

Key Points
Patients with spliceosome mutations have favorable overall outcomes with HMA+VEN therapy.
Patients with mutated U2AF1, especially with concurrent RAS mutations, appear more resistant to HMA+VEN.
Visual Abstract

Abstract
Spliceosome mutations (SRSF2, SF3B1, U2AF1, ZRSR2), are encountered in ∼50% of secondary acute myeloid leukemia cases (sAML) and define a molecular subgroup with outcomes similar to sAML in de novo AML patients treated with intensive chemotherapy. Outcomes in patients with spliceosome mutations treated with hypomethylating agents in combination with venetoclax (HMA+VEN) remains unknown. The primary objective was to compare outcomes in patients with spliceosome mutations vs wild-type patients treated with HMA+VEN. Secondary objectives included analysis of the mutational landscape of the spliceosome cohort and assessing the impact of co-occurring mutations. We performed a retrospective cohort analysis of patients treated with HMA+VEN–based regimens at The University of Texas MD Anderson Cancer Center. A total of 119 patients (spliceosome mutated n = 39 [SRSF2, n = 24; SF3B1, n = 8; U2AF1, n = 7]; wild-type, n = 80) were included. Similar responses were observed between spliceosome and wild-type cohorts for composite complete response (CRc; 79% vs 75%, P = .65), and measurable residual disease–negative CRc (48% vs 60%, P = .34). Median overall survival for spliceosome vs wild-type patients was 35 vs 14 months (P = .58), and was not reached; 35 months and 8 months for patients with SRSF2, SF3B1, and U2AF1 mutations, respectively. IDH2 mutations were enriched in patients with SRSF2 mutations and associated with favorable outcomes (1- and 2-year overall survival [OS] of 100% and 88%). RAS mutations were enriched in patients with U2AF1 mutations and associated with inferior outcomes (median OS, 8 months). Comparable outcomes were observed between patients with vs without spliceosome mutations treated with HMA+VEN regimens, with specific co-mutation pairs demonstrating favorable outcomes.
Introduction
Mutations in genes encoding components of the spliceosome complex (SRSF2, U2AF1, SF3B1, ZRSR2) are identified in approximately one-third of patients with myelodysplastic syndrome (MDS) and nearly 50% of secondary acute myeloid leukemia cases evolving from MDS (sAML).1-4 Though less commonly encountered in de novo AML (∼7%), splicing mutations (in particular SRSF2) correlate with inferior outcomes to standard induction therapy,1,5,6 mirroring a clinical course similar to patients with sAML.5 Contrary to these historically poor outcomes observed in patients with spliceosome mutations, mutations in genes involved in the cohesin complex (STAG1, STAG2, SMC3, SMC1A, RAD21), often considered alongside spliceosome mutations, appear to have an equivocal prognostic impact.7 The treatment landscape for AML now incorporates small molecule and molecularly targeted therapies, in which the differential influence of co-occurring mutations has resulted in increasingly challenging decision making in the management of AML. Understanding outcomes within these molecular subgroups is needed.
The incorporation of venetoclax (VEN) into low-intensity regimens has rapidly emerged as the standard of care for older, unfit patients with AML.8 Molecularly defined subgroups of patients with improved responses to VEN-based therapy have been defined.8,9 Certain molecular subgroups (NPM1, IDH1/2, or TP53 mutated) appear predictive of VEN sensitivity or resistance, yet outcomes among other subgroups have not been described in depth. Although spliceosome mutations traditionally associate with inferior outcomes, VEN-based therapy may abrogate the negative prognostic impact of this molecular subgroup.10 In a correlative analysis of molecular determinates of outcomes in patients treated with hypomethylating agents (HMA) combined with VEN, gene mutations in SRSF2 were the most commonly detected, and associated with a complete response (CR) rate of 65%.9 Additionally, patients with SRSF2 mutations treated in the phase 1b Chemotherapy and Venetoclax in Elderly Acute Myeloid Leukemia (CAVEAT) trial investigating the addition of VEN to “5+2” anthracycline-based chemotherapy achieved higher blast reductions (median, 47%), translating into a favorable 82% overall response rate (ORR; CR + complete response with incomplete hematologic recovery [CRi]), and a median overall survival (OS) of 31 months.11 Recent preclinical work similarly has suggested mutations in cohesin complex genes (in particular RAD21) result in increased VEN sensitivity in vitro.12
Understanding responses within specific subgroups of patients receiving VEN-based therapies is imperative for the identification of those patients most likely to benefit from novel combinations, in addition to those at increased risk of relapse. Although improved responses have been observed in prospective trials of VEN-based therapy, long-term outcomes in patients with spliceosome and/or cohesin mutations treated with VEN have yet to be fully reported. The primary aim of this study was to investigate outcomes in patients with spliceosome mutations treated with HMA+VEN at The University of Texas MD Anderson Cancer Center.
Methods
Study design and patient selection
Patients ≥18 years of age with a diagnosis of AML treated with frontline HMA+VEN–based therapy either on prospective clinical trial protocols (n = 98; NCT03404193, NCT02993523, NCT02203773) or off protocol (n = 21) at The University of Texas MD Anderson Cancer Center were included in this cohort analysis. Patients with acute promyelocytic leukemia were excluded. Patients with FLT3-ITD mutations receiving FLT3 inhibitors in combination with HMA+VEN on protocol were included. Patients were stratified into spliceosome (presence of SRSF2, SF3B1, U2AF1, ZRSR2 mutations) mutant and wild-type (ie, absence of spliceosome mutations) cohorts. Cohesin complex mutations (STAG1, STAG2, SMC3, SMC1A, RAD21) were considered as a subgroup within the 2 larger cohorts for outcomes analyses. All included subjects were authorized after review of the MD Anderson Cancer Center institutional review board, and conducted under the Declaration of Helsinki.
Data collection and analysis
Patient data were reviewed for age at diagnosis, AML disease group (de novo, sAML, or therapy-related AML [tAML]), date of treatment initiation, therapy received, date of last follow-up, and survival status (living vs deceased) at last follow-up. Cytogenetics were performed using standard metaphase karyotype analysis in our Clinical Laboratory Improvement Amendments–certified laboratory. Molecular analysis was performed using next-generation sequencing (NGS) using a targeted 81-gene panel of recurrently mutated genes in myeloid malignancies as previously described.13 Measurable residual disease (MRD) was assessed using 8-color multiparameter flow cytometry (FC) using leukemia-associated immunophenotype or different from normal assessment with a minimum sensitivity of 10−3 to 10−4 (0.1%-0.01%). Patients were stratified by European LeukemiaNet (ELN) risk group according to 2017 ELN guidelines,14 and response criteria were defined per International Working Group criteria for AML15 (ORR: CR + CRi + complete response with partial hematologic recovery [CRh] + partial response + morphologic leukemia-free state; composite complete response [CRc]: CR + CRi + CRh). Time-to-event end points included OS (C1D1 until death), event-free-survival (EFS; C1D1 until relapse or death), and duration of response (DOR; time from achieving morphologic CRc until death or relapse).
Comparisons between cohorts for treatment response and clinical variables were determined using the χ2 test or Fisher’s exact test, whereas continuous variables were assessed using independent t test or the Wilcoxon rank-sum test as appropriate. Time-to-event analysis was assessed using the log-rank method, with logistical regression analysis and Cox proportional hazard modeling to determine the effect of variables on response as appropriate. False discovery rate (FDR)-adjusted P values using the Benjamini-Hochberg procedure were reported for survival analyses with multiple comparisons.
Results
Patient demographics are shown in Table 1. A total of 119 patients treated with HMA+VEN therapy were identified (azacitidine + VEN, n = 14; 10-day decitabine + VEN, n = 100; 5-day decitabine + VEN, n = 5). Thirty-nine patients (33%) had mutations in spliceosome genes (SRSF2, n = 24; SF3B1, n = 8; U2AF1, n = 7). Median age of the entire cohort was 72 years. Patients with spliceosome mutations were significantly older (median age 75 vs 70 years, P = .047), and more likely to be male (85% vs 40%, P < .01). The incidence of spliceosome mutations was 34% in de novo AML, 40% in sAML, and 24% in tAML. No significant differences were observed regarding patients diagnosed with sAML (21% vs 15%) or tAML (18% vs 28%) in the spliceosome vs wild-type cohort, respectively. The majority of patients in both cohorts had adverse risk disease by 2017 ELN criteria (spliceosome, 54%; wild-type, 64%).
Patient demographics
| Demographic | All patients (N = 119) | Spliceosome (n = 39) | Wild-type (n = 80) | P |
|---|---|---|---|---|
| Age, median (range), y | 72 (40-89) | 75 (56-85) | 70 (40-89) | .047 |
| Male, n (%) | 65 (55) | 33 (85) | 32 (40) | <.01 |
| ECOG, median (range) | 1 (0-3) | 1 (0-3) | 1 (0-3) | .11 |
| Median WBC (range), ×103/μL | 3.1 (0.3-81) | 3.6 (0.3-81) | 2.9 (0.5-43) | .61 |
| Treatment duration, median (range), mo | 5.7 (0.1-43) | 5.5 (0.5-43) | 5.7 (0.1-32) | .90 |
| Treatment regimen, n (%) | ||||
| Azacitidine + venetoclax | 14 | 3 (8) | 11 (14) | .54 |
| 5-d decitabine + venetoclax | 5 | 2 (5) | 3 (4) | .66 |
| 10-d decitabine + venetoclax | 100 | 34 (87) | 66 (83) | .60 |
| FLT3 inhibitor | 14 (12) | 2 (5) | 12 (15) | .14 |
| Disease, n (%) | ||||
| De novo AML | 70 (59) | 24 (62) | 46 (58) | |
| sAML | 20 (17) | 8 (21) | 12 (15) | .44 |
| MDS | 17 | 7 | 10 | — |
| MPN | 3 | 1 | 2 | — |
| tAML | 29 (24) | 7 (18) | 22 (28) | .36 |
| ELN risk group, n (%) | ||||
| Favorable | 28 (24) | 11 (28) | 17 (21) | .49 |
| Intermediate | 19 (16) | 7 (18) | 12 (15) | .79 |
| Adverse | 72 (61) | 21 (54) | 51 (64) | .32 |
| Cytogenetic group, n (%) | ||||
| Diploid | 39 (33) | 14 (36) | 25 (31) | .68 |
| Other intermediate | 30 (25) | 14 (36) | 16 (20) | .07 |
| Adverse/complex | 50 | 11 (28) | 39 (49) | .047 |
| Demographic | All patients (N = 119) | Spliceosome (n = 39) | Wild-type (n = 80) | P |
|---|---|---|---|---|
| Age, median (range), y | 72 (40-89) | 75 (56-85) | 70 (40-89) | .047 |
| Male, n (%) | 65 (55) | 33 (85) | 32 (40) | <.01 |
| ECOG, median (range) | 1 (0-3) | 1 (0-3) | 1 (0-3) | .11 |
| Median WBC (range), ×103/μL | 3.1 (0.3-81) | 3.6 (0.3-81) | 2.9 (0.5-43) | .61 |
| Treatment duration, median (range), mo | 5.7 (0.1-43) | 5.5 (0.5-43) | 5.7 (0.1-32) | .90 |
| Treatment regimen, n (%) | ||||
| Azacitidine + venetoclax | 14 | 3 (8) | 11 (14) | .54 |
| 5-d decitabine + venetoclax | 5 | 2 (5) | 3 (4) | .66 |
| 10-d decitabine + venetoclax | 100 | 34 (87) | 66 (83) | .60 |
| FLT3 inhibitor | 14 (12) | 2 (5) | 12 (15) | .14 |
| Disease, n (%) | ||||
| De novo AML | 70 (59) | 24 (62) | 46 (58) | |
| sAML | 20 (17) | 8 (21) | 12 (15) | .44 |
| MDS | 17 | 7 | 10 | — |
| MPN | 3 | 1 | 2 | — |
| tAML | 29 (24) | 7 (18) | 22 (28) | .36 |
| ELN risk group, n (%) | ||||
| Favorable | 28 (24) | 11 (28) | 17 (21) | .49 |
| Intermediate | 19 (16) | 7 (18) | 12 (15) | .79 |
| Adverse | 72 (61) | 21 (54) | 51 (64) | .32 |
| Cytogenetic group, n (%) | ||||
| Diploid | 39 (33) | 14 (36) | 25 (31) | .68 |
| Other intermediate | 30 (25) | 14 (36) | 16 (20) | .07 |
| Adverse/complex | 50 | 11 (28) | 39 (49) | .047 |
ECOG, Eastern Cooperative Oncology Group; MPN, myeloproliferative neoplasm; WBC, white blood cell.
Cytogenetic and molecular characteristics
The cytogenetic class of the cohorts is shown in Table 1. The majority of patients in the spliceosome cohort had intermediate-risk cytogenetics (72%; diploid, 36%; other intermediate, 36%), with a minority (28%) having adverse risk or complex cytogenetic findings. A larger proportion of wild-type patients had adverse risk cytogenetics or complex karyotype (49%, P = .047).
The molecular landscape of the spliceosome cohort and specific amino acid alterations associated with spliceosome mutations are shown in Figure 1A-B. SRSF2 mutations predominantly resulted in well-described substitutions at proline residue 95 (n = 23, 96%),16 with only 1 patient demonstrating a p.A74G mutation (Figure 1B). SF3B1 mutations occurred within the common K700 and K666 residues.16 Of interest, the majority (75%) were K666N, which have recently been associated with inferior outcomes in myeloid malignancies.17 Similar to SRSF2, all U2AF1 mutations resulted in changes within the commonly identified S34 and Q157 residues within the first 2 zinc finger motifs of U2AF1.
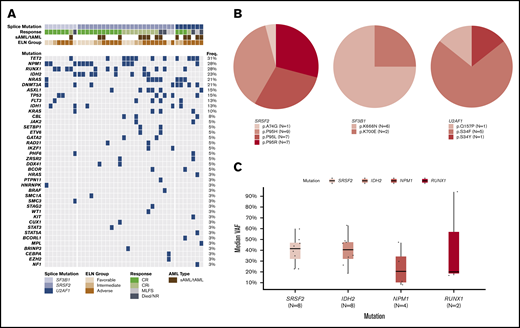
Molecular landscape of the spliceosome cohort. (A) Oncoprint of molecular mutations in patients with spliceosome mutations. (B) Frequency and associated amino acid substitution of included spliceosome mutations. (C) Variant allele frequency (VAF) of recurrently mutated genes in patients with SRSF2 and IDH2 co-mutations.
Mutation differences between cohorts are shown in Figure 2A. Mutations in DNMT3A, ASXL1, TET2, TP53, RUNX1, RAS, and NPM1 were prevalent at a frequency ≥15% in both cohorts. IDH mutations (36% vs 15%) significantly associated with spliceosome mutations (Figure 2B), whereas TP53, TET2, and EZH2 mutations were enriched in the wild-type cohort (P < .05).
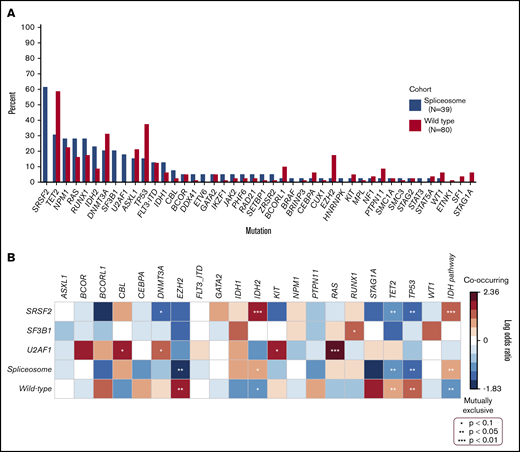
Co-mutation frequencies and enrichment in spliceosome-mutated and wild-type cohorts. (A) Mutation frequency in patients with vs without spliceosome mutations. (B) Heatmap demonstrating co-occurring mutations and mutually exclusive mutations between spliceosome mutations and cohorts, respectively.
Molecular differences between cohorts were also assessed by genetic class (ie, tumor suppressor, transcription factor, active signaling, methylation, and chromatin modifying). Within the spliceosome cohort, enrichment was observed for mutations in methylation genes (64% vs 33%, P < .01). Mutations in chromatin modifiers (ASXL1, BCOR, EZH2; 40% vs 20%, P = .04) and tumor suppressor mutations (TP53, WT1, PHF6) were enriched within the wild-type cohort (45% vs 23%, P = .03); active signaling mutations (FLT3, CBL, PTPN11, K/NRAS) demonstrated a trend toward enrichment in wild-type patients (55% vs 36%, P = .05). No significant difference was observed in transcription factor or cohesin complex mutations. Median mutation burden was not significantly different between cohorts (4 vs 4.5) or spliceosome mutations (SRSF2, 4; SF3B1, 3; U2AF1, 5; Figure 3A).
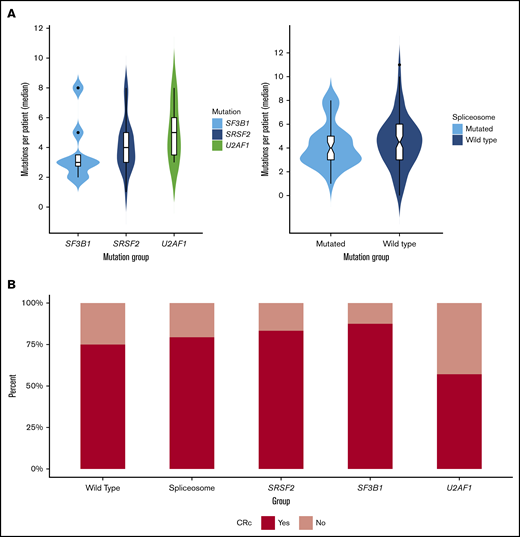
Mutation burden and treatment response in spliceosome-mutated and wild-type cohorts. (A) Median mutation burden in patients with SF3B1, SRSF2, and U2AF1 mutations and in patients with vs without spliceosome mutations. (B) Composite CR rates in wild-type and spliceosome cohorts, and by individual spliceosome mutation.
Mutational landscape within the spliceosome cohort
Co-mutation patterns in patients with SRSF2 (n = 24), SF3B1 (n = 8), and U2AF1 (n = 7) mutations were further explored (Figure 2B). SRSF2 mutations frequently co-occurred with mutations in IDH2 (33%), TET2 (29%), NPM1 (29%), and RUNX1 (25%). IDH2 mutations were found nearly exclusively with SRSF2 mutations compared with other spliceosome mutations (88% vs 11%) and were enriched compared with other mutational pairs (33% vs 9%, P < .01). SF3B1 mutations co-occurred with mutations in RUNX1 (50%), NPM1 (38%), DNMT3A (25%), TP53 (25%), and IDH1 (25%). U2AF1 mutations co-occurred with mutations in RAS (71%), TET2 (57%), and DNMT3A (57%). Compared with other spliceosome patients, RAS mutations were enriched in patients with U2AF1 mutations (P < .01).
Cohesin complex mutations have demonstrated preclinical sensitivity to VEN-based therapies12 and are often considered in the context of spliceosome mutations. Cohesin complex mutations were identified in 14% (n = 17) of the study cohort. 13% (n = 5) of patients in the spliceosome cohort had co-occurring cohesin complex mutations. Among the 12 wild-type patients with cohesin complex mutations (RAD21, n = 2; STAG1, n = 5; STAG2, n = 2; SMC1A, n = 2; SMC3, n = 1), frequent co-occurring mutations included TET2 (75%), TP53 (50%), NPM1 (33%), RUNX1 (33%), NOTCH1 (33%), ASXL1 (25%), DNMT3A (25%), and PTPN11 (25%).
Response to HMA+VEN
Response outcomes for the 2 cohorts are shown in Table 2 and Figure 3B. The ORR for the entire cohort was 81% (spliceosome, 89%; wild-type, 79%). CRc was achieved in 79% and 75% of patients in the spliceosome and wild-type cohorts. Spliceosome patients were more likely to achieve a CRi/CRh compared with wild-type patients (28% vs 11%, P = .03). Twenty-two patients (18%; spliceosome, n = 5 [13%] vs wild-type, n = 17 [21%]) were either refractory to therapy or deceased before disease assessment. Responses based on specific mutations are shown in Table 2. Patients with NPM1 mutations were more likely (93% vs 71%, P = .02), and those with TP53 mutations or adverse risk/complex cytogenetics less likely (TP53: 58% vs 84%, P = .01; adverse risk/complex cytogenetics: 64% vs 86%, P = .01) to achieve a CRc. Despite the differential response observed to treatment of these specific mutations, spliceosome mutations had no significant influence on response within the respective cohorts. CRc rates for patients with SRSF2, SF3B1, and U2AF1 mutations were 83%, 88%, and 57%.
Patient outcomes
| Response | All patients (N = 119) | Spliceosome (n = 39) | No spliceosome (n = 80) | P |
|---|---|---|---|---|
| ORR | 97 (81) | 34 (89) | 63 (79) | .32 |
| CRc | 91 (76) | 31 (79) | 60 (75) | .65 |
| CR | 71 (60) | 20 (51) | 51 (64) | .23 |
| CRi/CRh | 20 (17) | 11 (28) | 9 (11) | .03 |
| MLFS | 6 (5) | 3 (10) | 3 (4) | .39 |
| NR/died | 22 (18) | 5 (13) | 17 (21) | .32 |
| Composite CR by molecular group | P* | |||
| NPM1 (n = 29) | 27 (93) | 9/11 (82) | 18/18 (100) | .02 |
| IDH1 (n = 10) | 6 (60) | 3/5 (60) | 3/5 (60) | .24 |
| IDH2 (n = 16) | 14 (88) | 8/9 (89) | 6/7 (86) | .35 |
| FLT3-ITD/TKD (n = 20) | 16 (80) | 4/5 (80) | 12/15 (80) | .78 |
| TP53 (n = 36) | 21 (58) | 3/6 (50) | 18/30 (60) | .01 |
| Adverse/complex cytogenetics (n = 50) | 32 (64) | 5/11 (45) | 27/39 (69) | .01 |
| Response | All patients (N = 119) | Spliceosome (n = 39) | No spliceosome (n = 80) | P |
|---|---|---|---|---|
| ORR | 97 (81) | 34 (89) | 63 (79) | .32 |
| CRc | 91 (76) | 31 (79) | 60 (75) | .65 |
| CR | 71 (60) | 20 (51) | 51 (64) | .23 |
| CRi/CRh | 20 (17) | 11 (28) | 9 (11) | .03 |
| MLFS | 6 (5) | 3 (10) | 3 (4) | .39 |
| NR/died | 22 (18) | 5 (13) | 17 (21) | .32 |
| Composite CR by molecular group | P* | |||
| NPM1 (n = 29) | 27 (93) | 9/11 (82) | 18/18 (100) | .02 |
| IDH1 (n = 10) | 6 (60) | 3/5 (60) | 3/5 (60) | .24 |
| IDH2 (n = 16) | 14 (88) | 8/9 (89) | 6/7 (86) | .35 |
| FLT3-ITD/TKD (n = 20) | 16 (80) | 4/5 (80) | 12/15 (80) | .78 |
| TP53 (n = 36) | 21 (58) | 3/6 (50) | 18/30 (60) | .01 |
| Adverse/complex cytogenetics (n = 50) | 32 (64) | 5/11 (45) | 27/39 (69) | .01 |
Data are n (%) or n/N (%).
MLFS, morphologic leukemia-free state.
P value for all patients (spliceosome and wild-type) within a classified molecular group.
Seventy-four (81%; spliceosome, n = 24; wild-type, n = 50) of the 91 patients achieving a CRc had adequate samples for MRD analysis via FC, 17 (19%) had limited/inadequate samples. MRD negative (MRD−) CR was attained in 54% (n = 40), including 42% (n = 10) of spliceosome and 60% (n = 30) of wild-type patients. Among evaluable patients in the spliceosome cohort, MRD− CRc was observed in 56% (n = 9/16), 20% (n = 1/5), and 0% (n = 0/3) of those with SRSF2, SF3B1, and U2AF1 mutations.
Patients with isolated cohesin complex (n = 12) or co-occurring cohesin complex and spliceosome mutations (n = 5), demonstrated a 71% CRc rate (cohesin complex, 67%; spliceosome/cohesin co-mutated, 80%). Eight patients with cohesin complex mutations had evaluable MRD samples, with 38% (n = 3/8) attaining an MRD− CR.
Median variant allele frequency (VAF) at diagnosis in patients with SRSF2, SF3B1, and U2AF1 mutations was 38%, 33%, and 43%, respectively (supplemental Figure 1). A minority (26%) of patients had NGS analysis performed in remission. Median VAF of SRSF2, SF3B1, and U2AF1 mutations in remission were 17%, 6%, and 2%. In the 10 patients with spliceosome mutations who obtained an MRD− CRc, paired NGS analysis performed in remission in 5 patients with SRSF2 mutations demonstrated persistent SRSF2 mutations in 40% (n = 2), with a median remission SRSF2 VAF of 23% in these 2 patients. Both patients with persistent SRSF2 mutations demonstrated reductions in VAF on remission sampling. Notably, both patients (ages 71 and 72 years) had co-mutations in IDH2, which also persisted in remission. Patient 1 developed mutations in ASXL1, TET2, and GNAS in remission, consistent with emergence of novel clones. Patient 2 had a persistent NPM1 mutation in remission, likely a molecular indicator of a residual leukemic clone, despite negative FC (supplemental Figure 2).
OS
Survival outcomes are shown in Table 3. After a median follow-up of 24 months, the median EFS and OS for the entire cohort was 10 (95% confidence interval [95% CI], 9-15) and 14 months (95% CI, 12-not reached [NR]). Median DOR was 11 (95% CI, 7-NR) and 15 (95% CI, 10-24) months for spliceosome, and wild-type patients, respectively. No significant difference in OS was observed between patients with vs without spliceosome mutations (median OS, 35 [95% CI, 13-NR] vs 14 months [95% CI, 10-NR]; Figure 4A). One-year OS was 63% in the spliceosome cohort compared with 53% in the wild-type cohort. As shown in Figure 4B, median OS was NR (95% CI, 13-NR), 35 months (95% CI, 4-NR), and 8 months18 (95% CI, 3-NR) for patients with SRSF2, SF3B1, and U2AF1 mutations, respectively. Despite the spliceosome cohort being significantly older, no significant difference in OS was observed in patients age ≥70 or <70 years. Among responding patients with evaluable MRD (n = 74), achievement of MRD-negative CRc associated with improved DOR (median 19 vs 7 months, P < .01), EFS (median 20 vs 8 months, P < .01), and OS (median NR vs 12.6, P = .035; supplemental Figure 3). MRD-negativity demonstrated a trend toward improved OS in spliceosome patients (median, NR vs 14 months; P = .08), and resulted in numerically longer survival in wild-type patients (median, 20 vs 11 months; P = .16). The 2 patients described previously with persistent SRSF2 mutations identified on NGS despite flow MRD− CRc remain alive and in remission, after a median follow-up of 28 months.
Overall survival
| Demographic | Event-free survival (95% CI), mo | Overall survival (95% CI), mo | P* |
|---|---|---|---|
| All patients (N = 119) | 10 (9-15) | 14 (12-NR) | — |
| Spliceosome (n = 39) | 10 (6-NR) | 35 (13-NR) | .58 |
| ELN risk group | |||
| Favorable | NR (3-NA) | NR (3-NA) | .48 |
| Intermediate | 10 (8-NA) | NR (13-NA) | .97 |
| Adverse | 7 (3-NA) | 14 (5-NA) | .29 |
| Cytogenetic group | |||
| Diploid | NR (10-NA) | NR (−) | .57 |
| Other intermediate | 17 (9-NA) | 35 (13-NA) | .40 |
| Complex/adverse | 3 (2-NA) | 5 (3-NA) | .29 |
| AML type | |||
| De novo AML | 18 (8-NA) | NR (−) | .35 |
| Secondary AML | 7 (6-NA) | 10 (6-NA) | .80 |
| Therapy-related AML | 6 (5-NA) | 8 (6-NA) | .69 |
| Wild-type (n = 80) | 11 (9-19) | 14 (10-NA) | |
| ELN risk group | |||
| Favorable | 31 (13-NA) | NR (17-NA) | — |
| Intermediate | 32 (7-NA) | NR (12-NA) | — |
| Adverse | 9 (5.0-NA) | 10 (8-18) | — |
| Cytogenetic group | |||
| Diploid | 31 (20-NA) | NR (−) | — |
| Intermediate | 11 (7-NA) | 19 (9-NA) | — |
| Complex/adverse | 5 (5-10) | 9 (6-12) | — |
| AML type | |||
| De novo AML | 19 (11-NA) | 22 (14-NA) | — |
| Secondary AML | 5 (3.-NA) | 9 (6-NA) | — |
| Therapy-related AML | 6 (5.0-10) | 10 (5-NA) | — |
| Demographic | Event-free survival (95% CI), mo | Overall survival (95% CI), mo | P* |
|---|---|---|---|
| All patients (N = 119) | 10 (9-15) | 14 (12-NR) | — |
| Spliceosome (n = 39) | 10 (6-NR) | 35 (13-NR) | .58 |
| ELN risk group | |||
| Favorable | NR (3-NA) | NR (3-NA) | .48 |
| Intermediate | 10 (8-NA) | NR (13-NA) | .97 |
| Adverse | 7 (3-NA) | 14 (5-NA) | .29 |
| Cytogenetic group | |||
| Diploid | NR (10-NA) | NR (−) | .57 |
| Other intermediate | 17 (9-NA) | 35 (13-NA) | .40 |
| Complex/adverse | 3 (2-NA) | 5 (3-NA) | .29 |
| AML type | |||
| De novo AML | 18 (8-NA) | NR (−) | .35 |
| Secondary AML | 7 (6-NA) | 10 (6-NA) | .80 |
| Therapy-related AML | 6 (5-NA) | 8 (6-NA) | .69 |
| Wild-type (n = 80) | 11 (9-19) | 14 (10-NA) | |
| ELN risk group | |||
| Favorable | 31 (13-NA) | NR (17-NA) | — |
| Intermediate | 32 (7-NA) | NR (12-NA) | — |
| Adverse | 9 (5.0-NA) | 10 (8-18) | — |
| Cytogenetic group | |||
| Diploid | 31 (20-NA) | NR (−) | — |
| Intermediate | 11 (7-NA) | 19 (9-NA) | — |
| Complex/adverse | 5 (5-10) | 9 (6-12) | — |
| AML type | |||
| De novo AML | 19 (11-NA) | 22 (14-NA) | — |
| Secondary AML | 5 (3.-NA) | 9 (6-NA) | — |
| Therapy-related AML | 6 (5.0-10) | 10 (5-NA) | — |
NA, not available.
P value presented is comparison between spliceosome and wild-type cohorts for the respective demographic.
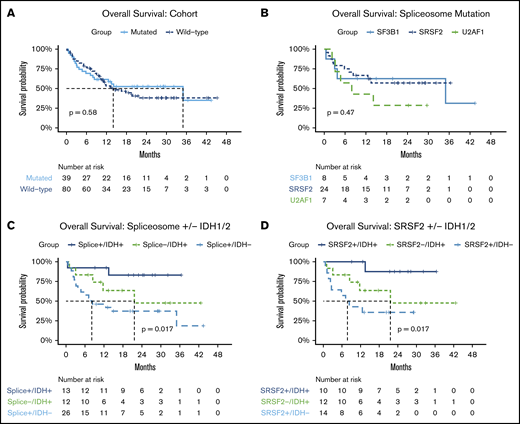
Survival outcomes by cohort and mutational subgroups. Overall survival in (A) patients with vs without spliceosome mutations; (B) by specific spliceosome mutation; (C) in patients with co-occurring spliceosome and IDH1/2 mutations, wild-type patients with IDH1/2 mutations, and spliceosome patients without IDH1/2 mutations; and (D) in patients with co-occurring SRSF2 and IDH1/2 mutations, wild-type patients with IDH1/2 mutations, and SRSF2-mutated patients without IDH1/2 mutations.
Median OS had not been reached in patients with ELN favorable or intermediate-risk disease in either cohort. Wild-type patients with adverse risk disease had significantly inferior survival with a median OS of 10 months (95% CI, 8-18; P < .01). In both cohorts, only adverse cytogenetics associated with an inferior outcome (median OS spliceosome: 5 months, P < .01; median OS wild-type: 9 months, P < .01) because spliceosome and wild-type patients with diploid or other intermediate cytogenetics had favorable OS (median OS spliceosome, diploid: NR vs other intermediate, 35 months; median OS wild-type, diploid: NR vs other intermediate, 19 months). Inferior OS was observed in patients with sAML or tAML compared with those with de novo disease in both cohorts (Table 3). No differential impact on survival was observed based on the presence or absence of spliceosome mutations in patients with sAML/tAML or within respective ELN or cytogenetic risk groups (supplemental Figure 4).
Molecular determinates of outcomes
Outcomes were assessed further by mutational class. NPM1 mutations (n = 29) were associated with a significant improvement in OS in the entire population (median OS, NR vs 12 months; P < .01). However, this survival difference was not significant in patients with spliceosome mutations (median OS, NR vs 14 months; P = .48) compared with wild-type (median OS, NR vs 10.4 months; P < .01) likely secondary to the low number of included patients. Methylation mutations (DNMT3A, TET2, IDH1, IDH2) demonstrated a trend toward improved OS in spliceosome patients (median OS, 35 vs 8 months; P = .09). Within the methylation group, this survival impact appeared largely driven by the presence of IDH mutations because no difference in OS was seen between those with or without methylation mutations when censoring patients with IDH mutations (median OS, 11 vs 8 months; P = .84). Tumor suppressor mutations (TP53, WT1, PHF6; n = 45) strongly associated with adverse outcomes in both spliceosome (median OS, 4 vs 35 months; P < .01) and wild-type patients (median OS, 8.5 vs NR; P < .01). No difference in OS was observed between patients with chromatin, active signaling, or transcription factor mutations irrespective of cohort.
Cohesin complex and spliceosome mutations
Patients with spliceosome mutations and/or cohesin complex mutations (n = 51) demonstrated a median OS of 35 months. When divided into by those with co-occurring cohesin and spliceosome mutations (n = 5), isolated spliceosome (n = 34), or isolated cohesin mutations (n = 12), no significant difference in OS was observed. Patients with co-occurring cohesin complex and spliceosome mutations had favorable 1-year survival of 80%, whereas those with isolated spliceosome or cohesin mutations had 1-year OS similar to that observed in wild-type patients (spliceosome, 58%; cohesin, 55%; wild-type, 53%; supplemental Figure 5).
Impact of co-mutations on outcomes to HMA+VEN
Patients with co-occurring spliceosome and IDH1/2 mutations (n = 13), including 7 patients with co-occurring NPM1 mutations, demonstrated particularly favorable OS compared with patients with spliceosome mutations in the absence of IDH1/2 (median OS NR vs 8 months, FDR-adjusted P = .024), with observed outcomes comparable to wild-type patients with IDH1/2 mutations without spliceosome mutations (median OS, NR vs 22 months; FDR-adjusted P = .18) (Figure 4C). Similar to wild-type patients with IDH2 mutations, 1- and 2-year OS in patients with spliceosome mutations in the context of co-occurring IDH1/2 mutations, was 92% and 83%, respectively. Interactions within this co-mutational subgroup were further explored. IDH1/2 mutations were enriched in patients with SRSF2 mutations compared with the entire cohort (42% vs 16%, P = .01). IDH2 mutations specifically were enriched in patients with SRSF2 mutations compared with the entire cohort (P < .01; Figure 2B). SRSF2/IDH1/2–mutated patients had 1- and 2-year OS rates of 100% and 88%, respectively (Figure 4D). SRSF2/IDH1/2–mutated patients had improved survival compared with SRSF2 mutated patients without co-occurring IDH mutations (median OS, NR vs 8 months; FDR-adjusted P = .02), and demonstrated a slight trend toward improved survival compared with wild-type IDH1/2–mutated patients without SRSF2 mutations (median OS, NR vs 22 months; FDR-adjusted P = .098). In patients with co-mutated SRSF2 and IDH2 with available NGS data for VAF analysis (n = 8), median VAF was most similar between SRSF2 and IDH2 mutations (42% and 41%, respectively), suggesting SRSF2 and IDH2 mutations may develop at similar points in time in the molecular hierarchy of AML (Figure 1C). RAS mutations were enriched in patients with U2AF1 mutations and associated with numerically lower CRc (and MRD− CR) rates. However, compared with wild-type patients with RAS mutations, no significant difference in survival was observed (median OS, 14 vs 13 months; P = .68; supplemental Figure 6).
Given the small numbers in our cohort, we also analyzed data from 892 samples obtained from 762 patients with AML from 2 previous studies19,20 to determine if the observed association between SRSF2 and IDH2 mutations in our cohort was retained in larger analyses. Although the frequency of identified SRSF2 mutations was lower (n = 65, 9%), IDH2 mutations were the most common identified co-mutation, were significantly enriched in patients with SRSF2 mutations (35%, P < .01) and were associated with improved survival compared with those with isolated SRSF2 mutations (20 vs 10 months, P = .02) treated with standard cytotoxic chemotherapy.
Discussion
Mutations within genes encoding the spliceosome complex (SRSF2, U2AF1, SF3B1, ZRSR2) historically have defined a high-risk AML subgroup associated with inferior outcomes with intensive chemotherapy.6 In this analysis of patients treated with HMA+VEN combinations, patients with spliceosome mutations had outcomes similar to a wild-type cohort of patients, suggesting HMA+VEN therapy may be an effective treatment regimen in this population. Patients with SRSF2 mutations were enriched for IDH2 mutations and appeared to particularly benefit from VEN-based treatment. Co-mutations in cohesin complex and spliceosome genes resulted in an 80% 1-year OS rate; however, this small molecular subgroup necessitates follow-up in larger cohorts.
Molecular classification is standard in the diagnostic workup of AML both for its prognostic value and to guide treatment in an era of molecularly targeted therapies.14,21 Spliceosome mutations are frequently identified in older patients with myeloid malignancies, a group more likely to be ineligible for intensive chemotherapy.4,6 In this cohort, spliceosome mutations were identified at a frequency of ∼33% in an older (median age, 75 years) population, consistent with prior analyses.6 Similar to prior investigations, patients with sAML had a numerically higher observed frequency of spliceosome mutations.2,5 Spliceosome-mutated patients demonstrated a heterogeneous molecular landscape, yet with a similar mutational burden compared with wild-type patients.5 Neither age nor mutation burden influenced OS with HMA+VEN treatment.
Molecular determinates of response to HMA+VEN have been reported.9 However, to our knowledge, the influence of co-occurring mutations in patients with spliceosome mutations has yet to be evaluated. In keeping with prior analyses, adverse cytogenetics and TP53 mutations associated with inferior OS, whereas NPM1 and IDH1/2 mutations associated with favorable outcomes in both spliceosome and wild-type cohorts.
Recent investigations have identified a differential prognostic impact imparted by specific spliceosome mutations and resultant amino acid residue changes occurring within the same gene,16,17 and identified mutant SF3B1 AML as a marker of VEN resistance.22 Although the small sample size precluded analysis of the differential impact of survival in patients with SF3B1 K666 vs K700 mutations, the median OS of 35 months in this patient population warrants further investigation to determine if HMA+VEN may overcome VEN resistance observed in SF3B1 mutated AML, and negate the negative impact associated with K666 mutations. The majority of SF3B1-mutated patients received decitabine in combination with VEN. Because SF3B1-mutated AML appears particularly sensitive to decitabine-based therapy,23 the reduction in VAF from diagnosis to remission (median VAF at diagnosis 33% vs remission 6%) may represent retained sensitivity to hypomethylating agents or synergistic activity of HMA+VEN within this molecular subgroup. SF3B1-mutated patients had lower rates of MRD-negative CRc compared with SRSF2 mutated patients, suggesting variable sensitivity to VEN between spliceosome mutations.
The enrichment of RAS mutations identified in patients with mutated U2AF1 appears consistent across studies.18 Because signaling mutations are a known primary and adaptive resistance mechanism to VEN,9 it is plausible the poor responses observed in these patients is in part attributable to the influence of RAS mutations as opposed to U2AF1 itself, resulting in increased resistance (no U2AF1 patients achieved an MRD-negative CR) and relapse to HMA+VEN–based therapy.
SRSF2 mutations have demonstrated sensitivity to VEN-based therapies.10,11 Within the spliceosome cohort, IDH2 mutations were enriched in patients with SRSF2 mutations, and associated with excellent 1- and 2-year OS (100% and 87%, respectively). Analysis of genomic data from larger independent patient cohorts confirmed this positive association.19,20 Single-cell DNA analysis of AML additionally demonstrated SRSF2/IDH2 frequently co-occur within the same leukemic clone,24 with the mutational pair implicated in leukemogenesis.25 Although limited by sample size, bulk NGS data found similar VAF frequencies of IDH2 and SRSF2, suggesting these mutations develop at similar time points in leukemic clonal evolution. This observation is further supported through the identification of persistent co-occurring SRSF2 and IDH2 mutations in 2 patients with MRD− remissions by FC. Prior work has highlighted the risk of leukemic progression imparted by spliceosome and IDH mutations when identified as components of clonal hematopoiesis, particularly within an older patient population.26,27 Thus, the persistence or emergence of mutations in remission representing preleukemic or novel leukemic clones highlights the importance of the proper identification and discrimination of such mutations from residual leukemia through expert hematopathological interpretation and incorporation of alternative methods of MRD assessment (ie, FC).
A clear explanation for the favorable outcomes in SRSF2/IDH2–mutated patients is not readily apparent; however, spliceosome mutations may influence BCL-2 family expression, and in turn modulate responses to VEN-based therapy.28 In our cohort, the improved survival in SRSF2 mutated patients treated with HMA+VEN appeared largely driven by the co-occurrence of IDH1/2 mutations with favorable survival to that seen in historic cohorts of SRSF2/IDH1/2–mutated patients treated with conventional chemotherapy. Survival in SRSF2-mutated patients without IDH mutations was a modest 8 months, a discordant finding from prior analyses.11 Although median follow-up time was adequate (24 months), these results must nonetheless be interpreted in the context of the small cohort sizes and retrospective nature of this analysis. Future investigations including larger patient cohorts are warranted to confirm these findings.
In conclusion, we demonstrate outcomes in patients with spliceosome mutations treated with HMA+VEN are comparable to those seen in wild-type patients. HMA+VEN therapy may overcome some of the adverse prognosis associated with specific spliceosome mutations or spliceosome mutations in the context of standard cytotoxic chemotherapy, with certain mutational pairs (SRSF2/IDH2) defining molecular groups imparting a favorable prognostic impact. Alternatively, other mutational pairs (U2AF1/RAS) may identify patients with suboptimal responses to HMA+VEN. Confirmation of these findings in prospective cohorts using molecularly targeted therapies will aid in the refinement of molecular subgroups of patients with increased sensitivity and response to venetoclax-based regimens.
Data will be made available on a case-by-case basis upon request of the corresponding author at cdinardo@mdanderson.org.
Authorship
Contribution: C.A.L., C.D.D., and S.L. performed data collection and analysis and prepared the manuscript; K.F. performed data analysis and assisted in manuscript preparation; S.P. performed data collection; and C.D.D., S.L., G.M.-B., A.M., T.K., N.D., G.B., N.P., K.S., Y.A., M.Y., N.J.S., K.C., M.O., K.P.P., E.J., F.R., H.M.K., G.G.-M., K.T., and M.Y.K. assisted in patient care, manuscript preparation, and review.
Conflict-of-interest disclosure: C.D.D. reports research support from AbbVie, Agios, Calithera, Cleave, BMS/Celgene, Daiichi-Sankyo, ImmuneOnc, and Loxo, and consultancy/advisory contributions for AbbVie, Agios, Celgene/BMS, Foghorn Therapeutics, Gilead, ImmuneOnc, Novartis, Takeda, and Notable Labs. N.J.S. reports research support from Takeda Oncology and Astellas Pharma Inc, consultancy support from Takeda Oncology and AstraZeneca, and honoraria from Amgen. N.D. reports research support from Daiichi-Sankyo, Bristol-Myers Squibb, Pfizer, Gilead, Sevier, Genentech, Astellas, Daiichi-Sankyo, AbbVie, Hanmi, Trovagene, FATE, Amgen, Novimmune, Glycomimetics, and ImmunoGen, and consultancy/advisory contributions to Daiichi-Sankyo, Bristol-Myers Squibb, Pfizer, Novartis, Celgene, AbbVie, Astellas, Genentech, Immunogen, Servier, Syndax, Trillium, Gilead, Amgen, and Agios. F.R. reports research support from AbbVie and advisory contributions and honoraria from Celgene and BMS. A.M. reports institutional research support from Celgene. T.K. reports research support from AbbVie, BMS, AstraZeneca, Cellenkos, Pfizer, Astellas, Ascentage, Pulmotec, Amgen, Genentech, Celgene, Incyte, Jazz, and Cyclacel, and honoraria from AbbVie, BMS, Pfizer, Novartis, Genentech, and Jazz. G.B. reports research contributions from AstraZeneca, GSK, BMS, AbbVie, Novartis, Incyte, Polaris, Xbiotech USA, Oncoceutics, PTC Therapeutics, Jannsen, Bioline Rx, Cyclacel, and consultancy contributions from PTC Therapeutics, Nkarta Therapeutics, Treadwell Therapeutics, BioTherix, Curio Science LLC, FTC Therapeutics, Argenx, and BioLine Rx. N.P. reports research contributions from Affymetrix, Stemline Therapeutics, AbbVie, Daiichi Sankyo, Plexxikon, Cellectis, Novartis, Samus Therapeutics; grant support from Affymetrix, SagerStrong; and honoraria from Stemline Therapeutics, LFB Biotechnologies, MustangBio, Incyte Corporation, Celgene, DAVA Oncology, Blueprint Medicines, Novartis, and Roche Diagnostics. K.S. reports research contributions from Novartis; consultancy contributions for Pfizer Japan, Novartis, Daiichi Sankyo; and honoraria from Otsuka. M.Y. reports research funding from Pfizer and Daiichi-Sankyo and honoraria from Pint Pharma. E.J. reports research contributions from Amgen, Adaptive Biotechnologies, AbbVie, Pfizer, Takeda, BMS, and Genentech, and advisory contributions for Amge, Adaptive Biotechnologies, AbbVie, Pfizer, Takeda, BMS, and Genentech. H.M.K. reports research contributions from AbbVie, Amgen, Jazz, Daiichi-Sankyo, BMS, Ascentage, Immunogen, Novartis, Pfizer, and Sanofi; advisory contributions for Actinium; and honoraria contributions from Aptitute Health, AbbVie, BioAscend, Δ Fly, Amgen, Daiichi-Sankyo, Adaptive Biotechnologies, Oxford Biomedical, Novartis, Actinium, and Pfizer. G.G.-M. reports research contributions from Celgene, Genentech, Novartis, Merck, Bristol-Myers Squibb, AbbVie, Helsinn Therapeutics, H3 Biomedicine, Amphivena Therapeutics, Onconova, Astex Pharmaceuticals; consultancy contributions from Celgene, Genentech, Bristol-Myers Squibb, Acceleron Pharmaceuticals, Helsinn Therapeutics, Astex Pharmaceuticals, and Jazz Pharmaceuticals; and honoraria contributions from Celgene, Acceleron Pharmaceuticals, AbbVie, Helsinn Therapeutics, and Astex Pharmaceuticals. M.Y.K. reports research contributions from Cellectis, Stemline Therapeutics, Ablynx, Forty-Seven, Genentech, Rafael Pharmaceutical, Agios, AstraZeneca, Calithera, Ascentage, AbbVie, Sanofi, Eli Lilly, F. Hoffmann-La Roche, and consultancy contributions to Stemline Therapeutics, Forty-Seven, Genentech, Kosji, AbbVie, Amgen, and F. Hoffmann-La Roche. The remaining authors declare no competing financial interests.
Correspondence: Courtney D. DiNardo, Department of Leukemia, The University of Texas MD Anderson Cancer Center, Unit 428, 1515 Holcombe Blvd, Houston, TX 77030; e-mail: cdinardo@mdanderson.org.

