cells are shown in recipient spleens on day 60 after BMT (top WT, bottom KO). Histograms for neuropilin 1 (Nrp1), PD-L1, and Bcl2 expression on gated H2Kb+CD45.2+Foxp3+ cells are shown (C). Data shown are from 1 representative of 3 individual experiments (n = 4-5 mice per group in each experiment). (D) CD4+CD25− T cells isolated from WT or miR-31 KO mice were stimulated with CD11c+ dendritic cells from BALB/c together with IL-2 (5 ng/mL), transforming growth factor β (5 μg/mL), and retinoic acid (40 nM). Frequency of Foxp3+ cells in gated live H2Kb+CD4+ cells is shown on day 5. (E-F) CD25hi-enriched induced Tregs (iTregs) were cultured in IL-2 (5 ng/mL), IL-12 (5 ng/mL), or IL-6 (10 ng/mL) in the presence of BALB/c APCs. Frequencies of Nrp1+ cells in gated live H2Kb+CD4+Foxp3+ cells (E) and Foxp3+ cells in gated live H2Kb+CD4+ cells (F) are shown. (G) Enriched iTregs (CD45.2+) were cocultured with B6 T cells (CD45.1+) at different ratios as indicated on the x-axis in the presence of BALB/c APCs. Frequencies of IFN-γ+ cells and live/dead dye+ cells in gated CD45.2+CD4+ cells and CFSElow cells in gated live CD45.1+ CD8 T cells are shown on day 4. (H) Enriched iTregs were injected together with BM cells (Rag1−/−) into lethally irradiated BALB/c mice followed by Teff cell (CD45.1+) transfer 3 days later. Expression of IFN-γ and CD25 on gated H2Kb+CD45.2+Foxp3+ cells is shown 4 days after Teff transfer. Data shown are from 1 representative of 2 individual experiments. *P < .05, **P < .01, ***P < .001, ****P < .0001. MFI, mean fluorescence intensity.
Tregs are essential for controlling excessive activity of Teff cells and inducing immune tolerance.33 Next, to understand how miR-31 regulates Treg function, we analyzed the phenotype of these cells and found similar expression levels of CTLA4, CD28, CD39, and ICOS on WT vs miR-31 KO Tregs in recipients post-BMT (data not shown). Importantly, KO Tregs expressed a higher level of Nrp1, which has been reported to interact with semaphorin-4a and enhance stability and function of Tregs34 (Figure 3C; supplemental Figure 12A). In addition, miR-31 KO Tregs expressed higher levels of PD-L1 and Bcl2, suggesting they may have better stability, suppressive function, and survival after BMT.
Adoptive transfer of ex vivo–generated iTregs is a promising therapeutic strategy for controlling inflammatory diseases, including GVHD.35 However, the instability of iTregs induced by inflammatory environments impedes successful iTreg immunotherapy. To elucidate how miR-31 regulates iTreg stability, we generated alloantigen-reactive CD4+ iTregs in vitro.36 We consistently found increased iTreg differentiation from miR-31 KO vs WT CD4 T cells (Figure 3D). Again, Nrp1 expression was found to be increased on KO vs WT iTregs before and after 3-day culturing in IL-2, -12, or -6 cytokine, favoring Treg, Th1, or Th17 differentiation, respectively (Figure 3E). Importantly, miR-31 KO iTregs had improved Foxp3 retention and stability after exposure to these cytokines (Figure 3F; supplemental Figure 12B). When cocultured with Teff cells (CD45.1+) and allogeneic antigen-presenting cells, KO iTregs produced significantly lower levels of IFN-γ than WT iTregs, indicating they were more stable during allogeneic response (Figure 3G). MiR-31 KO iTregs showed improved survival and superior suppressive function for controlling CD8 Teff cell proliferation (Figure 3G). Furthermore, KO iTregs significantly reduced IFN-γ production and increased CD25 expression compared with WT iTregs after transfer into allogeneic recipients, indicating a more stable and activated phenotype of KO iTregs (Figure 3H). Therefore, miR-31 negatively regulates Nrp1 expression on Tregs, and miR-31 deficiency increases the generation, stability, and function of iTregs.
Loss of miR-31 in donor T cells alleviates cGVHD while sparing GVL activity through maintaining CD8 T-cell function
Given that persistent GVL response is essential for successful allo-HCT, we further studied how miR-31 affects the T cell–mediated antileukemia response. We performed haploidentical BMT and transferred luciferase-transduced P815 leukemia cells into these recipients on the day of BMT. We observed that the recipients of WT T cells presented clinical features of cGVHD, including alopecia and skin abrasions (Figure 4A-B, gray arrows) starting on day ∼20 after BMT. Recipients of miR-31 KO T cells showed attenuated cGVHD scores and skin manifestations and had improved overall survival (Figure 4A-C). The recipients of BM-only grafts had uncontrolled tumor growth, as reflected by strong bioluminescence signaling, and 100% lethality within 15 days post-BMT (Figure 4D). Importantly, intact antileukemia ability persisted long term after allo-BMT in the recipients of miR-31 KO grafts, despite attenuated cGVHD in these mice (Figure 4D). Compared with WT controls, KO CD8 T cells expressed similar levels of cytolytic activity–related markers, including IFN-γ, tumor necrosis factor α, granzyme B, perforin, and CD107a after allo-BMT (Figure 4E; supplemental Figure 13). Therefore, our data strongly indicate that miR-31 expression in allogeneic donor T cells is crucial for T-cell induction of cGVHD but that it is dispensable in the T cell–mediated antileukemia effect after allo-BMT.
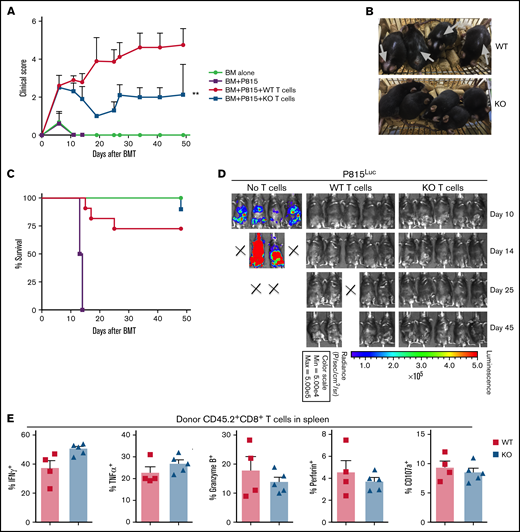
MiR-31 is dispensable in T cell–mediated GVL response. (A-E) WT or miR-31 KO T cells (2.75 × 106; CD45.2+) plus WT T cell–depleted BM (CD45.1+) were transferred into lethally irradiated B6D2F1 mice. On the day of BMT, 5000 luciferase-transduced P815 cells were IV injected into these recipients. Clinical scores (A), macroscopic photos of recipients on day 40 (B), and survival (C) are shown. P815 growth was monitored by bioluminescence imaging (D). Frequencies of IFN-γ+, tumor necrosis factor α–positive (TNFα+), granzyme B+, perforin+, and CD107a+ cells in gated live H2Kd-CD45.2+CD8+ cells (E) are shown in recipient spleens on day 50 post-BMT. Data shown are from 1 representative of 2 individual experiments (n = 4-5 mice per group for each experiment). **P < .01.
MiR-31 is dispensable in T cell–mediated GVL response. (A-E) WT or miR-31 KO T cells (2.75 × 106; CD45.2+) plus WT T cell–depleted BM (CD45.1+) were transferred into lethally irradiated B6D2F1 mice. On the day of BMT, 5000 luciferase-transduced P815 cells were IV injected into these recipients. Clinical scores (A), macroscopic photos of recipients on day 40 (B), and survival (C) are shown. P815 growth was monitored by bioluminescence imaging (D). Frequencies of IFN-γ+, tumor necrosis factor α–positive (TNFα+), granzyme B+, perforin+, and CD107a+ cells in gated live H2Kd-CD45.2+CD8+ cells (E) are shown in recipient spleens on day 50 post-BMT. Data shown are from 1 representative of 2 individual experiments (n = 4-5 mice per group for each experiment). **P < .01.
MiR-31 is critical for the activation, survival, and differentiation of CD4 T cells in a hypoxic environment via targeting of FIH1
We found that miR-31 KO CD4 T cells had compromised survival, elevated Treg generation, and attenuated Th17 differentiation during cGVHD development. It is known that HIF1α signaling enhances cell survival and promotes RORγt expression and Foxp3 degradation.37,38 Interestingly, FIH1, an asparaginyl hydroxylase that suppresses HIF1α activity, is negatively regulated by miR-31 in cancer cells.39,40 We hypothesized that miR-31 inhibits FIH1 expression, thus enhancing HIF1α signaling in T cells, which further augments their response. A highly conserved binding site of the seed sequence of miR-31 was presented in the 3′ untranslated region of FIH1 as predicted by TargetScan (Figure 5A). Given that FIH1 hydroxylates and controls HIF1α activity in an O2-dependent manner,41 we examined FIH1 expression in WT vs miR-31 KO CD4 T cells activated under normoxic or hypoxic conditions. WT T cells reduced FIH1 expression in 3% vs 21% O2 conditions as expected; however, KO T cells maintained FIH1 expression in 3% O2 (Figure 5B). Consistently, WT T cells upregulated HIF1α in 3% O2, whereas KO T cells failed to do so (Figure 5C). Consequently, miR-31 KO T cells exhibited impaired activation compared with WT cells in a 3% but not 21% O2 environment, reflected by reduced expression of CD69, ICOS, CD25, CD44, and Ki67 (Figure 5D; supplemental Figure 14).

MiR-31 is critical for the activation, survival, and differentiation of CD4 T cells in a hypoxic environment via targeting FIH1. (A) A highly conserved binding site of miR-31 seed sequence in the 3′ untranslated region (UTR) of FIH1 was identified using the TargetScan Web site. (B-E) CD4+CD25− WT or KO cells were isolated and stimulated with plate-bound anti-CD3 (4 μg/mL) and soluble anti-CD28 (2 μg/mL) in 3% or 21% oxygen (O2) conditions. Three days later, expression of FIH1 protein was determined by western blot (B), and expression of HIF1α (C) and CD69 and ICOS (D) in gated live CD4 T cells was determined by flow cytometry. CD4+CD25− WT or KO cells were stimulated with anti-CD3/CD28 in 3% O2 with 0.1 mM dimethyloxallyl glycine (DMOG) dissolved in phosphate-buffered saline (PBS) or vehicle control. Frequency of annexin V+7AAD+ dead cells is shown in gated CD4 T cells on day 3 (E). (F) CD4+CD25− WT of KO cells were stimulated with anti-CD3/CD28 together with anti–IFN-γ (1 μg/mL), transforming growth factor β (2 ng/mL), and IL-6 (1 ng/mL) in 3% O2. Expression of RORγt and Foxp3 in gated live CD4 T cells was determined by flow cytometry on day 3. Data shown are from 1 representative of 3 individual experiments. *P < .05, **P < .01, ***P < .001, ****P < .0001. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
MiR-31 is critical for the activation, survival, and differentiation of CD4 T cells in a hypoxic environment via targeting FIH1. (A) A highly conserved binding site of miR-31 seed sequence in the 3′ untranslated region (UTR) of FIH1 was identified using the TargetScan Web site. (B-E) CD4+CD25− WT or KO cells were isolated and stimulated with plate-bound anti-CD3 (4 μg/mL) and soluble anti-CD28 (2 μg/mL) in 3% or 21% oxygen (O2) conditions. Three days later, expression of FIH1 protein was determined by western blot (B), and expression of HIF1α (C) and CD69 and ICOS (D) in gated live CD4 T cells was determined by flow cytometry. CD4+CD25− WT or KO cells were stimulated with anti-CD3/CD28 in 3% O2 with 0.1 mM dimethyloxallyl glycine (DMOG) dissolved in phosphate-buffered saline (PBS) or vehicle control. Frequency of annexin V+7AAD+ dead cells is shown in gated CD4 T cells on day 3 (E). (F) CD4+CD25− WT of KO cells were stimulated with anti-CD3/CD28 together with anti–IFN-γ (1 μg/mL), transforming growth factor β (2 ng/mL), and IL-6 (1 ng/mL) in 3% O2. Expression of RORγt and Foxp3 in gated live CD4 T cells was determined by flow cytometry on day 3. Data shown are from 1 representative of 3 individual experiments. *P < .05, **P < .01, ***P < .001, ****P < .0001. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
To address whether miR-31 affects T-cell response through regulating HIF1α, CD4 T cells were activated in 3% O2 with the presence of DMOG, which enhances stability and activity of HIF1α by inhibiting enzymes that catalyze the hydroxylation reaction of HIF1α, including FIH1 and prolyl hydroxylase domain proteins.42 MiR-31 KO cells had significantly increased cell death than WT cells upon activation (Figure 5E). Addition of DMOG in culture partially restored survival of the KO cells but had no impact on WT cells. To evaluate whether HIF1α is involved in miR-31–mediated regulation of Th17/Treg differentiation, CD4 T cells were activated under 3% O2 in the presence of transforming growth factor β and IL-6, where they differentiated into RORγt+, Foxp3+, or Foxp3+RORγt+ subsets (Figure 5F). Fewer RORγt+, but more Foxp3+ and Foxp3+RORγt+, cells were observed in KO vs WT T cells. Adding DMOG increased RORγt+ but decreased Foxp3+ and Foxp3+RORγt+ subsets, leading to comparable RORγt and Foxp3 expression in WT vs KO cells. Furthermore, DMOG restored the activation of KO CD4 T cells under hypoxia (supplemental Figure 15). Taken together, these results demonstrate that miR-31 is critical for HIF1α upregulation through inhibiting FIH1 in low O2 conditions, which contributes to increased T-cell activation and survival and Th17/Treg imbalance.
Deficiency of miR-31 reduces HIF1α activity and glucose metabolism in allogeneic T cells after BMT
Hypoxic environments have been observed in a range of organs during inflammation damage, including the gastrointestinal tract, epidermis of skin, and thymus and even in regions of lymphoid organs.43 HIF1α signaling was reported to augment T-cell pathogenicity in murine GVHD.44 Hypoxia was reported to be involved in the pathogenesis of autoimmune disease by creating a proinflammatory environment.45 Having identified the critical role of miR-31 in regulating HIF1α expression and hypoxic response of T cells ex vivo, we further examined how miR-31 affects HIF1α expression in donor T cells during cGVHD development. We found that HIF1α expression was cell type and organ dependent; expression was higher in CD4 than in CD8 T cells and higher in skin-draining LNs than in spleens (Figure 6A-B). Importantly, KO donor T cells expressed significantly lower levels of HIF1α than WT T cells in recipient spleens and skin-draining LNs.
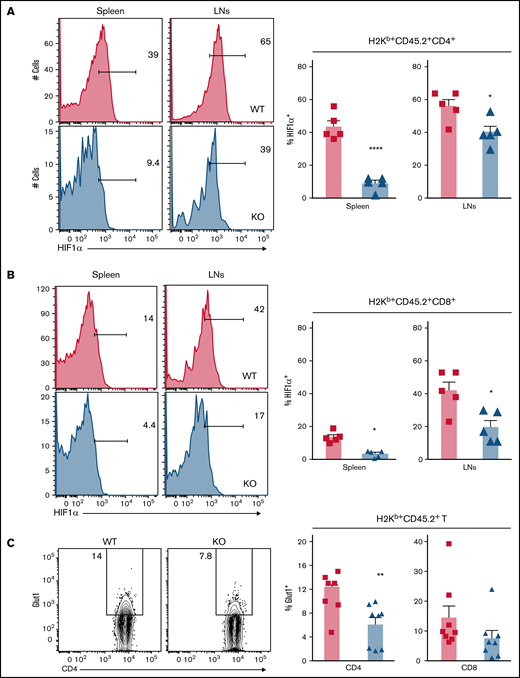

Deficiency of miR-31 reduces HIF1α activity and glucose metabolism in allogeneic T cells after BMT. Splenocytes (CD45.2+) from WT or miR-31 KO B6 mice were injected together with WT T cell–depleted (TCD) BM (CD45.1+) into lethally irradiated BALB/c mice. (A-B) Frequencies of HIF1α+ cells in gated live donor H2Kb+CD45.2+ CD4 (A) or CD8 (B) T cells in recipient spleens and skin-draining LNs are shown on day 50 to 60 post-BMT. Data shown are from 1 representative of 2 individual experiments. (C-D) Frequency of surface Glut1+ cells (C) and uptake of 2NBDG (D) in gated donor H2Kb+CD45.2+ CD4 or CD8 T cells are shown in recipient spleens. (E-F) CD4+CD25− WT or KO cells were stimulated with plate-bound anti-CD3 (4 μg/mL) and soluble anti-CD28 (2 μg/mL) in 3% O2 conditions. 2NBDG uptake in gated live CD4 T cells is shown on day 3 (E). In the same experimental setting, these activated CD4 T cells were subjected to Seahorse assay, and extracellular acidification rate (ECAR) was measured under basal conditions and after injection of 3 pharmacologic compounds: glucose (10 mM), oligomycin (1 μM), and 2-DG (100 mM). The diagram of ECAR and glycolysis calculated as increased ECAR after glucose injection are shown (F). *P < .05, **P < .01, ****P < .0001.
Deficiency of miR-31 reduces HIF1α activity and glucose metabolism in allogeneic T cells after BMT. Splenocytes (CD45.2+) from WT or miR-31 KO B6 mice were injected together with WT T cell–depleted (TCD) BM (CD45.1+) into lethally irradiated BALB/c mice. (A-B) Frequencies of HIF1α+ cells in gated live donor H2Kb+CD45.2+ CD4 (A) or CD8 (B) T cells in recipient spleens and skin-draining LNs are shown on day 50 to 60 post-BMT. Data shown are from 1 representative of 2 individual experiments. (C-D) Frequency of surface Glut1+ cells (C) and uptake of 2NBDG (D) in gated donor H2Kb+CD45.2+ CD4 or CD8 T cells are shown in recipient spleens. (E-F) CD4+CD25− WT or KO cells were stimulated with plate-bound anti-CD3 (4 μg/mL) and soluble anti-CD28 (2 μg/mL) in 3% O2 conditions. 2NBDG uptake in gated live CD4 T cells is shown on day 3 (E). In the same experimental setting, these activated CD4 T cells were subjected to Seahorse assay, and extracellular acidification rate (ECAR) was measured under basal conditions and after injection of 3 pharmacologic compounds: glucose (10 mM), oligomycin (1 μM), and 2-DG (100 mM). The diagram of ECAR and glycolysis calculated as increased ECAR after glucose injection are shown (F). *P < .05, **P < .01, ****P < .0001.
Our previous work demonstrates that glycolysis is the predominant metabolic pathway in T cells activated by alloantigens; glycolysis is essential for donor T cells to mediate GVHD.20 It was reported that HIF1α promotes T-cell glycolysis through regulating genes encoding glucose transporter and glycolytic enzymes.38 We found that miR-31 KO CD4 T cells reduced Glut1 expression and glucose uptake after allo-BMT (Figure 6C-D). However, similar expression levels of Glut1 and 2NBDG were observed in KO CD8 T cells compared with WT counterparts. We further examined how miR-31 affects glycolysis in CD4 T cells activated in 3% O2, close to the physiologic O2 levels in lymphoid and target organs.43 Consistently, KO T cells had significantly reduced glucose uptake, Glut-1 expression, extracellular acidification rate, and glycolysis (Figure 6E-F). Thus, miR-31 is important for Glut1 expression, glucose uptake, and glycolysis in CD4 T cells, which may provide essential metabolites and bioenergy required for increased T-cell expansion and Th17 differentiation during cGVHD development.
MiR-31 blockade by locked nucleic acid antagomir attenuates cGVHD after allo-BMT
Using genetic models, our data strongly support a key role for miR-31 in T-cell expansion and differentiation toward Th17 and away from Tregs, thus potentiating T-cell pathogenicity in cGVHD induction. To extend our finding, we tested this approach in a classic model of cGVHD after major histocompatibility complex–matched BMT (B10.D2 → BALB/c), in which cutaneous cGVHD is developed and mediated by donor IL-17–producing cells.46 Consistently, systemic administration of anti–miR-31 from day of BMT attenuated cGVHD severity (Figure 7A). Strikingly, delayed treatment started on day 21 post-BMT also effectively alleviated disease severity (Figure 7B). Similar to the observation in the genetic approach, pharmacologic blocking of miR-31 also attenuated pathologic damage and collagen deposition in recipient skin during cGVHD development (Figure 7C-D). Recipients with delayed anti–miR-31 treatment showed reduced frequencies of donor T cells and increased frequencies of B cells in spleens (Figure 7E). Increased numbers of B cells but comparable numbers of T cells were found in the recipients with delayed anti–miR-31 treatment compared with those with vehicle control (supplemental Figure 16A-B). In addition, delayed anti–miR-31 treatment improved Treg generation in recipients (Figure 7F). Blockade of miR-31 started on day 0, but not on day 21, of BMT attenuated Th17 differentiation in recipient skin-draining LNs (Figure 7F; data not shown). Pharmacologic inhibition of miR-31 could also increase FIH1 expression in T cells under hypoxic conditions (supplemental Figure 16C), consistent with the data observed in miR-31 KO T cells in the same experimental setting (Figure 5B). Furthermore, donor CD4 but not CD8 T cells expressed significantly lower HIF1α in recipients with anti–miR-31 treatment (Figure 7G). Our data demonstrate that systemic blockade of miR-31 can alleviate cGVHD. Similar to genetic deficiency, blocking miR-31 controls T-cell expansion, Treg differentiation, and HIF1α expression.
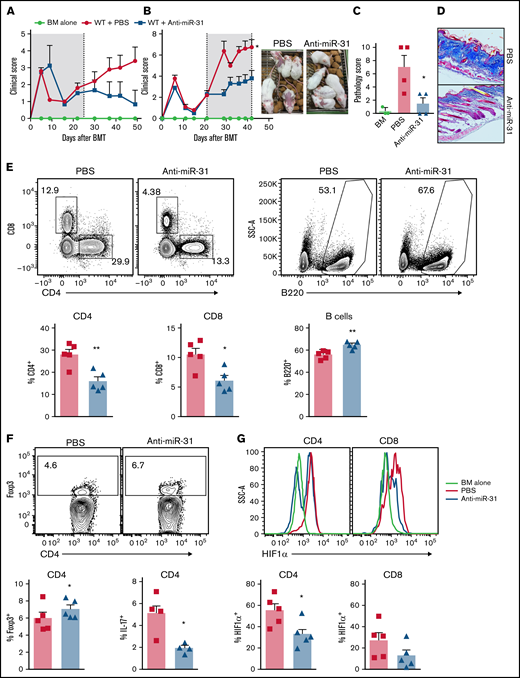
Blocking miR-31 alleviates cGVHD. (A-B) BALB/c mice were lethally irradiated and transferred with 5 × 106 T cell–depleted (TCD) BM plus 5 × 106 splenocytes from B10.D2 mice. Recipient mice were treated with anti–miR-31 starting on day 0 (A) or day 21 (B) after BMT. The clinical scores with macroscopic photos are shown on day 40 after BMT. (C-D) Cutaneous pathologic scores (C) and collagen deposition in skin evaluated with Masson’s trichrome stain (D) are shown on day 50. (E-G) In the experiments where anti–miR-31 treatment started at day 21, frequencies of CD4+, CD8+, and B220+ in gated donor Ly9.1− cells (E), Foxp3+ in gated donor Ly9.1− CD4 T cells (F), and HIF1α+ cells in gated donor Ly9.1− CD4 or CD8 T cells (G) are shown in recipient spleens on day 50. In the experiments where anti–miR-31 treatment started at day 0, frequency of IL-17+ in gated donor Ly9.1− CD4 T cells is shown in recipient skin-draining LNs on day 50 (F). *P < .05, **P < .01. PBS, phosphate-buffered saline; SSC, side scatter.
Blocking miR-31 alleviates cGVHD. (A-B) BALB/c mice were lethally irradiated and transferred with 5 × 106 T cell–depleted (TCD) BM plus 5 × 106 splenocytes from B10.D2 mice. Recipient mice were treated with anti–miR-31 starting on day 0 (A) or day 21 (B) after BMT. The clinical scores with macroscopic photos are shown on day 40 after BMT. (C-D) Cutaneous pathologic scores (C) and collagen deposition in skin evaluated with Masson’s trichrome stain (D) are shown on day 50. (E-G) In the experiments where anti–miR-31 treatment started at day 21, frequencies of CD4+, CD8+, and B220+ in gated donor Ly9.1− cells (E), Foxp3+ in gated donor Ly9.1− CD4 T cells (F), and HIF1α+ cells in gated donor Ly9.1− CD4 or CD8 T cells (G) are shown in recipient spleens on day 50. In the experiments where anti–miR-31 treatment started at day 0, frequency of IL-17+ in gated donor Ly9.1− CD4 T cells is shown in recipient skin-draining LNs on day 50 (F). *P < .05, **P < .01. PBS, phosphate-buffered saline; SSC, side scatter.
Discussion
Through miR profiling in murine T cells during alloreactive vs homeostatic responses, we identified miR-31 as a potential immunoregulator. We revealed a key role for miR-31 in augmenting donor T-cell pathogenicity in cGVHD induction but not in antileukemia response. MiR-31–deficient CD4 T cells had defects in proliferation and survival, differentiating more toward Tregs and less toward Th17 cells. MiR-31 promoted hypoxic adaptation of T cells by inhibiting FIH1 and therefore enhancing HIF1α signaling and glucose metabolism in T cells. To our knowledge, the current study is the first to uncover the biology of miR-31 in regulating hypoxic adaption of T-cell response through the FIH1/HIF1α pathway.
In multiple murine models of BMT, targeting miR-31 by genetic mutation or pharmacologic inhibition can significantly attenuate cGVHD symptoms, including sclerodermatous and pulmonary cGVHD manifestations. MiR-31 deficiency attenuates the expansion of mature alloreactive T cells, thus reducing the amount of pathogenic T cells, including IFN-γ– and IL-17A–producing and effector memory T cells in the recipients. MiR-31 influences the differentiation of mature/alloreactive T cells by increasing IL-17A production, which has been shown to not only drive dermal fibroblast proliferation but also enhance chemotaxis of macrophages that facilitate fibrosis by producing transforming growth factor β and platelet-derived growth factor α.47 Therefore, our data reveal a novel biology of miR-31 in promoting Th17 differentiation and fibrosis development in cGVHD. On the other hand, differentiation of Th17 and Tregs is considered reciprocal because Foxp3 antagonizes RORγt expression.48 MiR-31 limits Treg generation during cGVHD development, consistent with our previous report in EAE.17 Importantly, miR-31 negatively regulates the stability and function of Tregs during allogeneic response. In addition, miR-31 also affects the differentiation of a pathogenic extrafollicular CD4 T-cell population (PSGL-1loCD4+ T cells) that contributes to cGVHD pathogenesis by interacting with B cells in a GC-independent manner.31 It was reported that extrafollicular CD4 T cells express high levels of ICOS, IL-21, Stat3, and BCL6, which are required for their expansion. Furthermore, tissue-resident extrafollicular CD4 T cells augment autologous memory B-cell differentiation into plasma cells and antibody production during cGVHD development in humanized mice.32 Thus, extrafollicular T-B interaction is critical for pathogenic B-cell expansion and differentiation, especially where GC formation is inhibited when severe lymphopenia is presented during GVHD development.31,32
At molecular levels, miR-31 deficiency or inhibition hampers HIF1α expression in donor T cells. HIF1α enhances Th17 differentiation through promoting the transcriptional program enforcing glycolytic activity.38 HIF1α can directly bind to Foxp3, resulting in proteosomal degradation of Foxp3.37 HIF1α signaling in T cells can be induced by O2 limitation or T-cell receptor stimulation.43 We found that miR-31 was required for T-cell upregulation of HIF1α during activation in hypoxia and thus required for optimal activation, survival, and RORγt expression in low O2 conditions. We reason that through regulating the FIH1/HIF1α axis, miR-31 promotes alloreactive T-cell expansion and Th17/Treg imbalance in hypoxic environments, such as in GVHD target organs. Because HIF1α is reported to directly inhibit Nrp1,49 it is possible that attenuated HIF1α expression resulting from miR-31 deficiency may contribute to Nrp1 upregulation and Treg stabilization in miR-31 KO Tregs, although the mechanism remains to be further developed.
Metabolism is considered a basic housekeeping process that plays intricate roles in regulating T-cell response. Allogeneic T cells displayed higher extracellular acidification rates, glycolytic intermediates, and Glut1 and Gluts3 transcription in acute GVHD.20 Increased glycolysis as well as elevated glycolysis-driven OXPHOS were also shown in T cells in lupus.50 Because cGVHD shares autoimmune features with lupus, we infer that reduced Glut1 expression, glucose uptake, and glycolysis may be attributed to the attenuated pathogenicity of miR-31 KO CD4 T cells in cGVHD induction. Because HIF1α enhances the transcription of glucose transporters and glycolytic enzymes,38 we reason that, during cGVHD induction, miR-31 promotes glycolysis through upregulating HIF1α, which contributes to fulfilling the bioenergetic demand for T-cell expansion and promoting Th17 while limiting Treg transcriptional programing.
Interestingly, miR-31 deficiency seems to enhance the potency of CD8 T cell–mediated antiviral responses18 while reducing the pathogenicity of T cells, especially CD4+, in cGVHD and EAE.17 We found HIF1α was downregulated in miR-31–deficienct CD8 T cells. However, HIF1α seems nonessential for metabolic reprograming of CD8 T cells, consistent with lower expression of HIF1α in CD8 vs CD4 T cells after allo-BMT. MiR-31 expression was much higher in CD8 vs CD4 T cells after activation,18 suggesting miR-31 possibly targets different downstream messenger RNAs. Importantly, the largely maintained CD8 T-cell glucose metabolism and cytolytic molecules likely account for the preservation of the GVL effect.
Clinical studies evaluating miRs as noninvasive biomarkers (registered at www.clinicaltrials.gov as #NCT01521039)51,52 and/or therapeutic targets53 highlight the importance of miR biology in immune regulation and the potential of miR-based interference in allo-HCT. Our study reveals a novel biology of miR-31 in modulating T-cell adaptation to low O2 and their pathogenicity in cGVHD induction and therefore provides a rationale for targeting miR-31 to control cGVHD in patients after allo-HCT in the foreseeable future.
Acknowledgments
The authors appreciate the technical and equipment support provided by the staff in the Department of Laboratory Animal Research, Bioenergetics Core, Flow Cytometry Core, and Histological Core at the Medical University of South Carolina (MUSC). The authors acknowledge Gyda Beeson and Craig Beeson at MUSC for their support with bioenergetic analysis. The authors thank Viswanathan Palanisamy, Shikhar Mehrotra, Besim Ogretmen, and Carsten Krieg at MUSC for advisory suggestions and Rachel Magnin for assistance with techniques as well as Hongjun Wang for providing the trigas hypoxic chamber and Wenyu Gou and Hua Wei for assistance in hypoxia experiments.
This work was supported in part by the Hollings Cancer Center Fellowship (Y.W.) and National Institutes of Health grants R01s NIH/NIAID AI118305, NIH/NHLBI HL137373, and NIH/NHLBI HL140953 (X.-Z.Y.).
Authorship
Contribution: Y.W. designed and performed research, collected, analyzed, and interpreted data, and drafted and revised the manuscript; C.M. collected and analyzed data and edited the manuscript; C.L.W. performed lung function tests and revised the manuscript; S.S., D.B., M.H.S., M.Z., H.-J.C., K.Y., L.T., and H.N. assisted in collecting data; Z.L. analyzed microarray data; K.H. read and scored pathologic slides; L.M.S. provided the flexiVent system and edited the manuscript; H.W. provided miR-31flox mice and participated in experimental design; and X.-Z.Y. designed the research, interpreted data, and revised the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for L.M.S. and C.L.W. is Department of Medicine, University of Wisconsin School of Medicine and Public Health, Madison, WI.
Correspondence: Xue-Zhong Yu, Department of Microbiology & Immunology, Medical College of Wisconsin, 8701 Watertown Plank Rd, Milwaukee, WI 53226; e-mail: xuyu@mcw.edu; and Yongxia Wu, Department of Microbiology & Immunology, Medical College of Wisconsin, 8701 Watertown Plank Rd, Milwaukee, WI 53226; e-mail: wyongxia@mcw.edu.

