

Key Points
EL cells show activated glycolysis and high expression of LSD1 among the AML subtypes, which represent unique metabolic phenotypes.
LSD1 promotes glycolysis and heme synthesis by stabilizing GATA1 and maintains GATA1-target metabolic genes by downregulating C/EBPα.

Abstract
Acute myeloid leukemia (AML) is a heterogenous malignancy characterized by distinct lineage subtypes and various genetic/epigenetic alterations. As with other neoplasms, AML cells have well-known aerobic glycolysis, but metabolic variations depending on cellular lineages also exist. Lysine-specific demethylase-1 (LSD1) has been reported to be crucial for human leukemogenesis, which is currently one of the emerging therapeutic targets. However, metabolic roles of LSD1 and lineage-dependent factors remain to be elucidated in AML cells. Here, we show that LSD1 directs a hematopoietic lineage-specific metabolic program in AML subtypes. Erythroid leukemia (EL) cells particularly showed activated glycolysis and high expression of LSD1 in both AML cell lines and clinical samples. Transcriptome, chromatin immunoprecipitation–sequencing, and metabolomic analyses revealed that LSD1 was essential not only for glycolysis but also for heme synthesis, the most characteristic metabolic pathway of erythroid origin. Notably, LSD1 stabilized the erythroid transcription factor GATA1, which directly enhanced the expression of glycolysis and heme synthesis genes. In contrast, LSD1 epigenetically downregulated the granulo-monocytic transcription factor C/EBPα. Thus, the use of LSD1 knockdown or chemical inhibitor dominated C/EBPα instead of GATA1 in EL cells, resulting in metabolic shifts and growth arrest. Furthermore, GATA1 suppressed the gene encoding C/EBPα that then acted as a repressor of GATA1 target genes. Collectively, we conclude that LSD1 shapes metabolic phenotypes in EL cells by balancing these lineage-specific transcription factors and that LSD1 inhibitors pharmacologically cause lineage-dependent metabolic remodeling.
Introduction
Although aerobic glycolysis has been thought of as a common hallmark of cancer,1 emerging evidence suggests the existence of metabolic heterogeneity within and between tumor types, and this could be a potential barrier in targeting metabolic vulnerability in cancer therapies.2-7 Acute myeloid leukemia (AML) is a group of hematopoietic malignancies comprising many subtypes with different lineage identities and genetic/epigenetic lesions.8,9 Although the characteristic differences among subtypes have been described, variable metabolic phenotypes and their regulatory mechanisms remain unexplored. A previous report, using an MLL-AF9 AML model in mice, showed that leukemic cells are more vulnerable to perturbations of glycolytic genes than normal hematopoietic cells.10 Another report has shown a similar glycolysis dependency in AML cells harboring internal tandem repeats of the FLT3 gene.11 In addition, mutations in the isocitrate dehydrogenase gene generate a rare metabolite that causes epigenetic disruption in AML.12 Because these observations are limited to subtypes with specific genotypes, it remains unclear whether lineage differences are linked to metabolic properties in AML. In addition, the availability of nutrients such as glucose and glutamine exerts a profound influence on the cell fate decision during normal hematopoiesis.13 These observations raise the possibility that metabolic phenotypes and/or nutrient requirements vary among AML subtypes depending on lineage identities. Despite remarkable clinical advances, there is considerable variability in the success of therapy among AML subtypes.8,14 Thus, targeting of subtype-specific metabolic features could provide a powerful tool for next-generation AML therapy.
Lysine-specific demethylase-1 (LSD1) was first identified as a histone H3 lysine 4 (H3K4) demethylase and later as a demethylase for transcription factors (TFs) such as p53 and STAT3.15,16 LSD1 has been implicated in diverse biological processes, including cellular differentiation, tumor development, and metabolism.17,18 We previously reported that, in hepatocellular carcinoma cells, LSD1 represses mitochondrial respiration-associated genes such as PPARGC1A through H3K4 demethylation, while promoting the expression of glycolytic genes by facilitating hypoxia-inducible factor-1α (HIF-1α)–mediated transcription.19 In addition, high expression of LSD1 is associated with enhanced glucose uptake in human esophageal cancer.20 In hematopoietic cells, LSD1 physically interacts and cooperates with growth factor independence-1 and growth factor independence-1b, TFs that are involved in multiple steps of hematopoiesis.21 The depletion of LSD1 in the hematopoietic system results in defects in stem and progenitor cells, thereby impeding the differentiation of multiple lineages.22 Increased expression of LSD1 has been observed in many different types of human hematopoietic neoplasms, implying significant involvement in leukemogenesis.23 Indeed, small compound inhibitors of LSD1 have been shown to eradicate leukemic cells effectively.24-27
In this study, we investigated the role of LSD1 in metabolic regulation in human AML subtypes and found that erythroid leukemia (EL) cells have activated glycolysis and high expression of LSD1. Using transcriptomic and epigenomic approaches, we identified that LSD1 facilitates the function of the erythroid-specific factor GATA1, while suppressing the granulo-monocytic factor C/EBPα. In addition, we found that GATA1 and C/EBPα work in a mutually exclusive manner in EL cells, emphasizing a functional balance of these lineage-dependent TFs by LSD1. We therefore concluded that LSD1 plays essential roles in the metabolic heterogeneity of AML and especially in metabolic phenotypes of EL cells.
Methods
Cell culture
AML cell lines (HEL, TF1a, SET-2, NB4, and HL60) and K562 cells were grown in RPMI 1640 medium (Sigma), supplemented with 10% heat-inactivated fetal bovine serum, 50 U/mL penicillin, and 50 μg/mL streptomycin at 37°C with 95% air and 5% carbon dioxide. Detailed information on cell lines is provided in supplemental Table 1. Hemoglobin synthesis in HEL cells was induced by the addition of 30 μM hemin for 4 days and visualized by benzidine staining as described previously.28
Lentiviral expression of short hairpin RNA
Lentiviral vectors for tetracycline-inducible short hairpin RNA expression were obtained from the RIKEN BioResource Research Center (http://cfm.brc.riken.jp/Lentiviral_Vectors_J). The short hairpin RNA target sequences were as follows: shLSD1_#1, 5′-CACAAGGAAAGCTAGAAGA-3′; shLSD1_#2, 5′-AACAATTAGAAGCACCTTA-3′; shGATA1, 5′-GGATGGTATTCAGACTCGA-3′; shCEBPA, 5′-ACGAGACGTCCATCGACAT-3′; shALAS2_#1, 5′-GATGTGAAGGCTTTCAAGA-3′; shALAS2_#2, 5′-AGGCTTCATCTTTACCACT-3′; shGATA2, 5′-GAAGTGTCTCCTGACCCTA-3′; and shGFI1 5′-GCTCGGAGTTTGAGGACTTCT-3′.
Measurement of cellular glucose uptake
To measure glucose uptake, cells were incubated in the culture medium containing 100 μM 2-NBDG (Peptide Institute) for 2 hours, as previously described.29
Real-time measurement of glycolytic activity
Monitoring of cellular glycolytic activity was performed by using the XFe24 Extracellular Flux analyzer (Seahorse Bioscience). For floating cells, we used Cellbed (Japan Vilene), a high-purity silica fiber that can be used as a three-dimensional cell culture scaffold sheet on each well of the assay plate (300 000 cells per well). To evaluate glycolysis capacity, cells were cultured in glucose-free medium for 1 hour before the assay. Real-time measurement was initiated under glucose-free conditions, followed by the addition of 25 mM glucose and then a glycolysis inhibitor 2-deoxy-glucose at 100 mM. Glycolytic flux was determined by measuring the extracellular acidification rate.
Metabolomic analyses
Metabolomic analyses were conducted by using capillary electrophoresis time of flight mass spectrometry at Human Metabolome Technologies. Cluster analyses were performed as previously described.30 The amount of metabolite was normalized for hierarchical clustering by the average linking method using Cluster 3.0. The heatmap was visualized by Java Tree view. In supplemental Figure 4L, 57 metabolites were detected in our experiment and registered in the Cancer Cell Encyclopedia (CCLE) database.7
Poly (A) RNA-sequencing analysis
Total RNA from HEL cells was extracted by using the RNeasy Mini Kit (Qiagen). Messenger RNA (mRNA) was purified by using an NEBNext Poly (A) mRNA Magnetic Isolation Module (NEB). A complementary DNA library was synthesized by using a NEBNext Ultra DNA Library Prep Kit for Illumina (NEB) and was sequenced on a NextSeq 500 sequencer (Illumina) with 75 bp single-end reads. The resulting reads were aligned to the UCSC hg19 reference genome by using TopHat (version 1.4.1).31 Normalization, differential analysis, gene ontology analysis, and construction of the Venn diagram were performed by using Strand NGS (Strand Genomics). The primers used for quantitative reverse transcription polymerase chain reaction (RT-qPCR) to confirm gene expression are listed in supplemental Table 2.
Chromatin immunoprecipitation
About 8 × 106 and 4 × 106 HEL cells were used to detect methylated histone H3K4 and LSD1 enrichment, and GATA1 enrichment, respectively. Crosslinking, fragmentation, and chromatin immunoprecipitation (ChIP) experiments were performed with some modifications (supplemental Methods) based on previous reports.32,33 The primers used are listed in supplemental Table 2.
ChIP-sequencing analysis
Using 8 × 106 HEL cells, ChIP experiments were performed as noted earlier, and each protein-bound chromatin fraction was collected for DNA purification. Library preparation was done by using the NEBNext Ultra II DNA Library Kit for Illumina (New England Biolabs). Adapter-ligated DNA fragments were purified by using Agencourt AMPure XP. High-throughput sequencing was performed by using a NextSeq 500 Sequencer with 75 bp single-end reads. The qualified reads were aligned onto the human reference genome hg19 by using the Burrows-Wheeler Alignment algorithm.34 Duplicate reads and the reads with low overall quality or low mapping quality were excluded. The final numbers of mapped reads are listed in supplemental Table 3. Peak detection was done by using the MACS2 algorithm. For LSD1/ChIP-sequencing, sequencing data from duplicate samples were separately mapped to the genome, merged, and then subjected to peak calling with MACS2 algorithm. Motif analyses and peak annotations were done with HOMER.35 Peaks within intergenic regions were associated with nearby genes if they were within a 100-kb window from the transcriptional start site of a specific gene. The cooccupancy of LSD1 with other proteins was analyzed by using ChIP-Atlas (http://chip-atlas.org/). Visualization of ChIP-sequencing data was done by using the Integrative Genomics Viewer (http://software.broadinstitute.org/software/igv/) after converting BAM files into bigWig files. ChIP-sequencing for H3K9me3 in K562 cells was obtained from the ENCODE/Broad Institute via the UCSC Genome Browser Web site (http://genome.ucsc.edu/index.html).
Clinical data sets
Transcriptome data set from clinical AML cases were obtained from The Cancer Genome Atlas (TCGA) database (https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga). Transcriptome data from EL (AML-M6) cases were obtained from the St. Jude PeCan Data Portal88 (https://pecan.stjude.cloud/proteinpaint/study/ael).36 These data sets use different algorithms for the normalization of expression levels (TCGA, transcripts per million; St. Jude PeCan Data Portal88, counts per million). Therefore, to analyze these data sets collectively, we normalized the expression level of each gene to an internal control. A gene encoding TATA-binding protein (TBP) was selected for this purpose because it showed relatively low sample-to-sample variation compared with other candidates such as RPLP0 and ACTB.
Statistical analysis and reproducibility
Data are presented as the mean ± standard deviation. Equality of variance was examined by using an F test. All statistical analyses were performed by using a two-tailed Student’s t test. A value of P < .05 was considered statistically significant. Representative data/images were replicated in at least 3 independent experiments.
Results
High expression of LSD1 is directly linked to enhanced glucose metabolism in EL
To examine whether LSD1 contributes to enhanced glycolysis in leukemic cells, we analyzed the gene expression profiles of AML cell lines and clinical samples using publicly available data sets from the TCGA and the CCLE.37,38 Expression of LSD1 and GLUT1 (SLC2A1), a major regulator of glucose flux, was significantly and positively correlated in both cell lines and clinical samples (Figure 1A-B; supplemental Figure 1A). In particular, these genes were highly expressed in EL, which is classified as a unique subtype of AML (FAB M6).39,40 An analysis of a published single-cell RNA-sequencing data41 from normal hematopoietic cells revealed enhanced expression of LSD1 in the erythroid lineage (e-ery and l-ery) (supplemental Figure 1B). Notably, LSD1-high EL cells with different genetic backgrounds exhibited higher levels of medium acidification and GLUT1 expression than did other AML cells (Figure 1C; supplemental Figure 1C-D; supplemental Table 1), indicating that enhanced glycolysis is a characteristic of erythroid lineage. To test the function of LSD1 in metabolic regulation, we generated doxycycline-inducible LSD1-knockdown (KD) cells using distinct EL cell lines, HEL and TF1a. LSD1-KD downregulated the expression of GLUT1 as well as PKLR, an essential glycolytic enzyme in erythroid cells (Figure 1D-E; supplemental Figure 1E-H).42 Consistent with the gene expression profiles, LSD1-KD cells showed reduced glucose uptake and glycolytic capacity (Figure 1F-G; supplemental Figure 1I-K).

High levels of LSD1 expression and glycolytic activity in erythroleukemia cells. Scatter plots showing positive correlation between LSD1 and GLUT1 expression in AML in TCGA clinical samples (n = 173) (A) and CCLE cell lines (n = 37) (B). Pearson product correlation coefficient and P values are indicated. (C) Glycolysis/OXPHOS balance of HEL and HL60 cells, determined by using an extracellular flux analyzer. Values indicate the ratio of extracellular acidification rate (ECAR) and oxygen consumption rate (OCR). Values are mean ± standard deviation (SD) of 10 wells. (D) Expression changes of glycolytic genes in HEL cells expressing short hairpin RNA against LSD1 (shLSD1#1). Full descriptions of gene symbols are provided in supplemental Table 2. qRT-PCR values, which were normalized to the expression levels of the 36B4 gene, are shown as the fold difference against control (ctrl) samples. (E) Decrease of GLUT1 protein was confirmed in LSD1-KD HEL cells. Scanned images of unprocessed blots are shown in supplemental Figure 10. (F) Reduction of glucose uptake in LSD1-KD HEL cells. 2-NBDG incorporation was determined by flow cytometry. Mean fluorescence intensities are shown. (G) Reduced glycolytic activity in LSD1-KD HEL cells. Values are mean ± SD of 5 assays. (H) Expression changes of glycolytic genes under the treatment with the LSD1 inhibitor S2101. (I) Decrease of GLUT1 protein in S2101-treated HEL cells. (J) Reduction of glucose uptake by S2101 treatment. (K) Reduction of glycolytic activity in S2101-treated HEL cells. Values are mean ± SD of 5 assays. All samples were collected at day 4 unless indicated otherwise. All histogram data are mean ± SD of triplicate results unless indicated otherwise. *P < .05, **P < .01 vs control. CMP, common myeloid progenitor; FPKM, fragments per kilobase of transcript per million mapped reads; GMP, granulocyte erythroid progenitor; HSC, hematopoietic stem cell; MEP, megakaryocyte erythroid progenitor.
High levels of LSD1 expression and glycolytic activity in erythroleukemia cells. Scatter plots showing positive correlation between LSD1 and GLUT1 expression in AML in TCGA clinical samples (n = 173) (A) and CCLE cell lines (n = 37) (B). Pearson product correlation coefficient and P values are indicated. (C) Glycolysis/OXPHOS balance of HEL and HL60 cells, determined by using an extracellular flux analyzer. Values indicate the ratio of extracellular acidification rate (ECAR) and oxygen consumption rate (OCR). Values are mean ± standard deviation (SD) of 10 wells. (D) Expression changes of glycolytic genes in HEL cells expressing short hairpin RNA against LSD1 (shLSD1#1). Full descriptions of gene symbols are provided in supplemental Table 2. qRT-PCR values, which were normalized to the expression levels of the 36B4 gene, are shown as the fold difference against control (ctrl) samples. (E) Decrease of GLUT1 protein was confirmed in LSD1-KD HEL cells. Scanned images of unprocessed blots are shown in supplemental Figure 10. (F) Reduction of glucose uptake in LSD1-KD HEL cells. 2-NBDG incorporation was determined by flow cytometry. Mean fluorescence intensities are shown. (G) Reduced glycolytic activity in LSD1-KD HEL cells. Values are mean ± SD of 5 assays. (H) Expression changes of glycolytic genes under the treatment with the LSD1 inhibitor S2101. (I) Decrease of GLUT1 protein in S2101-treated HEL cells. (J) Reduction of glucose uptake by S2101 treatment. (K) Reduction of glycolytic activity in S2101-treated HEL cells. Values are mean ± SD of 5 assays. All samples were collected at day 4 unless indicated otherwise. All histogram data are mean ± SD of triplicate results unless indicated otherwise. *P < .05, **P < .01 vs control. CMP, common myeloid progenitor; FPKM, fragments per kilobase of transcript per million mapped reads; GMP, granulocyte erythroid progenitor; HSC, hematopoietic stem cell; MEP, megakaryocyte erythroid progenitor.
Treatment with S2101, a selective inhibitor of LSD1, led to similar gene expression and metabolic changes (Figure 1H-K; supplemental Figure 2A-D). These results were reproduced by the use of alternative LSD1 inhibitors, T-3775440 and GSK-LSD1 (supplemental Figure 2E-J). Moreover, LSD1 inhibition selectively inhibited the growth of EL and megakaryoblastic leukemia cells, which exhibited high LSD1 expression (Figure 1B; supplemental 3). In non-EL AML cells, GLUT1 expression was mostly unaffected by LSD1 inhibition (supplemental Figure 2K). The data show that increased LSD1 in the EL subtype is directly linked to enhanced glycolytic activities.
LSD1 facilitates metabolic pathways associated with erythroid lineage
To further identify LSD1-dependent metabolic pathways in EL cells, we analyzed the gene expression changes in LSD1-inhibited HEL cells by RNA-sequencing. We identified that 334 genes were commonly downregulated by both LSD1-KD and S2101 treatment (Figure 2A). Of note, genes involved in heme biosynthesis were commonly downregulated by LSD1 inhibition. By qRT-PCR, we confirmed that key genes in heme metabolism such as ALAS2 and SLC25A37 (encoding 5′-aminolevulinate synthase-2 and mitoferrin-1, respectively) were significantly downregulated in LSD1-inhibited HEL and TF1a cells (Figure 2C; supplemental Figure 4A-F). To test the essentiality of heme synthesis in EL cells, we generated ALAS2-KD HEL cells. ALAS2-KD resulted in reduced cell growth, when combined with Fe2+ depletion by deferoxamine, an iron chelator, suggesting that the heme synthetic pathway is involved in survival and/or growth (supplemental Figure 4G-I). Because heme synthesis is intimately associated with erythrocyte function, we hypothesized that LSD1 directs a lineage-linked metabolic phenotype. To test this theory, we analyzed metabolic properties in LSD1-inhibited cells after treatment with hemin, a chemical that promotes erythroid characteristics. Consistent with the gene expression changes, hemin-induced hemoglobin production was downregulated by the loss of LSD1 (Figure 2D; supplemental Figure 4J). It is important to note that LSD1-inhibited cells did not exhibit morphologic changes (supplemental Figure 4K), suggesting that differentiation status was not altered. Enhancement of erythroid-type metabolism by hemin was confirmed by a metabolomic examination, in which hemin treatment led to increases in the EL-enriched metabolites (supplemental Figure 4L), based on the CCLE metabolome database.7 We found that such hemin-induced increases of metabolites were mostly canceled by treatment with S2101 (Figure 2E). Collectively, these data suggest that LSD1 directs an erythroid-associated metabolic phenotype in EL cells.

LSD1 facilitates erythroid lineage-linked metabolic phenotype. RNA-sequencing analysis. Venn diagrams of genes upregulated (A) and downregulated (B) more than 1.2-fold by LSD1-KD and S2101 treatment (10 µM) are indicated (left). Gene ontology (GO) analysis of the gene sets significantly upregulated and downregulated by LSD1 inhibition (right). Top 10 pathways with statistical significance are indicated with log10P values. Pathways related to heme synthesis (upregulated) and membrane-bound proteins (downregulated) are highlighted in red and blue, respectively. (C) Schema of hemoglobin synthesis pathway (left). qRT-PCR data showing expression changes of hemoglobin biosynthesis genes in LSD1-KD HEL cells (right). (D) Light microscopy image of benzidine-stained HEL cells (left). Arrows represent benzidine-positive cells. Proportions of benzidine-positive cells (right). The experiment was triplicated by calculating 1000 cells in each assay. (E) Metabolomic analysis of LSD1-inhibited HEL cells. Cells were treated with hemin and S2101 and were subjected to capillary electrophoresis time of flight mass spectrometry–based metabolomic analysis. (F-G) Expression changes of non-erythroid hematopoietic markers in LSD1-KD– and S2101-treated HEL cells. All histogram data are mean ± standard deviation of triplicate results. *P < .05, **P < .01 vs control. ALA, 5′-aminolevulinic acid; CPgen III, coproporphyrinogen III; PP IX, protoporphyrin IX.
LSD1 facilitates erythroid lineage-linked metabolic phenotype. RNA-sequencing analysis. Venn diagrams of genes upregulated (A) and downregulated (B) more than 1.2-fold by LSD1-KD and S2101 treatment (10 µM) are indicated (left). Gene ontology (GO) analysis of the gene sets significantly upregulated and downregulated by LSD1 inhibition (right). Top 10 pathways with statistical significance are indicated with log10P values. Pathways related to heme synthesis (upregulated) and membrane-bound proteins (downregulated) are highlighted in red and blue, respectively. (C) Schema of hemoglobin synthesis pathway (left). qRT-PCR data showing expression changes of hemoglobin biosynthesis genes in LSD1-KD HEL cells (right). (D) Light microscopy image of benzidine-stained HEL cells (left). Arrows represent benzidine-positive cells. Proportions of benzidine-positive cells (right). The experiment was triplicated by calculating 1000 cells in each assay. (E) Metabolomic analysis of LSD1-inhibited HEL cells. Cells were treated with hemin and S2101 and were subjected to capillary electrophoresis time of flight mass spectrometry–based metabolomic analysis. (F-G) Expression changes of non-erythroid hematopoietic markers in LSD1-KD– and S2101-treated HEL cells. All histogram data are mean ± standard deviation of triplicate results. *P < .05, **P < .01 vs control. ALA, 5′-aminolevulinic acid; CPgen III, coproporphyrinogen III; PP IX, protoporphyrin IX.
In our RNA-sequencing data, 409 genes were upregulated by LSD1-KD and S2101 treatment (Figure 2B). Many of these genes were associated with membrane-bound proteins. Among these genes, CD48 and CD34, which are surface markers of lymphocytes and hematopoietic stem cells, respectively, were prominently upregulated by LSD1 inhibition (Figures 2F-G, 4A). These data indicate that LSD1 suppresses non-erythroid features in EL cells.

LSD1 cooperates with GATA1 to promote glycolysis. (A) ChIP-sequencing (ChIP-seq) analysis. Distribution of LSD1-enriched peaks in HEL cells, relative to annotated regions in the genome. (B) Motif analysis of 8280 LSD1-enriched peaks. Motif and statistical analyses were performed by using HOMER software. (C) Coimmunoprecipitation of LSD1 and GATA1. Input lane contains the 10% of the total amount of whole cell extract, relative to IP lanes. (D) Gene expression changes in GATA1-KD HEL cells. (E) Decreased GLUT1 protein in GATA1-KD HEL cells. Samples were collected at indicated days after doxycycline (DOX) induction. (F) Reduction of glucose uptake in GATA1-KD HEL cells, as determined by 2-NBDG incorporation. (G) Reduction of glycolytic activity in GATA1-KD HEL cells. Values are mean ± standard deviation of 5 assays. (H) LSD1-dependent enrichment of GATA1 at ALAS2 and GLUT1 gene enhancers. ChIP-qPCR analyses were performed in control (ctrl) and LSD1-KD HEL cells. Enrichment values were calculated as percentage of input DNA. (I) Growth curve of ctrl and GATA1-KD HEL cells. (J) Downregulation of GATA1 protein in LSD1-KD HEL cells. Samples were collected at indicated days after DOX induction. (K) Effect of proteasome inhibition on GATA1 protein in LSD1-KD cells. The cells were treated with MG132 (10 µM) for 6 hours before harvest. Values are mean ± standard deviation of triplicate results except for those in panel G. *P < .05, **P < .01. 5′UTR, 5′ untranslated region; IgG, immunoglobulin G; TSS, transcriptional start site; TTS: transcriptional termination site.
LSD1 cooperates with GATA1 to promote glycolysis. (A) ChIP-sequencing (ChIP-seq) analysis. Distribution of LSD1-enriched peaks in HEL cells, relative to annotated regions in the genome. (B) Motif analysis of 8280 LSD1-enriched peaks. Motif and statistical analyses were performed by using HOMER software. (C) Coimmunoprecipitation of LSD1 and GATA1. Input lane contains the 10% of the total amount of whole cell extract, relative to IP lanes. (D) Gene expression changes in GATA1-KD HEL cells. (E) Decreased GLUT1 protein in GATA1-KD HEL cells. Samples were collected at indicated days after doxycycline (DOX) induction. (F) Reduction of glucose uptake in GATA1-KD HEL cells, as determined by 2-NBDG incorporation. (G) Reduction of glycolytic activity in GATA1-KD HEL cells. Values are mean ± standard deviation of 5 assays. (H) LSD1-dependent enrichment of GATA1 at ALAS2 and GLUT1 gene enhancers. ChIP-qPCR analyses were performed in control (ctrl) and LSD1-KD HEL cells. Enrichment values were calculated as percentage of input DNA. (I) Growth curve of ctrl and GATA1-KD HEL cells. (J) Downregulation of GATA1 protein in LSD1-KD HEL cells. Samples were collected at indicated days after DOX induction. (K) Effect of proteasome inhibition on GATA1 protein in LSD1-KD cells. The cells were treated with MG132 (10 µM) for 6 hours before harvest. Values are mean ± standard deviation of triplicate results except for those in panel G. *P < .05, **P < .01. 5′UTR, 5′ untranslated region; IgG, immunoglobulin G; TSS, transcriptional start site; TTS: transcriptional termination site.
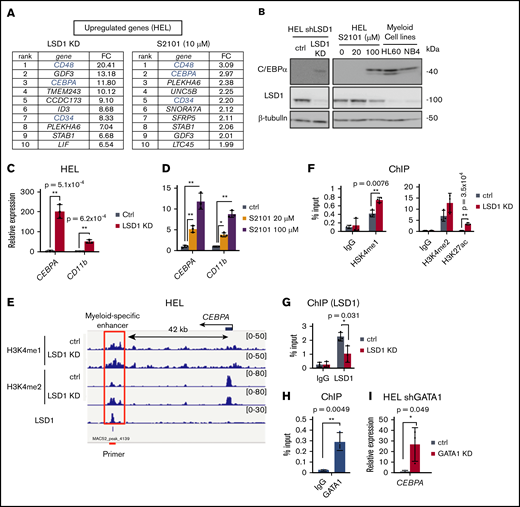
LSD1 represses a myeloid-specific enhancer of the CEBPA gene via H3K4 demethylation. (A) Top 10 upregulated genes by LSD1 inhibition. Fold change (FC) values relative to untreated control are indicated. (B) Expression of C/EBPα protein in LSD1-KD– and S2101-treated HEL cells. (C-D) Expression changes of myeloid-related genes in LSD1-KD– and S2101-treated HEL cells. (E) Enrichment of H3K4me1, H3K4me2, and LSD1 at the CEBPA gene locus by ChIP-sequencing analysis. LSD1-KD data are also indicated. Red line indicates the ChIP-qPCR primer at the myeloid-specific enhancer. Data were visualized by using Integrative Genomics Viewer (http://software.broadinstitute.org/software/igv/). (F) Increase of H3K4me1, H3K4me2, and H3K27ac at the CEBPA enhancer by LSD1-KD. ChIP-qPCR signals were calculated relative to that of histone H3. (G) Enrichment of LSD1 at CEBPA enhancer examined by ChIP-qPCR. (H) Enrichment of GATA1 at theCEBPA enhancer in HEL cells tested by ChIP-qPCR. Enrichment values at the enhancer were normalized to input DNAs for panels F to H. (I) Upregulation of CEBPA in GATA1-KD HEL cells. Values are mean ± standard deviation of triplicate results. *P < .05, **P < .01.
LSD1 represses a myeloid-specific enhancer of the CEBPA gene via H3K4 demethylation. (A) Top 10 upregulated genes by LSD1 inhibition. Fold change (FC) values relative to untreated control are indicated. (B) Expression of C/EBPα protein in LSD1-KD– and S2101-treated HEL cells. (C-D) Expression changes of myeloid-related genes in LSD1-KD– and S2101-treated HEL cells. (E) Enrichment of H3K4me1, H3K4me2, and LSD1 at the CEBPA gene locus by ChIP-sequencing analysis. LSD1-KD data are also indicated. Red line indicates the ChIP-qPCR primer at the myeloid-specific enhancer. Data were visualized by using Integrative Genomics Viewer (http://software.broadinstitute.org/software/igv/). (F) Increase of H3K4me1, H3K4me2, and H3K27ac at the CEBPA enhancer by LSD1-KD. ChIP-qPCR signals were calculated relative to that of histone H3. (G) Enrichment of LSD1 at CEBPA enhancer examined by ChIP-qPCR. (H) Enrichment of GATA1 at theCEBPA enhancer in HEL cells tested by ChIP-qPCR. Enrichment values at the enhancer were normalized to input DNAs for panels F to H. (I) Upregulation of CEBPA in GATA1-KD HEL cells. Values are mean ± standard deviation of triplicate results. *P < .05, **P < .01.
LSD1 cooperates with GATA1 to promote metabolic gene expression
To gain mechanistic insight into metabolic gene regulation by LSD1, we examined the distribution of LSD1-bound sites in HEL cells by ChIP-sequencing analysis. Among the 8280 LSD1 ChIP peaks, 81% were located in introns and intergenic regions (Figure 3A). Motif analysis of LSD1-bound sites identified enrichment of consensus motifs for transcription factors such as the GATA family and RUNX family proteins (Figure 3B). The coexistence of LSD1- and GATA-bound sites was confirmed by comparing our ChIP-sequencing data with publicly available data sets using ChIP-Atlas (http://chip-atlas.org/) (supplemental Figure 5A).43 The GATA family consists of GATA1-6, each having a unique expression pattern and function. In particular, deregulation of GATA1 function has been linked to impaired erythroid differentiation, and thus to leukemogenesis.44 Interestingly, GATA1 was abundantly expressed in HEL cells (supplemental Figure 5B) and physically interacted with LSD1 (Figure 3C; supplemental Figure 5C), implying an active role of GATA1 in EL cells. To explore the role of GATA1 in metabolic regulation, we knocked down GATA1 in HEL and TF1a cells and found remarkably reduced expression of GLUT1 as well as a known GATA1-regulated gene, ALAS2 (Figure 3D-E; supplemental Figure 5D-E). Consistently, GATA1-KD led to a downregulation of glucose uptake and extracellular acidification rate (Figure 3F-G). By ChIP-qPCR analyses, we found that GATA1 occupies putative enhancers of GLUT1 and ALAS2 genes (Figure 3H; supplemental Figure 5G-H). We also observed a growth reduction in GATA1-KD cells (Figure 3I; supplemental Figure 5F), indicating an essential role of GATA1 in cell maintenance. These data collectively suggest that GATA1 promotes glycolysis by directly regulating GLUT1 expression in EL cells.
Notably, we found that the enrichment of GATA1 at GLUT1 and ALAS2 enhancers was abolished by LSD1 depletion (Figure 3H; supplemental Figure 5G-H). In addition, GATA1 protein decreased under LSD1 inhibition, while GATA1 mRNA remained unaffected (Figure 3J; supplemental Figure 4E-F; supplemental Figure 5I-M), suggesting a posttranscriptional control of GATA1 by LSD1. Indeed, when a proteasome inhibitor (MG132) was used in combination with LSD1 inhibition, the reduction of GATA1 was canceled (Figure 3K; supplemental Figure 5K-M). Taken together, these data show that LSD1 protects GATA1 from proteasomal degradation, which in turn enhances glycolysis and heme synthesis in EL cells.
LSD1 and GATA1 repress the myeloid-specific enhancer of CEBPA
Because LSD1 inhibition led to a derepression of non-erythroid hematopoietic genes (Figure 2B,F,G), we next investigated whether the suppression of nonerythroid features is important for metabolic phenotypes. Among the genes upregulated by LSD1 inhibition, the CEBPA gene, which encodes a key granulo-monocytic transcription factor C/EBPα, showed a dramatic increase (Figure 4A; supplemental Figure 6A-D). We confirmed that C/EBPα increased to levels comparable to those of myeloblastic leukemia cells (Figure 4B). Consistently, CD11b, a well-known myeloid marker, was markedly upregulated by LSD1 inhibition, indicative of a compromised erythroid identity (Figure 4C-D; supplemental Figure 6A-B). To gain mechanistic insights, by ChIP-sequencing analysis, we examined the effects of LSD1 inhibition on mono- and di-methylated H3K4 (H3K4me1 and me2) that are hallmarks of enhancer function (supplemental Figure 6E-G). Combination analyses of the ChIP-sequencing data revealed that LSD1-enriched sites were located close to the H3K4me1 and H3K4me2 peaks but not to the H3K9me3 peaks (data from K562 cells) (supplemental Figure 6E-F). Consistent with the H3K4 demethylating activity, LSD1-KD elevated the levels of H3K4me1 and H3K4me2 near the LSD1-enriched sites (supplemental Figure 6G). An LSD1-KD–induced increase of H3K4me1 and me2 was found at 42 kb downstream of the CEBPA transcription start site, a region that has been reported as a myeloid-specific enhancer of this gene (Figure 4E).45 By ChIP-qPCR, we confirmed the increase of methylated H3K4 as well as H3K27ac, a mark of active enhancers, by LSD1-KD (Figure 4F; supplemental Figure 6H-J). Importantly, we found an occupancy of LSD1 in this region (Figure 4E-G), suggesting that LSD1 directly represses the CEBPA enhancer through H3K4 demethylation. Notably, GATA1 was also present in this region (Figure 4H; supplemental Figure 6K), and its KD led to a remarkable upregulation of CEBPA expression (Figure 4I). The data collectively revealed that LSD1 and GATA1 together repress the CEBPA gene, contributing to the maintenance of erythroid cell identity.
LSD1 expression is correlated positively with metabolic and GATA1 genes and negatively with theCEBPA gene in EL cells
We found that LSD1 promotes glycolysis and heme synthesis by facilitating GATA1 function in EL cells (Figure 3). To gain a broader view of the LSD1-dependent metabolic program in AML, we analyzed publicly available transcriptome data sets. In clinical AML samples (data from TCGA), LSD1 and its downstream metabolic genes such as ALAS2 and PKLR were coexpressed with significant correlation, and M6/M7 leukemic cells particularly showed high expression of both genes (Figure 5A-B; red and yellow dots). In addition, LSD1 and GATA1 genes were highly coexpressed in M6/M7 cells among AML subtypes (Figure 5C-D), suggesting the cooperative control of LSD1 and GATA1 in EL cells at both protein and mRNA levels. Furthermore, consistent with our experimental data in Figure 4, the expression of CEBPA was negatively correlated with those of LSD1 and GATA1, particularly in M6/M7 cells among AML cell lines (data from CCLE) (Figure 5E-F).

LSD1 expression is correlated positively with metabolic and GATA1 genes and negatively with CEBPA gene in AML cells of erythroid and megakaryocytic lineages. Scatter plots showing positive correlation of LSD1 and ALAS2 (A), LSD1 and PKLR (B), and LSD1 and GATA1 (C) in AML clinical samples in the TCGA database (n = 173). (D) Scatter plots showing positive correlation between LSD1 and GATA1 in AML cell lines (CCLE, n = 37). Scatter plots showing inverse correlation between LSD1 and CEBPA (E) and between GATA1 and CEBPA (F) in AML cell lines (CCLE, n = 37). Pearson product correlation coefficient and P values are indicated.
LSD1 expression is correlated positively with metabolic and GATA1 genes and negatively with CEBPA gene in AML cells of erythroid and megakaryocytic lineages. Scatter plots showing positive correlation of LSD1 and ALAS2 (A), LSD1 and PKLR (B), and LSD1 and GATA1 (C) in AML clinical samples in the TCGA database (n = 173). (D) Scatter plots showing positive correlation between LSD1 and GATA1 in AML cell lines (CCLE, n = 37). Scatter plots showing inverse correlation between LSD1 and CEBPA (E) and between GATA1 and CEBPA (F) in AML cell lines (CCLE, n = 37). Pearson product correlation coefficient and P values are indicated.
To further evaluate the relevance of LSD1 in metabolic traits in EL, we made use of a published data set that consists of transcriptome profiles of 141 AML-M6 cases.36 Expression of GLUT1 and LSD1 was mildly correlated, whereas LSD1 expression did not correlate with ALAS2 within the M6 cases studied (supplemental Figure 7A-B). We found significant positive correlations of LSD1 with PKLR and GATA1, consistent with our in vitro observations (supplemental Figure 7C-D). In addition, CEBPA was negatively correlated with both LSD1 and GATA1 (supplemental Figure 7E-F). We then sought to compare these M6 cases with the non-M6 cases in the TCGA data set (n = 171). Consistent with their lineage identities, the 2 groups showed distinct expression of GATA1, CEBPA, and PU.1 (normalized to an internal control, TBP) (supplemental Figure 7G). Scatter plot analyses revealed that enhanced expression of LSD1 coexisted with high expression of GLUT1, ALAS2, and GATA1 in the M6 group (supplemental Figure 7H-J), whereas CEBPA expression was low in the same group (supplemental Figure 7K).
We next explored the possible role of LSD1 in metabolic variation at the single-cell level using a published single-cell RNA-sequencing data set from patients with AML; malignant cells in this data set were classified by hallmark AML mutations.41 Of interest, we found that when leukemic cells were divided into subpopulations according to genotypes (mutations in FLT3, NPM1, DNMT3, and p53 genes), there were no obvious differences in the metabolic gene expression (supplemental Figure 8A). Using the same data set, we found that the expression of LSD1 was correlated with those of lineage and metabolic genes in both normal and malignant cell populations (supplemental Figure 8B-C). These data further emphasize the essential roles of LSD1 in shaping lineage-dependent metabolic phenotypes in AML cells.
C/EBPα interferes with GATA1-dependent metabolic pathways
Although C/EBPα is known to primarily contribute to myeloid differentiation, the role of this protein in leukemic metabolism has not been investigated. To test this, we knocked down CEBPA in TF1a cells that expressed a detectable basal amount of the endogenous C/EBPα among EL cell lines (Figure 6A). Interestingly, the loss of C/EBPα increased GATA1 protein about twice (Figure 6B). GATA1-targeted metabolic genes such as ALAS2 and GLUT1 were upregulated by CEBPA-KD, with enhanced enrichment of GATA1 at its target enhancers (Figure 6C-D). Consistent with the gene expression changes, glucose uptake was small but significantly augmented by CEBPA-KD (Figure 6E).

C/EBPα interferes with GATA1-dependent metabolic program. (A) Expression of C/EBPα protein in TF1a and HEL. It is noteworthy that C/EBPα was detectable in TF1a but not in HEL cells under basal conditions (left). Intensities of C/EBPα bands were quantified by densitometry and normalized to those for β-tubulin (right). (B) Increased GATA1 in CEBPA-KD TF1a cells. CEBPA-KD upregulated GATA1 protein. (C) Expression changes of LSD1 target genes in CEBPA-KD TF1a cells. (D) Increased enrichment of GATA1 at the ALAS2 and GLUT1 enhancers in CEBPA-KD TF1a cells. (E) Increased glucose uptake in CEBPA-KD TF1a cells. (F) Expression changes of LSD1 target genes in S2101 treated and CEBPA-KD TF1a cells. Concentration of S2101 is 50 µM. (G) GATA1 enrichments at ALAS2 and GLUT1 enhancers in S2101-treated and CEBPA-KD TF1a cells. Enrichment values at the enhancers were normalized to input DNA for panels D and G. (H) Restored glucose uptake by CEBPA-KD in S2101-treated TF1a cells. S2101 was used at 20 µM. Values are mean ± standard deviation of triplicate results. *P < .05, **P < .01.
C/EBPα interferes with GATA1-dependent metabolic program. (A) Expression of C/EBPα protein in TF1a and HEL. It is noteworthy that C/EBPα was detectable in TF1a but not in HEL cells under basal conditions (left). Intensities of C/EBPα bands were quantified by densitometry and normalized to those for β-tubulin (right). (B) Increased GATA1 in CEBPA-KD TF1a cells. CEBPA-KD upregulated GATA1 protein. (C) Expression changes of LSD1 target genes in CEBPA-KD TF1a cells. (D) Increased enrichment of GATA1 at the ALAS2 and GLUT1 enhancers in CEBPA-KD TF1a cells. (E) Increased glucose uptake in CEBPA-KD TF1a cells. (F) Expression changes of LSD1 target genes in S2101 treated and CEBPA-KD TF1a cells. Concentration of S2101 is 50 µM. (G) GATA1 enrichments at ALAS2 and GLUT1 enhancers in S2101-treated and CEBPA-KD TF1a cells. Enrichment values at the enhancers were normalized to input DNA for panels D and G. (H) Restored glucose uptake by CEBPA-KD in S2101-treated TF1a cells. S2101 was used at 20 µM. Values are mean ± standard deviation of triplicate results. *P < .05, **P < .01.
We next investigated whether C/EBPα participates in metabolic reprogramming under LSD1 inhibition, using a combination of S2101 and CEBPA-KD. In this setting, we confirmed that the induction of CEBPA expression by S2101 was completely abolished by CEBPA-KD (Figure 6F). Importantly, CEBPA-KD reversed downregulation of metabolic genes, including GLUT1 under LSD1 inhibition, with a partial recovery of GATA1 binding at target enhancers (Figure 6F-G). Consistent with this finding, glucose uptake in LSD1-inhibited cells was restored by CEBPA-KD (Figure 6H). The results suggest that C/EBPα suppresses GATA1 function and thereby remodels the metabolic phenotype in EL cells.
Finally, we tested the involvement of other transcriptional regulators that are associated with erythroid lineage and LSD1 function. GATA2 is known to operate at an early phase of erythroid maturation before GATA1 expression.46 In our GATA2-KD experiments in HEL cells, there were no changes in glucose uptake, glycolytic activity, or the expression of heme synthesis genes (supplemental Figure 9A-D), suggesting that these metabolic pathways are specifically regulated by GATA1. We then performed KD of GFI1, a transcriptional repressor known to cooperate with LSD1.47 Although some of the metabolic genes were affected by GFI1-KD, glucose uptake and glycolytic activity remained unchanged (supplemental Figure 9E-H). In our preliminary proteomic study in HEL cells, we did not find bindings of LSD1 with TAL1, JARID1A, LDB1, or LMO2, which are known to cooperate with LSD1 and/or GATA1 (data not shown).48,49 Taken together, these data suggest a unique mode of metabolic gene regulation by the LSD1/GATA1 axis.
Discussion
In the current study, we showed that LSD1 regulates both GATA1 and C/EBPα, which directly control lineage-specific and key metabolic genes in EL cells among AML subtypes. Specifically, LSD1 facilitates the function of an erythroid TF, GATA1, while repressing a granulo-monocytic TF, C/EBPα (Figure 7). As a consequence, dominance of GATA1 over C/EBPα drives the expression of EL-dependent metabolic genes such as those involved in glycolysis and heme synthesis. Because expression of LSD1 was highly correlated with that of metabolic genes across AML subtypes, balancing of lineage TFs by LSD1 likely has a broad impact on the metabolic diversification in AML. A recent report showed that biallelic CEBPA mutation combined with heterozygous mutation in GATA2 induce EL in mice.50 In these mutant mice, myeloid progenitors, which ectopically expressed erythroid TFs including GATA1, underwent malignant transformation. Thus, during EL development, disrupted balance of lineage TFs (possibly induced by LSD1) may trigger metabolic reprogramming.

Schematic model: control of lineage TF balance by LSD1 defines the metabolic phenotype in EL cells. Overexpressed LSD1 stabilizes GATA1, which activates glycolysis and heme biosynthesis genes while LSD1 suppresses CEBPA expression via H3K4 demethylation. Lineage-specific transcription factors GATA1 and C/EBPα are mutually restraining. Therefore, LSD1 inhibition derepresses CEBPA expression and downregulates GATA1 function, leading to metabolic remodeling and growth arrest.
Schematic model: control of lineage TF balance by LSD1 defines the metabolic phenotype in EL cells. Overexpressed LSD1 stabilizes GATA1, which activates glycolysis and heme biosynthesis genes while LSD1 suppresses CEBPA expression via H3K4 demethylation. Lineage-specific transcription factors GATA1 and C/EBPα are mutually restraining. Therefore, LSD1 inhibition derepresses CEBPA expression and downregulates GATA1 function, leading to metabolic remodeling and growth arrest.
Enhanced glucose utilization is a hallmark of many tumor cells. In fact, myc and HIF-1α, TFs that are commonly activated in tumors, have been implicated in glycolytic activation.51 We and others have previously shown that LSD1 promotes glycolysis by increasing the stability of HIF-1α.19,52 In EL cells, we found no involvement of LSD1 in stabilizing HIF-1α (data not shown). Instead, we showed that LSD1 cooperates with GATA1 to transcriptionally activate key glucose metabolism genes. These observations suggest that LSD1 is widely involved in aerobic glycolysis by using distinct sets of TFs in various cancer types.
We found that GATA1 directly controls EL-associated metabolism and is essential for cell growth. Our findings may be seemingly contradictory to a previous report showing that attenuated expression of GATA1 induces erythroid leukemia.53 However, the authors in the same paper described that residual expression of GATA1 contributed to malignancy. In recent reports, either a gain of GATA1-interfering factor or a loss of GATA1-collaborator impeded normal erythropoiesis and generated EL-like states in mice,54,55 suggesting that, while losing erythropoietic capacity, GATA1 acquires an alternative function during EL development. Thus, a reasonable explanation of our findings is that LSD1 potentiates the leukemogenic function of GATA1 to generate a unique metabolic phenotype in EL. Mechanistically, we found that LSD1 binds to and stabilizes GATA1 at the protein level. Previous reports showed that GATA1 is susceptible to caspase-mediated proteolysis and is protected by HSP70,56 and that HSP27 protects GATA1 from proteasomal degradation.57 Because we showed that proteasome inhibition restored GATA1 under LSD1 inhibition, LSD1 may modulate GATA1 status via the function of THE HSP pathway. Because we observed increased GATA1 protein by CEBPA-KD (Figure 6B), C/EBPα might also be involved in these pathways via transcriptional regulation.
C/EBPα is essential for the differentiation of granulocytes and monocytes.58 In a subset of AML expressing RUNX1-fusion oncoproteins, forced expression of C/EBPα is sufficient to reverse aberrant epigenetic programs associated with the undifferentiated leukemic state.59 In leukemic B-cell lines, forced expression of C/EBPα induced trans-differentiation into the macrophage lineage and reduced the tumorigenicity.60 Furthermore, inactivating mutations in the CEBPA gene have been detected at a high frequency in AML,61 suggesting that C/EBPα acts as a tumor suppressor in hematopoietic cells. Here, we found that the induction of C/EBPα in LSD1-inhibited cells led to the downregulation of glycolysis and heme synthesis genes. Thus, C/EBPα deficiency may induce metabolic reprogramming associated with differentiation blockade and malignant phenotypes in AML.
It is important to note that EL cells with different genetic mutations exhibited enhanced glycolytic activities. Notably, our transcriptome and metabolome data from different AML subtypes indicated that glycolytic activity was highly dependent on LSD1 and GATA1 but not on the gene mutation types. This evidence indicates that lineage identity has a significant impact on shaping the metabolic phenotypes in AML. Nonetheless, accumulating data suggests that genetic background is a major determinant of AML characteristics, and that specific mutations have been implicated in metabolic features.62,63 Thus, lineage identity and genetic lesions may cooperatively direct metabolic reprogramming that allows AML cells to develop optimal survival strategies.
Clinical trials for the treatment of AML with LSD1 inhibitors have been implemented with promising outcomes.64 EL cases with enhanced LSD1 expression may be highly sensitive to this treatment. In this study, we showed that disruption of heme synthesis by ALAS2-KD enhanced the toxicity of an iron chelator, deferoxamine (supplemental Figure 4G). Because ALAS2 expression is regulated by LSD1/GATA1 axis, LSD1 inhibition combined with disruption of iron metabolism may effectively attack metabolic vulnerability in EL. Targeting lineage-specific metabolic features combined with LSD1 inhibition may contribute to a highly selective eradication of AML cells.
The data reported in this article have been deposited in the Gene Expression Omnibus database (accession numbers GSM145397 [RNA-seq], GSE145399 [histones], and GSE145401 [LSD1]).
Requests by other investigators regarding materials, data sets, and protocols may be submitted to the corresponding authors (Shinjiro Hino [s-hino@kumamoto-u.ac.jp] or Mitsuyoshi Nakao [mnakao@gpo.kumamoto-u.ac.jp]).
Acknowledgments
This work was supported by the following funding sources: JSPS KAKENHI (grants JP18H02618 and 18K19479 [M.N.]; 20H04108, JP16K07215, and 25430178 [S.H.]; and 20K16335 [K.K.]), Takeda Science Foundation (M.N. and S.H.), The SGH Foundation (S.H.), and the Japan Agency for Medical Research and Development (JP16gm0510007) (M.N.).
Authorship
Contribution: K.K., S.H., and M.N. designed research; K.K., S.H., A.S., K. Anan, R.T., H.A., K. Araki, and Y.H. performed research; K.K., S.H., and T.S. analyzed data; K.K., S.H., and M.N. wrote the paper; and K.N. and M.N. supervised the study.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Shinjiro Hino, Department of Medical Cell Biology, Institute of Molecular Embryology and Genetics, Kumamoto University 2-2-1 Honjo, Chuo-ku, Kumamoto 860-0811, Japan; e-mail: s-hino@kumamoto-u.ac.jp; or Mitsuyoshi Nakao, Department of Medical Cell Biology, Institute of Molecular Embryology and Genetics, Kumamoto University 2-2-1 Honjo, Chuo-ku, Kumamoto 860-0811, Japan; e-mail: mnakao@gpo.kumamoto-u.ac.jp.

