

Key Points
Loss of CYLD, NFKBIA, TRAF2, TRAF3, KCTD5, AMBRA1, or RFX7 reduced the anti-DLBCL activity of CC-122 independent of IKZF1/3 degradation.
Ablation of KCTD5 promotes the stabilization of its cognate substrate, GNG5, resulting in attenuated response to CC-122.

Abstract
CC-122 is a next-generation cereblon E3 ligase–modulating agent that has demonstrated promising clinical efficacy in patients with relapsed or refractory diffuse large B‐cell lymphoma (R/R DLBCL). Mechanistically, CC-122 induces the degradation of IKZF1/3, leading to T-cell activation and robust cell-autonomous killing in DLBCL. We report a genome-wide CRISPR/Cas9 screening for CC-122 in a DLBCL cell line SU-DHL-4 with follow-up mechanistic characterization in 6 DLBCL cell lines to identify genes regulating the response to CC-122. Top-ranked CC-122 resistance genes encode, not only well-defined members or regulators of the CUL4/DDB1/RBX1/CRBN E3 ubiquitin ligase complex, but also key components of signaling and transcriptional networks that have not been shown to modulate the response to cereblon modulators. Ablation of CYLD, NFKBIA, TRAF2, or TRAF3 induces hyperactivation of the canonical and/or noncanonical NF-κB pathways and subsequently diminishes CC-122–induced apoptosis in 5 of 6 DLBCL cell lines. Depletion of KCTD5, the substrate adaptor of the CUL3/RBX1/KCTD5 ubiquitin ligase complex, promotes the stabilization of its cognate substrate, GNG5, resulting in CC-122 resistance in HT, SU-DHL-4, and WSU-DLCL2. Furthermore, knockout of AMBRA1 renders resistance to CC-122 in SU-DHL-4 and U-2932, whereas knockout of RFX7 leads to resistance specifically in SU-DHL-4. The ubiquitous and cell line–specific mechanisms of CC-122 resistance in DLBCL cell lines revealed in this work pinpoint genetic alternations that are potentially associated with clinical resistance in patients and facilitate the development of biomarker strategies for patient stratification, which may improve clinical outcomes of patients with R/R DLBCL.
Introduction
Diffuse large B‐cell lymphoma (DLBCL), the most common type of non‐Hodgkin lymphoma, is a heterogeneous group of diseases with variable outcomes that are differentially characterized by clinical and molecular features, cell of origin, and frequently recurring mutations.1,2 The most common up-front treatment of DLBCL is chemoimmunotherapy with (cyclophosphamide, doxorubicin, vincristine, and prednisone with rituximab), which leads to a cure in ∼50% of DLBCL patients.2 Despite recent advances in treatment options, most patients with relapsed or refractory diffuse large B‐cell lymphoma (R/R DLBCL) has a poor prognosis with a median survival of 4 to 6 months.3 Novel therapeutics for R/R DLBCL are therefore critical for the improvement of clinical outcomes.
In the past 2 decades, several cereblon modulators, including immunomodulatory (IMiD) drugs and next-generation cereblon E3 ligase modulating (CELMoD) agents, have been developed to target disease-driving proteins for degradation via a molecular glue mechanism.4-7 CC-122 (avadomide) is a novel CELMoD agent with immunomodulatory and cell-autonomous antitumor activities in DLBCL. Mechanistically, CC-122 redirects cereblon, the substrate receptor of CUL4/DDB1/RBX1/CRBN E3 ubiquitin ligase complex (CRL4CRBN), to induce the ubiquitination and degradation of ikaros (IKZF1) and aiolos (IKZF3) proteins, resulting in T-cell activation, as well as induction of apoptosis and growth inhibition in malignant B cells in DLBCL cell lines and patients.8 CC-122 has shown promising clinical activity as a single agent or in combination with anti-CD20 antibodies in patients with R/R DLBCL.9,10 An overall response rate (ORR) of 29%, including 11% with a complete response, was observed in 84 patients with de novo R/R DLBCL treated with CC-122 monotherapy.11 A 26-gene classifier identifying tumors with infiltration of T cells and macrophages was significantly enriched for responders (ORR, 44% vs 19% in classifier-positive vs -negative patient subgroups).11,12 However, the unresponsiveness to CC-122 treatment was evident in most patients with DLBCL, most likely owing to preexisting or acquired genetic or epigenetic lesions in malignant B cells conferring resistance.
Herein, we present the delineation of the mechanisms of CC-122 resistance in DLBCL cell lines through a genome-wide CRISPR/Cas9 knockout screening in the DLBCL cell line SU-DHL-4 with follow-up validation of genes and pathways in a panel of DLBCL cell lines. The identifications of genetic alternations affecting tumor-intrinsic activity of CC-122 in DLBCL cell lines may inform the design of patient stratification strategies and rational combinations for future clinical trials of CC-122 or other CELMoDs in R/R DLBCL.
Methods
Genome-wide CRISPR screening
A total of 1 × 109 SU-DHL-4 cells stably expressing Cas9 protein were inoculated with a single guide RNA (sgRNA) library containing lentiviral supernatant at a multiplicity of infection of 0.5 according to the Cellecta CRISPR screening guide. Cells were grown in 2-L flasks with agitation and at least 2 × 107 cells were kept after each passage to exceed the 150-k library complexity by more than 1000-fold, maintaining library representation. CC-122 was replenished every 4 or 5 days. For genomic DNA isolation and sequencing library preparation, cell pellets (9 × 107) were collected in technical duplicates at days 3 and 14 after infection.
Detailed “Materials and methods” can be found in the supplemental Data. The study has been approved by Bristol Myers Squibb.
Results
Targeted degradation of IKZF1 and IKZF3 solely mediates the antiproliferative activity of CC-122 in DLBCL
IKZF1 and IKZF3 are well-characterized targets of many cereblon modulators, including thalidomide, lenalidomide, pomalidomide, iberdomide, and CC-122.4,5,8,13 We have reported that the anti-DLBCL activity of CC-122 is directly linked to the degradation of IKZF1 and IKZF3.4,5,8,13 However, in addition to IKZF1 and IKZF3, CC-122 can effectively induce the degradation of ZFP91 and, to a much lesser extent, PDE-6 and WIZ.9 To determine the individual role of IKZF1, IKZF3, and ZFP91 in mediating the antitumor effect of CC-122 in DLBCL, we evaluated the growth inhibitory effect of CC-122 in DLBCL cell lines with stable overexpression of NanoLuc luciferase (as a negative control) or degradation-resistant mutated IKZF1, IKZF3, or ZFP91, using the CellTiter-Glo cell viability assay (Figure 1A-B; supplemental Figure 1A). We have shown that IKZF1 or IKZF3 interacts with cereblon through a β-hairpin degron motif residing in the second C2H2 zinc finger domain (IKZF1 ZnF-2, residues 145-167; IKZF3 ZnF-2, residues 146-168).14 As expected, replacement of the key glycine residue in the degron motif of IKZF1/3 with alanine (IKZF1 G151A; IKZF3 G152A) prevented CC-122–induced degradation in DLBCL cell lines KARPAS-422, SU-DHL-4, HT, and RIVA (Figure 1C-D; supplemental Figure 1B). Similar to IKZF1/3, the recruitment of ZFP91 to cereblon by CC-122 is mediated by a degron motif in its fourth C2H2 zinc finger domain (ZFP91 ZnF-4, residues 400-422),15 and mutation of glycine 405 to alanine (G405A) in the ZnF4 completely abolished the CC-122–induced degradation (Figure 1C-D; supplemental Figure 1B). Consistent with our observation that degradation of IKZF1 and IKZF3 is directly associated with the antiproliferative effects of CC-122 in DLBCL cell lines,9 overexpression of degradation-resistant mutations of IKZF1 or IKZF3 (IKZF1-G151A or IKZF3-G152A) conferred various degrees of significant resistance to CC-122 in all 4 tested cell lines (Figure 1A-B; supplemental Figure 1A). On the other hand, overexpression of the ZFP91 degradation-resistant mutation G405A did not show any protection against CC-122 (Figure 1A-B; supplemental Figure 1A), ruling out the involvement of ZFP91 in CC-122–induced cell-autonomous activity in DLBCL.
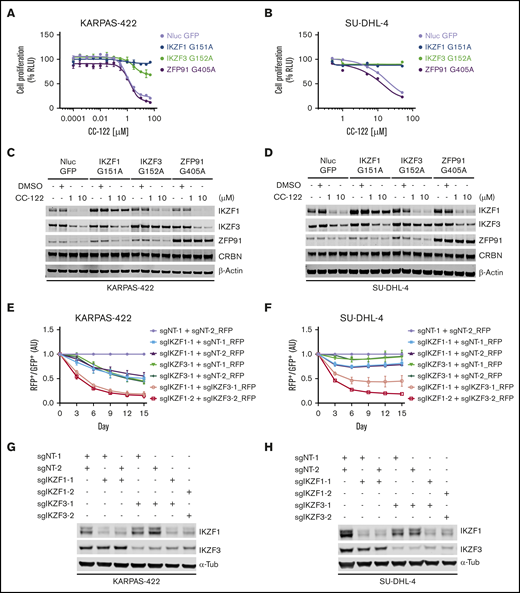
The antiproliferative effect of CC-122 is solely mediated by the degradation of IKZF1 and IKZF3 in DLBCL. (A-D) Dose response curves of CC-122 in KARPAS-422 (A) and SU-DHL-4 (B) cells ectopically expressing NanoLuc luciferase (NLuc) or degradation-resistant mutants of IKZF1, IKZF3, or ZFP91. Cells were treatment with DMSO or CC-122 at the indicated concentrations for 5 days, followed by cell viability assessment using CellTiter-Glo (CTG). Shown are percentages of CTG luminescence signals relative to DMSO controls. Data are shown as the mean ± standard error of the mean (SEM). KARPAS-422, 2 technical replicates; SU-DHL-4, 3 technical replicates. (C-D) Immunoblot analysis of KARPAS-422 (C) and SU-DHL-4 (D) cells ectopically expressing NLuc or degradation-resistant mutants of IKZF1, IKZF3, or ZFP91. Cells were treated with CC-122 at the indicated concentrations for 24 hours. (E-F) Cell fitness assessment of KARPAS-422 (E) or SU-DHL-4 (F) cells with knockout of IKZF1 alone, IKZF3 alone, or both, compared with control cells using a flow cytometry–based CRISPR competition assay. KARPAS-422 or SU-DHL-4 cells stably expressing Cas9 were transduced with a lentiviral vector coexpressing GFP and sgNT-1, or with lentiviral vectors coexpressing RFP and the indicated sgRNAs targeting IKZF1, IKZF3, or both. Three days after infection, RFP and GFP cells were mixed at a 1:1 ratio, and the change in the RFP+/GFP+ ratio was monitored by flow cytometry every 3 days thereafter. RFP+/GFP+ ratios of cells after transduction with the indicated sgRNA were first normalized to their RFP+/GFP+ ratios on day 0 and then to the sgNT-1/sgNT-2 controls at each time point. Data are shown as the mean ± SEM; 3 biological replicates. (G-H) Immunoblot analysis of KARPAS-422 (G) or SU-DHL-4 (H) cells used for the completion assay described in panels E-F.
The antiproliferative effect of CC-122 is solely mediated by the degradation of IKZF1 and IKZF3 in DLBCL. (A-D) Dose response curves of CC-122 in KARPAS-422 (A) and SU-DHL-4 (B) cells ectopically expressing NanoLuc luciferase (NLuc) or degradation-resistant mutants of IKZF1, IKZF3, or ZFP91. Cells were treatment with DMSO or CC-122 at the indicated concentrations for 5 days, followed by cell viability assessment using CellTiter-Glo (CTG). Shown are percentages of CTG luminescence signals relative to DMSO controls. Data are shown as the mean ± standard error of the mean (SEM). KARPAS-422, 2 technical replicates; SU-DHL-4, 3 technical replicates. (C-D) Immunoblot analysis of KARPAS-422 (C) and SU-DHL-4 (D) cells ectopically expressing NLuc or degradation-resistant mutants of IKZF1, IKZF3, or ZFP91. Cells were treated with CC-122 at the indicated concentrations for 24 hours. (E-F) Cell fitness assessment of KARPAS-422 (E) or SU-DHL-4 (F) cells with knockout of IKZF1 alone, IKZF3 alone, or both, compared with control cells using a flow cytometry–based CRISPR competition assay. KARPAS-422 or SU-DHL-4 cells stably expressing Cas9 were transduced with a lentiviral vector coexpressing GFP and sgNT-1, or with lentiviral vectors coexpressing RFP and the indicated sgRNAs targeting IKZF1, IKZF3, or both. Three days after infection, RFP and GFP cells were mixed at a 1:1 ratio, and the change in the RFP+/GFP+ ratio was monitored by flow cytometry every 3 days thereafter. RFP+/GFP+ ratios of cells after transduction with the indicated sgRNA were first normalized to their RFP+/GFP+ ratios on day 0 and then to the sgNT-1/sgNT-2 controls at each time point. Data are shown as the mean ± SEM; 3 biological replicates. (G-H) Immunoblot analysis of KARPAS-422 (G) or SU-DHL-4 (H) cells used for the completion assay described in panels E-F.
Overexpression of IKZF1-G151A completely abolished the antiproliferative effect of CC-122 in KARPAS-422, SU-DHL-4 and HT cells, whereas In RIVA cells, the observed protection was less pronounced. IKZF3-G152A exhibited similar effect on CC-122 response as IKZF1-G151A in KARPAS-422, SU-DHL-4, and RIVA cells. In contrast, in HT cells, IKZF3-G152A overexpression conferred partial protection to CC-122, whereas IKZF1-G151A overexpression eliminated the activity of CC-122. IKZF1 and IKZF3 are highly homologous transcription factors that can form homodimers or heterodimers to regulate lymphocyte differentiation.16 Therefore, it is likely that IKZF1 and IKZF3 can redundantly drive the survival of malignant B cells in DLBCL, providing an explanation for the complete rescue, with overexpression of either IKZF1-G151A or IKZF3-G152A.
To further characterize the role of IKZF1 and IKZF3 in mediating the response to CC-122 in DLBCL, we used a flow cytometry-based CRISPR competition assay to assess the effect of genetic inactivation of IKZF1, IKZF3, or both on cell fitness in 6 DLBCL cell lines KARPAS-422, SU-DHL-4, U-2932, RIVA, HT, and SU-DHL-16 (Figure 1E-F; supplemental Figure 1C-D). Expression of 2 different single guide RNAs (sgRNAs) targeting IKZF1- or IKZF3-induced specific and efficient downregulation of the intended target protein in all 6 DLBCL cell lines (Figure 1G-H; supplemental Figure 1E). Knockout of IKZF1 reduced cell proliferation by 25% to 50%, compared with control cells expressing nontargeting sgRNAs, (sgNT-1 and sgNT-2) in all 6 cell lines except U-2932. Deletion of IKZF3 reduced cell proliferation by 50% to 75% in KARPAS-422, SU-DHL-16, and HT cells, with minimal effect in SU-DHL-4, U-2932, and RIVA cells. Compared with single knockout of IKZF1 or IKZF3, simultaneous elimination of both IKZF1 and IKZF3 produced synergistic inhibition of cell proliferation in SU-DHL-4, U-2932, and RIVA cells, and additive growth suppression in KARPAS-422, SU-DHL-16, and HT cells (Figure 1E-F; supplemental Figure 1D). These results suggest that IKZF1 and IKZF3 play a redundant role in maintaining cell survival and growth in DLBCL cell lines, and that loss of both IKZF1 and IKZF3 is sufficient to account for the antiproliferative effects of CC-122 in DLBCL cells.
Genome-wide CRISPR screening reveals genes and pathways governing the response to CC-122 in DLBCL
CC-122 has shown robust efficacy in a significant fraction of patients with R/R DLBCL; however, inherent or therapy-related resistance independent of the degradation level of IKZF1/3 occurred in most of the patients.11 To delineate genetic alterations in patients with DLBCL that might attenuate the response to CC-122, we performed a genome-wide CRISPR/Cas9 screening to identify genes and pathways that affect the response to CC-122 in the DLBCL cell line SU-DHL-4.
SU-DHL-4 cells stably expressing Cas9 were transduced with a pooled lentiviral library targeting 19 011 protein-encoding genes with 4 to 8 different sgRNAs for each gene. At day 3 after transduction (time 0; T0), cells were treated with 10 µM CC-122 or dimethyl sulfoxide (DMSO) vehicle control for an additional 11 days to allow for 10 more doublings of the DMSO-treated cells (time 10; T10), followed by amplification of sgRNA coding regions from genomic DNA collected from viable cells at T0 and T10, and next-generation sequencing to identify and quantify the abundance of sgRNAs (Figure 2A; supplemental Table 1). CC-122 treatment significantly inhibited the proliferation of SU-DHL-4 cells (supplemental Figure 2A), and consistently affected the sgRNA read count distribution in 2 technical replicates, compared with vehicle-treated cells (supplemental Figure 2B). Next, the log2-fold change (log2FC) in read count for each sgRNA in the CC-122-treated cells vs DMSO control at T10 was calculated, and the average log2FC value of all sgRNAs targeting a gene of interest was designated as the gene enrichment score (Figure 2A).

A Genome-wide CRISPR screening reveals candidate CC-122 resistance genes in DLBCL. (A) The genome-wide CRISPR screening of SU-DHL-4 cells. (B) Scatter plot of genes with a log2FC (T10_CC-122 vs T10_DMSO) >0 and false discovery rate (FDR) <0.05; x-axis: gene essentiality score shown as log2FC (T10_DMSO vs T0_DMSO); y-axis: resistance score shown as log2FC (T10_CC-122 vs T10_DMSO). Brown open circles mark resistance genes common to other cereblon modulators. Red solid circles mark novel resistance genes unique to CC-122. (C) Log2FC (T10_CC-122 vs T10_DMSO) values of sgRNAs targeting CC-122–enriched genes. Blue represents the background distribution of the sgRNA library, and each vertical line represents an individual sgRNA (5-8 per gene). (D) Pathway enrichment analysis of genes enriched by CC-122 treatment, with log2 FC >1 and FDR <0.05 relative to DMSO control. The color and size of the dots represent the adjusted significance level and gene ratio, respectively. Gene ratio refers to the number of input genes annotated to an individual pathway as a ratio of all input genes annotated to any Reactome database pathway.
A Genome-wide CRISPR screening reveals candidate CC-122 resistance genes in DLBCL. (A) The genome-wide CRISPR screening of SU-DHL-4 cells. (B) Scatter plot of genes with a log2FC (T10_CC-122 vs T10_DMSO) >0 and false discovery rate (FDR) <0.05; x-axis: gene essentiality score shown as log2FC (T10_DMSO vs T0_DMSO); y-axis: resistance score shown as log2FC (T10_CC-122 vs T10_DMSO). Brown open circles mark resistance genes common to other cereblon modulators. Red solid circles mark novel resistance genes unique to CC-122. (C) Log2FC (T10_CC-122 vs T10_DMSO) values of sgRNAs targeting CC-122–enriched genes. Blue represents the background distribution of the sgRNA library, and each vertical line represents an individual sgRNA (5-8 per gene). (D) Pathway enrichment analysis of genes enriched by CC-122 treatment, with log2 FC >1 and FDR <0.05 relative to DMSO control. The color and size of the dots represent the adjusted significance level and gene ratio, respectively. Gene ratio refers to the number of input genes annotated to an individual pathway as a ratio of all input genes annotated to any Reactome database pathway.
Among the 52 top-ranked genes with a more than twofold enrichment at the gene level (log2FC >1), we have identified many genes encoding well-characterized proteins that mediate the degradation of IKZF1 and IKZF3 induced by cereblon modulators. These include the CRL4CRBN subunits CRBN and CUL-4A,4,5 the ubiquitin conjugation enzymes UBE2G1 and UBE2D3,17,18 and multiple members of the COP-9 signalosome, including GPS1, COPS2, COPS4, COPS5, COPS6, COPS7B, and COPS818 (Figure 2B-C; supplemental Table 2). In addition, pathway enrichment analysis of top-ranked resistant hits identified 4 genes encoding well-known repressors of the canonical or noncanonical NF-κB signaling pathways: CYLD, NFKBIA, TRAF2, and TRAF3 (Figure 2B-D; supplemental Figure 2C; supplemental Table 3). Moreover, we identified a few CC-122-resistant genes such as KCTD5, AMBRA1, and RFX7 that had not been reported to modulate the response to other cereblon modulators (Figure 2B-C).
NF-κB activation confers resistance to CC-122 in DLBCL
CYLD is a deubiquitinase that removes K63-linked ubiquitination and negatively regulates the canonical NF-κB pathway.19 IκBα (encoded by NFKBIA) prevents the activation of the canonical NF-κB pathway by sequestering the NF-κB dimers in the cytoplasm.20 TRAF2 and TRAF3 form a complex with an ubiquitin E3 ligase cIAP-1/2 to promote the ubiquitination and degradation of NIK and the subsequent inhibition of the noncanonical NF-κB pathway.21 In the CRISPR screening, almost all sgRNAs targeting NF-κB repressor genes CYLD, NFKBIA, TRAF2, and TRAF3 were significantly enriched in SU-DHL-4 cells in response to CC-122 treatment (Figure 2C), indicating that hyperactivation of the NF-κB pathway abrogated the antitumor activity of CC-122 in DLBCL. To test this hypothesis, we used the CRISPR competition assay to evaluate the activity of CC-122 in SU-DHL4-Cas9 cells transduced with a lentiviral vector expressing a nontargeting sgRNA (sgNT-1), or 1 of 2 different sgRNAs specifically targeting CYLD, NFKBIA, TRAF2, or TRAF3. SU-DHL4-Cas9 cells expressing a CRBN-specific sgRNA, sgCRBN-8, and cells expressing a sgRNA targeting an intronic noncoding region, sgNC-8, were used as a positive control and a negative control, respectively (Figure 3A; supplemental Figure 3A-B). Consistent with the CRISPR screening result, CC-122 treatment induced a time- and concentration-dependent enrichment of cells with depletion of CYLD, NFKBIA, TRAF2, TRAF3, or CRBN over control cells, and CRBN elimination conferred more resistance to CC-122 than inactivation of CYLD, NFKBIA, TRAF2, or TRAF3 did. In addition, the gene knockout effects of CYLD and NFKB1A were also more pronounced than those of TRAF2 and TRAF3 (Figure 3A; supplemental Figure 3B).

Inactivation of NF-κB repressors confers resistance to CC-122. (A) Cell fitness assessment of SU-DHL-4 cells with knockout of CRBN, CYLD, NFKBIA, TRAF2, or TRAF3 in the presence or absence of CC-122, compared with control cells using a flow cytometry–based CRISPR competition assay. SU-DHL-4 cells stably expressing Cas9 were transduced with a lentiviral vector coexpressing GFP and sgNT-1, or with lentiviral vectors coexpressing RFP and the indicated sgRNAs. Three days after infection, RFP and GFP cells were mixed at a 1:1 ratio and treated with DMSO or CC-122 at the indicated concentrations. The change in the RFP+/GFP+ ratio was then monitored by flow cytometry at the indicated time points. RFP+/GFP+ ratios of cells after transduction with the indicated sgRNA were first normalized to their RFP+/GFP+ ratios on day 0 and then to DMSO controls at each time point. Data are shown as mean ± standard deviation (SD); 2 biological replicates. Data were analyzed by 2-tailed unpaired Student t test. *P < .05; **P < .01; ***P < .001; NS (not significant), P > .05. (B) Immunoblot analysis of SU-DHL-4-Cas9 cells transduced with lentiviral vectors expressing the indicated sgRNAs. Three days after transduction, cells were treated with 2 µg/mL puromycin for 3 additional days to select sgRNA-expressing cells. (C) Immunoblot analysis of nuclear protein extractions of SU-DHL-4 cells transduced with the indicated sgRNAs. The histone H3 level was used as the loading control. (D) Heat map showing the CC-122–induced enrichment of DLBCL cells with loss of CRBN or NF-κB repressors in the CRISPR competition assay. The enrichment score is calculated as the FC of the RFP+/GFP+ ratios between cells treated with 50 µM CC-122 vs their DMSO controls on day 10. (E) Immunoblot analysis of SU-DHL-4-Cas9 cells transduced with lentiviral vectors expressing the indicated sgRNAs. Cells were treated with either DMSO or 10 µM CC-122 for 24 hours. (F) Flow cytometry analysis of SU-DHL-4 cells with expression of the indicated sgRNA. Cells were continuously treated with DMSO or 50 µM CC-122 for 10 days, followed by staining with annexin V and 7-AAD.
Inactivation of NF-κB repressors confers resistance to CC-122. (A) Cell fitness assessment of SU-DHL-4 cells with knockout of CRBN, CYLD, NFKBIA, TRAF2, or TRAF3 in the presence or absence of CC-122, compared with control cells using a flow cytometry–based CRISPR competition assay. SU-DHL-4 cells stably expressing Cas9 were transduced with a lentiviral vector coexpressing GFP and sgNT-1, or with lentiviral vectors coexpressing RFP and the indicated sgRNAs. Three days after infection, RFP and GFP cells were mixed at a 1:1 ratio and treated with DMSO or CC-122 at the indicated concentrations. The change in the RFP+/GFP+ ratio was then monitored by flow cytometry at the indicated time points. RFP+/GFP+ ratios of cells after transduction with the indicated sgRNA were first normalized to their RFP+/GFP+ ratios on day 0 and then to DMSO controls at each time point. Data are shown as mean ± standard deviation (SD); 2 biological replicates. Data were analyzed by 2-tailed unpaired Student t test. *P < .05; **P < .01; ***P < .001; NS (not significant), P > .05. (B) Immunoblot analysis of SU-DHL-4-Cas9 cells transduced with lentiviral vectors expressing the indicated sgRNAs. Three days after transduction, cells were treated with 2 µg/mL puromycin for 3 additional days to select sgRNA-expressing cells. (C) Immunoblot analysis of nuclear protein extractions of SU-DHL-4 cells transduced with the indicated sgRNAs. The histone H3 level was used as the loading control. (D) Heat map showing the CC-122–induced enrichment of DLBCL cells with loss of CRBN or NF-κB repressors in the CRISPR competition assay. The enrichment score is calculated as the FC of the RFP+/GFP+ ratios between cells treated with 50 µM CC-122 vs their DMSO controls on day 10. (E) Immunoblot analysis of SU-DHL-4-Cas9 cells transduced with lentiviral vectors expressing the indicated sgRNAs. Cells were treated with either DMSO or 10 µM CC-122 for 24 hours. (F) Flow cytometry analysis of SU-DHL-4 cells with expression of the indicated sgRNA. Cells were continuously treated with DMSO or 50 µM CC-122 for 10 days, followed by staining with annexin V and 7-AAD.
To confirm the activation of the canonical and/or noncanonical NF-κB pathways in SU-DHL-4 cells with knockout of CYLD, NFKBIA, TRAF2, or TRAF3, we assessed the nuclear translocation and transcriptional activity of NF-κB/Rel family proteins. As expected, knockout of CYLD, NFKBIA, TRAF2, or TRAF3 promoted the accumulation of p50 and p52 in the nucleus (Figure 3B-C), and the increase of nuclear p52 protein levels was much higher in cells with knockout of TRAF2 or TRAF3, compared with cells with loss of CYLD or NFKBIA (Figure 3C). Knockout of CYLD or NFKBIA significantly augmented the transcriptional activity of p50, c-Rel and Rel-B, whereas deletion of TRAF2 or TRAF3 resulted in a substantial increase in the activity of p52 and Rel-B (supplemental Figure 3C). Interestingly, inactivation of CYLD, but not NFKBIA, TRAF2, or TRAF3, showed a significant increase in Rel-A activity, which is linked to more resistance to CC-122 as described above (Figure 3A; supplemental Figure 3B-C).
Next, we sought to determine whether knockout of CYLD, NFKBIA, TRAF2, TRAF3, or CRBN can also confer resistance in 5 additional DLBCL cell lines HT, KARPAS-422, Pfeiffer, WSU-DLCL2, and RIVA, using the CRISPR competition assay. Knockout of CRBN abrogated the response to CC-122 in all tested lines (Figure 3D); however, differential responses to inactivation of CYLD, NFKBIA, TRAF2, or TRAF3 were observed in a cell line–specific manner. Notably, none of the 4 NF-κB repressor genes, when knocked out, affected the response to CC-122 in KARPAS-422, whereas inactivation of 1 or several NF-κB repressors conferred resistance in the 4 remaining cell lines (Figure 3D). For instance, knockout of CYLD or TRAF3, by using 2 different sgRNAs, attenuated the response to CC-122 in WSU-DLCL2, RIVA, and Pfeiffer cells, whereas TRAF2 only showed consistent CC-122 resistance with both sgRNAs in RIVA cells (Figure 3D; supplemental Figure 3D; supplemental Table 4). These data suggest that NF-κB activation is likely to reduce the antitumor activity of CC-122 in most DLBCL cell lines, and additional genetic and/or epigenetic alteration(s) may modulate this desensitization effect.
To understand the mechanism of CC-122 resistance rendered by NF-κB activation, we evaluated the CC-122–induced degradation of IKZF1 and IKZF3 and apoptosis in SU-DHL-4 cells with inactivation of CYLD, NFKBIA, TRAF2, TRAF3, or CRBN. Continuous treatment with 50 µM CC-122 for 10 days drastically increased the percentage of annexin V+/7-AAD+ cells in SU-DHL-4 cells expressing a control sgRNA, sgNC-8 (Figure 3F). Knockout of CRBN completed eliminated this effect through blockage of CC-122–induced IKZF1/3 degradation (Figure 3F; supplemental Figure 3E-F). Interestingly, loss of CYLD, NFKBIA, TRAF2, or TRAF3 displayed inhibition on annexin V+/7-AAD+ cells without affecting the degradation or expression of IKZF1 and IKZF3 (Figure 3E-F; supplemental Figure 3F).
The NF-κB pathway is critical for normal B-cell development and survival. It is possible that the anti-DLBCL activity of CC-122 is partially mediated by the direct inhibition of NF-κB activity, and NF-κB hyperactivation after the loss of CYLD, NFKBIA, TRAF2, or TRAF3 abrogated this effect. To test this hypothesis, we measured the activity of NF-κB/Rel family proteins in SU-DHL-4 and RIVA cells treated with DMSO, 5 µM CC-122, or 100 ng/mL LPS (as a positive control) for 24 hours. LPS treatment increased the activity of several NF-κB/Rel family proteins in both SU-DHL-4 and RIVA cells. However, no significant difference was observed between samples treated with DMSO or CC-122, indicating that CC-122 treatment may not directly modulate the activity of NF-κB members (supplemental Figure 3G). To further test whether CC-122 could affect the NF-κB signaling pathway, the abundance of messenger RNA (mRNA) of several NF-κB signature targets, such as TNF,22 NFKBIA,23 CXCL8 (IL-8),24 and CCL5 (RANTES),25 was monitored by using quantitative polymerase chain reaction (qPCR) in SU-DHL-4 cells treated with DMSO or 10 µM CC-122 for 0.5, 1, 2, 4, 8, and 24 hours. As shown in supplemental Figure 3H, there were no statistically significant changes in mRNA levels of all 4 NF-κB signature genes at all time points, except small changes in TNF and CCL5 levels at 8 hours.
To gain a better understanding of the role of the NF-κB pathway in mediating the CC-122 response in DLBCL, we performed mRNA sequencing on SU-DHL-4 parental cells and cells with loss of CYLD, NFKBIA, TRAF2, or TRAF3 after treatment with DMSO or 10 µM CC-122. Gene Set Enrichment Analysis on the hallmark NF-κB pathway are shown in supplemental Figure 3I. Knockout of NF-κB repressors show significant enrichment in the NF-κB pathway, and CC-122 treatment also showed a subtle positive enrichment of the NF-κB pathway, most likely related to the increase in overlapping type 1 interferon target genes, such as DDX58. No significant negative enrichment was identified for the NF-κB pathway gene sets. The top 100 differentially expressed genes upon NF-κB repressor genetic perturbation were singled out (supplemental Figure 3J). As shown in the heat map, no substantial differences in most of those genes were observed in control cells upon CC-122 treatment, confirming again that CC-122 may not have a substantial effect on the NF-κB signaling pathway. Furthermore, a correlation analysis showed that the effect of CC-122 was largely abolished or even reversed in cells with genetic ablation of CYLD, NFKBIA, TRAF2, or TRAF3 (supplemental Figure 3K-L).
To further clarify whether a reactive NF-κB activation or inactivation can be triggered by prolonged exposure to CC-122, we assessed the effect of CC-122 on apoptosis and NF-κB activity in SU-DHL-4 cells after 3, 5, and 10 days of treatment (supplemental Figure 3M-P). A significant increase in annexin V+ cells was observed at days 5 and 10 (supplemental Figure 3M-N). In contrast, no significant change in nuclear or cytosolic p50, p52, Rel A, Rel B, c-Rel, IκΒα, and Bcl-2 were detected at all 3 time points tested (supplemental Figure 3O). Furthermore, we assessed the mRNA abundance of several NF-κB target genes TNF, NFKBIA, CCL5, and CXCL8, using qPCR (supplemental Figure 3P). Interestingly, prolonged CC-122 treatment significantly increased the mRNA level of TNF and reduced the level of CCL5. Further investigation is needed to evaluate whether this alteration is linked to immunomodulatory effects, as it is with other IMiDs. However, no significant change in NFKBIA or CXCL8 was observed at all 3 time points.
Together, these data suggest that CC-122 does not directly modulate the central regulators of the NF-κB signaling pathway. Therefore, we speculated that the constitutive activation of the NF-κB signal pathway upon depletion of NF-κB repressors in DLBCL cells would counteract the CC-122–induced cytotoxicity by eliciting a strong prosurvival signal.
GNG5 upregulation mediates the attenuated response to CC-122 in KCTD5–deficient DLBCL
In the CRISPR screening, multiple KCTD5-specific sgRNAs were significantly enriched in SU-DHL-4 cells after treatment with CC-122 for 10 days (Figure 2B-C). Aligning with this result, knockout of KCTD5 by using 2 different sgRNAs significantly diminished the response to CC-122 in the CRISPR competition assay (Figure 4A; supplemental Figure 4A-B). Ablation of KCTD5 also conferred resistance to CC-122 in 3 of 5 additional DLBCL cell lines tested (Figure 4B; supplemental Table 5). Similar to the mechanism of CC-122 resistance elicited by NF-κB activation, depletion of KCTD5 protected cells against CC-122 without affecting the degradation of IKZF1 and IKZF3 in SU-DHL-4 cells (Figure 4C). Furthermore, the basal protein levels of KCTD5 and NF-κB repressors (CYLD, IκBa, TRAF3, and TRAF2) were examined in 11 DLBCL cell lines (supplemental Figure 5F). However, no clear correlation was identified between the expression levels and the CC-122 resistance patterns in the competition assays.
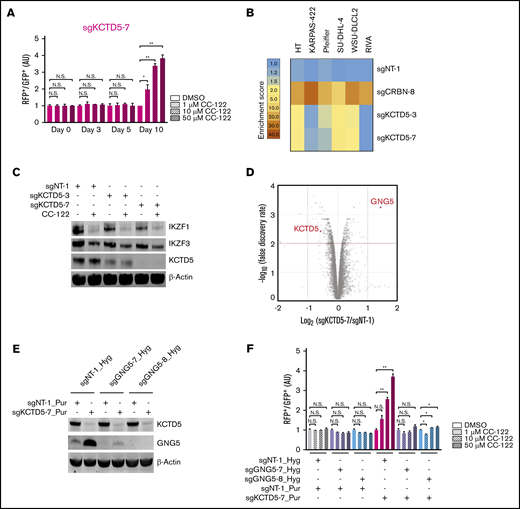
GNG5 upregulation mediates the CC-122 resistance rendered by KCTD5 inactivation. (A) Cell fitness assessment of SU-DHL-4 cells with knockout of KCTD5 in the presence or absence of CC-122, compared with control cells by a flow cytometry–based CRISPR competition assay, as described in Figure 3A. Data are shown as the mean ± standard deviation (SD); 2 biological replicates. (B) Heat map showing the CC-122–induced enrichment of DLBCL cells with loss of CRBN or KCTD5 in the CRISPR competition assay. Enrichment score is calculated as the FC of the RFP+/GFP+ ratios of cells treated with 50 µM CC-122 vs their DMSO controls on day 10. (C) Immunoblot analysis of SU-DHL-4-Cas9 cells transduced with lentiviral vectors expressing the indicated sgRNAs. Cells were treated with either DMSO or 10 µM CC-122 for 24 hours. (D) A volcano plot of differentially abundant proteins in response to CC-122 treatment relative to DMSO control. SU-DHL-4-Cas9 cells transduced with lentiviral vectors expressing sgNT-1 or sgKCTD5-7 were subjected to TMT proteomics analysis. The x-axis indicates the log2-FC of each protein in cells expressing sgKCTD5-7 vs cells expressing sgNT-1. P values were corrected for multiple hypothesis testing using the Benjamini-Hochberg method to arrive at an adjusted P value (Padj, also known as an FDR). The y-axis shows the log10 (FDR) values indicating statistical significance, such that proteins lying above the dotted red line are statistically significant findings with Padj < .01. (E) Immunoblot analysis of SU-DHL-4-Cas9 cells transduced with lentiviral vectors expressing the indicated sgRNAs. SU-DHL-4-Cas9 cells were first transduced with a lentiviral vector expressing sgNT-1, sgGNG5-7, or sgGNG5-8, and then selected by 400 µg/mL hygromycin for 10 days. The selected cells were then transduced with a lentiviral vector expressing sgNT-1 or sgKCTD5-7 followed by selection with puromycin (2 µg/mL) for 3 days. (F) Cell fitness assessment of SU-DHL-4 cells with knockout of KCTD5, GNG5, or both in the presence or absence of CC-122, compared with control cells, by flow cytometry–based CRISPR competition assay. SU-DHL-4-Cas9 cells stably expressing sgNT-1, sgGNG5-7, or sgGNG5-8 were transduced with a lentiviral vector coexpressing GFP and sgNT-1 or with a lentiviral vector coexpressing RFP and sgNT-1 or sgKCTD5-7. Three days after infection, RFP+ and GFP+ cells were mixed at a 1:1 ratio and treated with DMSO or CC-122 at the indicated concentrations. The change in the RFP+/GFP+ ratio was then monitored by flow cytometry at day 7. RFP+/GFP+ ratios of cells after transduction with the indicated sgRNA were first normalized to their RFP+/GFP+ ratios on day 0 and then to that of DMSO controls at day 7. (A,F) Data were analyzed by 2-tailed unpaired Student t test. *P < .05; **P < .01; NS, P > .05.
GNG5 upregulation mediates the CC-122 resistance rendered by KCTD5 inactivation. (A) Cell fitness assessment of SU-DHL-4 cells with knockout of KCTD5 in the presence or absence of CC-122, compared with control cells by a flow cytometry–based CRISPR competition assay, as described in Figure 3A. Data are shown as the mean ± standard deviation (SD); 2 biological replicates. (B) Heat map showing the CC-122–induced enrichment of DLBCL cells with loss of CRBN or KCTD5 in the CRISPR competition assay. Enrichment score is calculated as the FC of the RFP+/GFP+ ratios of cells treated with 50 µM CC-122 vs their DMSO controls on day 10. (C) Immunoblot analysis of SU-DHL-4-Cas9 cells transduced with lentiviral vectors expressing the indicated sgRNAs. Cells were treated with either DMSO or 10 µM CC-122 for 24 hours. (D) A volcano plot of differentially abundant proteins in response to CC-122 treatment relative to DMSO control. SU-DHL-4-Cas9 cells transduced with lentiviral vectors expressing sgNT-1 or sgKCTD5-7 were subjected to TMT proteomics analysis. The x-axis indicates the log2-FC of each protein in cells expressing sgKCTD5-7 vs cells expressing sgNT-1. P values were corrected for multiple hypothesis testing using the Benjamini-Hochberg method to arrive at an adjusted P value (Padj, also known as an FDR). The y-axis shows the log10 (FDR) values indicating statistical significance, such that proteins lying above the dotted red line are statistically significant findings with Padj < .01. (E) Immunoblot analysis of SU-DHL-4-Cas9 cells transduced with lentiviral vectors expressing the indicated sgRNAs. SU-DHL-4-Cas9 cells were first transduced with a lentiviral vector expressing sgNT-1, sgGNG5-7, or sgGNG5-8, and then selected by 400 µg/mL hygromycin for 10 days. The selected cells were then transduced with a lentiviral vector expressing sgNT-1 or sgKCTD5-7 followed by selection with puromycin (2 µg/mL) for 3 days. (F) Cell fitness assessment of SU-DHL-4 cells with knockout of KCTD5, GNG5, or both in the presence or absence of CC-122, compared with control cells, by flow cytometry–based CRISPR competition assay. SU-DHL-4-Cas9 cells stably expressing sgNT-1, sgGNG5-7, or sgGNG5-8 were transduced with a lentiviral vector coexpressing GFP and sgNT-1 or with a lentiviral vector coexpressing RFP and sgNT-1 or sgKCTD5-7. Three days after infection, RFP+ and GFP+ cells were mixed at a 1:1 ratio and treated with DMSO or CC-122 at the indicated concentrations. The change in the RFP+/GFP+ ratio was then monitored by flow cytometry at day 7. RFP+/GFP+ ratios of cells after transduction with the indicated sgRNA were first normalized to their RFP+/GFP+ ratios on day 0 and then to that of DMSO controls at day 7. (A,F) Data were analyzed by 2-tailed unpaired Student t test. *P < .05; **P < .01; NS, P > .05.
KCTD5 is a BTB/POZ domain–containing protein, which has been proposed to act as a substrate adaptor of cullin-3 RING E3 ligase.26 Recently, Brockmann et al reported that the CRL3KCTD5 ligase complex destabilizes the Gβγ subunits GNB1 and GNG5 to reduce the phosphorylation of AKT at Ser 473, leading to suppression of the AKT pathway.27 To test whether the diminished growth inhibitory effect of CC-122 in KCTD5-deficient SU-DHL-4 cells is attributable to the stabilization of Gβγ proteins and consequent activation of the AKT signaling, we performed quantitative proteomic profiling of SU-DHL4-Cas9 cells expressing a nontargeting sgRNA (sgNT-1) or a KCTD5-specific sgRNA (sgKCTD5-7) (supplemental Figure 4C). GNG5 was the only protein that showed a more than twofold increase in abundance with statistical significance (Figure 4D; supplemental Table 6), and stabilization of GNG5 was confirmed by immunoblot analysis (Figure 4E). Moreover, GNG5 accumulation upon depletion of KCTD5 was observed in HT cells, another DLBCL cell line that displayed resistance to CC-122 after KCTD5 inactivation (supplemental Figure 4D). In contrast, KCTD5 knockout did not affect the response to CC-122 in KARPAS-422 cells, which lack the expression of endogenous GNG5 protein (supplemental Figure 4E), suggesting that upregulation of GNG5 in response to KCTD5 depletion decreases the antitumor activity of CC-122 in DLBCL. Indeed, elimination of GNG5 via CRISPR-mediated gene editing restored the response to CC-122 in KCTD5-deficient SU-DHL-4 cells (Figure 4E-F).
To verify whether KCTD5 knockout inhibits the AKT signaling pathway, we measured the level of phospho-Ser 473 AKT in HT, KARPAS-422, and SU-DHL-4 cells, with or without KCTD5 inactivation. However, a significant change in phosphorylation of AKT at Ser 473 was not detected in any of the 3 DLBCL cell lines tested (supplemental Figure 4D-F) or in the presence or absence of CC-122 treatment (supplemental Figure 4G). In contrast, knockout of PTEN with 2 different sgRNAs substantially increased the level of phospho-Ser 473 AKT in SU-DHL-4 cells (supplemental Figure 4F), suggesting that it is unlikely that CC-122 resistance is conferred by KCTD5 loss owing to the inhibition of the AKT signaling, as previously reported in other human cell lines.27
Inactivation of AMBRA1 or RFX7 renders cell line–specific resistance to CC-122
RFX7 and AMBRA1 were among 52 top-ranked genes with multiple sgRNAs that showed significant enrichment by CC-122 in the CRISPR screening (Figure 2B-C). In the CRISPR competition assay, knockout of RFX7 or AMBRA1 using 2 different sgRNAs significantly reduced the growth inhibitory effect of CC-122 in SU-DHL-4 cells, aligning with the CRISPR screening result (Figure 5A; supplemental Figure 5A-C). Further validation of this finding in 3 additional DLBCL cell lines HT, RIVA, and U-2932, showed that AMBRA1 knockout also diminished the activity of CC-122 in U-2932 cells, whereas ablation of RFX7 failed to display any effect on CC-122 response in all 3 tested cell lines (Figure 5B; supplemental Figure 4D; supplemental Table 7).

Loss of RFX7 or AMBRA1 attenuates the response to CC-122. (A) Cell fitness assessment of SU-DHL-4 cells with knockout of RFX7 or AMBRA1 in the presence or absence of CC-122, compared with control cells by flow cytometry–based CRISPR competition assay as described in Figure 3A. Data are shown as the mean ± SD; 2 biological replicates. Data were analyzed by 2-tailed unpaired Student t test. *P < .05; **P < .01; ***P < .001; NS, P > .05. (B) Heat map showing the CC-122–induced enrichment of DLBCL cells with loss of CRBN, RFX7, or AMBRA1 in the CRISPR competition assay. Enrichment score is calculated as FC of the RFP+/GFP+ ratios between cells treated with 50 µM CC-122 vs their DMSO controls on day 9, 10, or 11, as indicated. (C) Immunoblot analysis of SU-DHL4-Cas9 cells transduced with lentiviral vectors expressing the indicated sgRNAs. Cells were treated with either DMSO or CC-122 at the indicated concentrations for 24 hours. (D) Flow cytometry of SU-DHL-4 cells with expression of the indicated sgRNAs. Cells were continuously treated with DMSO or 50 µM CC-122 for 11 days, followed by staining with annexin V and 7-AAD.
Loss of RFX7 or AMBRA1 attenuates the response to CC-122. (A) Cell fitness assessment of SU-DHL-4 cells with knockout of RFX7 or AMBRA1 in the presence or absence of CC-122, compared with control cells by flow cytometry–based CRISPR competition assay as described in Figure 3A. Data are shown as the mean ± SD; 2 biological replicates. Data were analyzed by 2-tailed unpaired Student t test. *P < .05; **P < .01; ***P < .001; NS, P > .05. (B) Heat map showing the CC-122–induced enrichment of DLBCL cells with loss of CRBN, RFX7, or AMBRA1 in the CRISPR competition assay. Enrichment score is calculated as FC of the RFP+/GFP+ ratios between cells treated with 50 µM CC-122 vs their DMSO controls on day 9, 10, or 11, as indicated. (C) Immunoblot analysis of SU-DHL4-Cas9 cells transduced with lentiviral vectors expressing the indicated sgRNAs. Cells were treated with either DMSO or CC-122 at the indicated concentrations for 24 hours. (D) Flow cytometry of SU-DHL-4 cells with expression of the indicated sgRNAs. Cells were continuously treated with DMSO or 50 µM CC-122 for 11 days, followed by staining with annexin V and 7-AAD.
To discern the underlying mechanisms by which RFX7 and AMBRA1 modulate the response to CC-122, we assessed the gene knockout effect of RFX7 and AMBRA1 on IKZF1/3 degradation and apoptosis induction in SU-DHL-4 cells upon exposure to CC-122. Ablation of neither RFX7 nor AMBRA1 appeared to affect CRBN expression level or CC-122–induced IKZF1/3 degradation (Figure 5C; supplemental Figure 5B). However, a significant reduction of annexin V+/7-AAD+ cell populations after loss of RFX7 or AMBRA1 was evident in SU-DHL-4 cells (Figure 5D; supplemental Figure 5E), suggesting that the alleviation of CC-122–induced apoptosis may at least partially contribute to CC-122 resistance mediated by RFX7 or AMBRA1.
Discussion
In this work, we demonstrated that the degradation of IKZF1 and IKZF3 is both necessary and sufficient to explain the cell-autonomous activity of CC-122 in DLBCL cell lines. In addition, through a genome-wide CRIPSR screening, we elucidated the mechanism of CC-122 resistance in DLBCL cell lines and identified genes and pathways that, when they go awry, may affect the response to CC-122 independent of IKZF1/3 degradation in patients with DLBCL.
Unbiased genome-wide CRISPR/Cas9 screens have been used to define the mechanisms of resistance of multiple cereblon modulators targeting IKZF1 and IKZF3.18,28,29 Our CC-122 CRISPR/Cas9 screening in SU-DHL-4 cells identified several genes that have been found to confer resistance to IKZF1/3 degraders in multiple myeloma and primary effusion lymphoma cell lines. Of the 13 overlapping hits from our work and the lenalidomide resistance screening in MM1S myeloma cell line reported by Sievers et al,18 12 are known to mediate or modulate the degradation of IKZF1/3 induced by cereblon modulators, which demonstrates the robustness of CRISPR screens for identifying common resistance mechanisms of cereblon modulators and supports the reproducibility and authenticity of novel resistance mechanisms specific to CC-122 in DLBCL that were revealed in this screening.
Hyperactivation of the canonical and noncanonical NF-κB signaling pathways upon loss of CYLD, NFKBIA, TRAF2, and TRAF3 significantly blunted the antiproliferative effects of CC-122 without any effect on IKZF1/3 degradation in most of the DLBCL cell lines tested. Assessment of the NF-κB activity after a short-term (0.5-24 hours) or long-term (days 3-10) treatment with CC-122 indicated that CC-122–induced IKZF1/3 degradation may not directly or indirectly modulate the activity of the central NF-κB signaling regulators. NF-κB activation significantly reduced the induction of apoptosis by CC-122, most likely owing to the general prosurvival effects of the NF-κB signaling. Ablation of TRAF2 was also reported to cause resistance to lenalidomide in MM1S cells without an exploration of the potential mechanisms of resistance.26 Based on the results of our work, it is possible that NF-κB prosurvival activity can directly counteract the apoptotic signal induced by lenalidomide in myeloma.
It was reported that the activation of the NF-κB transcription complex was detected in more than 95% of activated B-cell–like (ABC) and ∼47% germinal center B-cell–like (GCB) DLBCL cases, some of which are associated with somatic mutations in NF-κB pathway genes.30 Loss-of-function lesions in the NF-κB–repressor genes CYLD, NFKBIA, TRAF3, TRAF2, TNFAIP3, and NFKBIE, as well as highly recurrent gain-of-function mutations in MYD88, CD79, and CARD11, were commonly found in DLBCL biopsy sspecimens,31 and roughly 15% of the DLBCL biopsy specimens and cell lines were reported to carry either deletion or mutations in TRAF3.32 However, CC-122 monotherapy for R/R DLBCL showed potent immune modulation and clinical activity in both the ABC and GCB subtypes.11,12 These findings suggest that NF-κB hyperactivation alone in malignant B cells may not be sufficient to confer complete resistance to CC-122 in patients with DLBCL, and additional genetic lesion(s), when combined with NF-κB activation mutations, could result in more substantial resistance to CC-122, more specifically in GCB-DLBCL than in ABC-DLBCL. It is also likely that the antitumor activity of CC-122 in ABC-DLBCL is mainly driven by immunomodulatory effects, as opposed to direct cell-autonomous killing in GCB-DLBCL, thereby masking the resistance associated with NF-κB hyperactivation. Stratification of DLBCL patients by using a multigene classifier composed of genetic alternations including NF-κB activation mutations, ABC vs GCB signature genes, and genes associated with immunomodulation may be applicable for the enrichment of CC-122 responders.
The CRISPR screening hits also identified several genes, including KCTD5, RFX7, and AMBRA1, which, when knocked out, abrogated the response to CC-122 in a cell line–specific manner. Accumulation of Gβγ subunit GNG5 upon depletion of KCTD5 diminished the growth inhibitory effect of CC-122 in several DLBCL cell lines. Although KCTD5 was shown to suppress the AKT signaling in multiple solid tumor cell lines, we did not observe any obvious effect of KCTD5 loss on AKT signaling in DLBCL cell lines. Further investigation is needed to identify additional downstream component(s) of the GNG5–mediated GPCR signaling that regulate(s) the CC-122 response in DLBCL. Loss of RFX7 conferred resistance to CC-122 in SU-DHL-4 but not in the other DLBCL cell lines tested. RFX7, a member of the regulatory factor X family of winged helix transcription factors, was found to suppress lymphomagenesis in mice, and its expression level was substantially downregulated in malignant cells of human DLBCL.33 Rare truncation mutations in RFX7 were also identified in Burkitt lymphoma.34 It remains to be determined whether and how RFX7 modulates the CC-122 response in patients with DLBCL or other B-cell lymphomas. Last, elimination of the proautophagic protein AMBRA135 partially blocked the CC-122–induced apoptosis specifically in SU-DHL-4 and U-2932 cells. AMBRA1 was found to promote the dephosphorylation and degradation of c-Myc, thereby suppressing cell proliferation and tumorigenesis.36 It will be interesting to explore whether the modulation of CC-122 response by AMBRA1 is also mediated by c-Myc or other downstream targets.
In summary, we identified general and cell line–specific genes that are associated with CC-122 resistance in DLBCL. Characterization of additional candidate genes identified in our CC-122 CRISPR screening may reveal other novel resistance mechanisms. Integration of DNA, RNA, and protein expression profiling in patients with DLBCL with the findings shown in this work will shed further light on the molecular underpinnings of CC-122 response and resistance.
The data reported in this article have been deposited in the Gene Expression Omnibus database (accession number GSE158303).
For data sharing requests, please contact the corresponding author (zhongying.mo@bms.com).
Acknowledgments
The authors thank Chin-Chun Lu, Yifeng Xia, Xinde Zheng, Andy Christoforou, Antonia Lopez-Girona, Michael Pourdehnad, Marc Casillas, Depinder Khaira, and Rajkumar Balakrishnan for helpful discussion and/or technical assistance.
Authorship
Contribution: G.L. and Z.M. designed the experiments and analyzed the data; Z.M., R.M., S.N., and A.S.-T. performed the cell cultures, flow cytometry competition assay, immunoblot analyses, and qRT-PCR; S. Wood and S. Weng performed the genome-wide CRISPR-Cas9 screening; I.S.J. processed and analyzed the CRISPR screening data; Z.M. and S.N. performed the NF-κB activity assay; R.M. and S. Weng performed the cell proliferation assays; Z.M., S.N., and R.M. prepared the mRNA sequencing samples; C.F. and M.K. analyzed the mRNA sequencing data; S. Wood, R.M., and A.S.-T. prepared the plasmids and lentivirus; Z.M. prepared the proteomic profiling samples; J.M.B. and M.S. performed the mass spectrometry proteomic profiling analysis; G.L. conceived the work; G.L., K.W., and M.R. supervised the work; Z.M. wrote the first draft of the manuscript; and all authors edited and reviewed the manuscript.
Conflict-of-interest disclosure: All authors are, or have been, employees and/or equity holders of Bristol Myers Squibb.
Correspondence: Zhongying Mo, Bristol Myers Squibb, 10300 Campus Point Dr, Suite 100, San Diego, CA 92121; e-mail: zhongying.mo@bms.com; and Gang Lu, Bristol Myers Squibb, 10300 Campus Point Dr, Suite 100, San Diego, CA 92121; e-mail: gang.lu@bms.com.

