

Abstract
Since the early days of vaccination, targeted immunotherapy has gone through multiple conceptual changes and challenges. It now provides the most efficient and up-to-date strategies for either preventing or treating infections and cancer. Its most recent and successful weapons are autologous T cells carrying chimeric antigen receptors, engineered purposely for binding cancer-specific antigens and therefore used for so-called adoptive immunotherapy. We now face the merger of such achievements in cell therapy: using lymphocytes redirected on purpose to bind specific antigens and the clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) revolution, which conferred genome-editing methodologies with both safety and efficacy. This unique affiliation will soon and considerably expand the scope of diseases susceptible to adoptive immunotherapy and of immune cells available for being reshaped as therapeutic tools, including B cells. Following the monumental success story of passive immunotherapy with monoclonal antibodies (mAbs), we are thus entering into a new era, where a combination of gene therapy/cell therapy will enable reprogramming of the patient’s immune system and notably endow his B cells with the ability to produce therapeutic mAbs on their own.
Introduction
Various strategies are currently available for passive immunotherapy, notably with monoclonal antibodies (mAbs) mimicking endogenous immunoglobulins for targeting an antigen (Ag), and thus well tolerated. Active immunotherapy is, on the contrary, based on the patient’s own immune system, as after vaccination. Finally, adoptive immunotherapy reshapes autologous Ag-specific cells on purpose and is now to be boosted by new genetic engineering methods. Although future chimeric antigen receptors (CAR) T-cell protocols will likely replace lentiviral expression by clustered regularly interspaced short palindromic repeats (CRISPR)–mediated retailoring of T-cell receptor (TCR) genes, gene editing could also be applied to other cell lineages, especially B cells.
B cells provide the best suited immunoglobulin factory for producing either membrane-bound or secreted immunoglobulin in lymphocytes or plasma cells (PCs). In lymphocytes, membrane immunoglobulin provides the Ag-binding component of the B-cell receptor, which, on Ag sensing and presentation, triggers immunoglobulin class switching and affinity maturation before activated cells differentiate into immunoglobulin-secreting PCs.
The modular architecture of immunoglobulin was synthetically remodeled under multiple formats: single-chain (sc) fragments, minibodies, bi-specific Abs, and immunotoxins. Gene engineering methodologies now make it doable to express such retailored immunoglobulin in primary B cells, the in vivo use of which might then address multiple unmet health needs. Endogenously synthesized mAbs would notably be valuable in situations needing either (1) lifelong treatment (autoimmune, inflammatory, infectious, genetic, or residual cancer diseases), (2) permanent infusion (circumventing the issues of pharmacodynamic variations seen with intravenous injection and of rapid in vivo catabolism seen with bi- and trispecific mAbs), (3) local delivery in sites where PCs are homing, and/or (4) efficient expression despite unfit structure (for mAbs affected by chemistry, manufacturing, and control issues because of nonoptimal structures).
This review provides an overview of such recent promising advances for adoptive immunotherapy.
Immunotherapy from the origins
Active immunotherapy began centuries ago with variolation to immunize people against smallpox and led to the concept of vaccination with viruses closely related to a pathogen but attenuated or nonpathogenic (Figure 1). Many vaccines now consist of purified or synthetic microbial components or simply nucleic acids encoding them. Recombinant viruses also provide platforms for developing new vaccines against emerging pathogens such as the recently arisen SARS-CoV-2.

A timeline of the history of immunotherapy. Although based on old medical practices such as variolation, the progresses of immunotherapy methods have strongly accelerated in the recent years with multiple discoveries concerning mostly antibodies (in blue), antigens and cellular immunity (in orange), and more recently genome edition (in red). Although this figure mentions important milestones and notably Nobel Prizes, all these progresses have clearly resulted from the joint and incremental efforts of multiple scientists and medical teams throughout the world.
A timeline of the history of immunotherapy. Although based on old medical practices such as variolation, the progresses of immunotherapy methods have strongly accelerated in the recent years with multiple discoveries concerning mostly antibodies (in blue), antigens and cellular immunity (in orange), and more recently genome edition (in red). Although this figure mentions important milestones and notably Nobel Prizes, all these progresses have clearly resulted from the joint and incremental efforts of multiple scientists and medical teams throughout the world.
Passive immunotherapy reached an initial milestone with a Nobel Prize in Medicine (1901), awarded to Behring for serotherapy of diphtheria, based on the administration of serum from convalescent patients. Besides infections, anti-rhesus D immunoglobulin G (IgG) from immunized donors are also widely administrated to mothers after delivery to prevent alloimmunization. Finally, passive immunotherapy strategies now include a huge array of recombinant mAbs targeting tumor or microbial Ag for specifically treating multiple disorders.
Cytokines can also be used to modulate immune responses, and inversely, mAbs are available for counteracting the action of tumor necrosis factor α or interleukin-6 (IL-6) in inflammatory conditions, notably those resulting from adoptive immunotherapy.
Cell therapy began in the 1950s for treating leukemia with bone marrow transplantation, which became safer after the discovery of the human leukocyte antigen system. Such allogenic transplants often associate with graft-versus-host disease, which can now be controlled and used for its graft-versus-tumor effects.
Cancer therapy can also make use of autologous tumor-infiltrating lymphocytes,1 which notably proved efficient for treating melanomas. Recently, it became possible to engineer T cells expressing CAR T cells),2 and use of immune cells generated from induced pluripotent stem cells also emerged.3,4 Although such therapies become a new standard, using other lineages could expand the spectrum of immunotherapies, and B cells are specifically attractive in this regard, given their capacities to produce large amounts of immunoglobulin and to support immune memory.
Recent developments in immunotherapy
Although antibodies and immune cells are the most specific tools for immunotherapy, new strategies for manipulating their production are further expanding the spectrum of their applications (Figure 2).
Recent developments in mAb therapy
mAbs provide the largest class of biomedicines for treating cancers, infections, and autoimmunity, and their efficacy constantly improves. Murine mAbs have been largely replaced by less immunogenic chimeric, humanized, or even entirely human mAbs. Abs can be conjugated with cytotoxic drugs and with other functional proteins for conveying them to specific targets. The improved targeting of antibody-drug conjugates translates into lower toxicity of the attached drug. Antibody-drug conjugate specificity can even be increased by making use of bispecific mAbs. Strategies for enhancing mAb stability are also available either through optimized binding to the neonatal Fc receptor (FcRn) or through conjugation with hydrophilic polymers such as polyethylene glycol. Reciprocally, a classical strategy to extend the half-life of recombinant proteins and eventually strengthen their immunomodulating properties is to fuse them with an IgG Fc domain that notably results in their recycling by the FcRn. Etanercept, a soluble tumor necrosis factor receptor (TNFR)-Fc; abatacept, a soluble cytotoxic T-lymphocyte–associated protein 4 (CTLA4)-Fc; and luspatercept (activinRIIb-Fc) are such immunomodulatory fusion proteins.

New-generation mAb structures. Because of biological engineering, multiple structures of antibodies exist today. Therapeutic mAb structures are increasingly humanized, some are only fragments (ScFv, nanobody), and others are combined with drugs such as toxins, enzymes, radioelements, or with chemicals like polyethylene glycol. There are also Abs able to bind 2 or 3 different targets at the same time named BiTE (bispecific T-cell engager), BiKE (bispecific NK-cell engager), TriKE (trispecific NK-cell engager), quadroma, Knob-into-hole, CrossMAbVH-VL, CrossMAbCH1-CL, CrossMAbFab, and dual variable domain IgG (DVD-IgG).
New-generation mAb structures. Because of biological engineering, multiple structures of antibodies exist today. Therapeutic mAb structures are increasingly humanized, some are only fragments (ScFv, nanobody), and others are combined with drugs such as toxins, enzymes, radioelements, or with chemicals like polyethylene glycol. There are also Abs able to bind 2 or 3 different targets at the same time named BiTE (bispecific T-cell engager), BiKE (bispecific NK-cell engager), TriKE (trispecific NK-cell engager), quadroma, Knob-into-hole, CrossMAbVH-VL, CrossMAbCH1-CL, CrossMAbFab, and dual variable domain IgG (DVD-IgG).
There are still some limitations with mAb therapy, such as the treatment escape or the formation of aggregates,5 and we thus need next-generation strategies delivering Abs with modulated half-life, effector properties, biodistribution, and toxicity. This also includes functional Ab fragments, monovalent Fabs or bivalent F(ab′)2, single-domain Abs (nanobodies), and single-chain variable fragments (ScFv). Nanobodies composed of VL, VH, or VHH (ie, the type of V domain naturally found in some sc camelid Abs) are small-size molecules that remain as specific as conventional Abs. They are highly soluble, do not aggregate, and efficiently reach poorly vascularized tissues, and, in the absence of effector domains, they mostly act as antagonists or allosteric inhibitors.6
ScFvs are composed of linked VH and VL domains, eventually combined as dimers (diabody), trimers (tribody), or even tetramers (tetrabody), to increase their avidity for the target. Associating different ScFvs can cumulate their specificities, as for bispecific T-cell engagers (BiTEs) aimed at bridging target with effector cells. Blinatumomab, for example, bridges CD19+ target cells with cytotoxic T cells, and a trispecific Ab was proposed to target myeloma cells together with CD3 on T cells and the costimulatory CD28.7 Similarly, natural killer cell engagers (NKCEs) associate anti-CD16 binding NK cells, with 1 (BiKE) or 2 (TriKE) other scFvs specific for cancer cells. Some TriKEs additionally bind a cytokine, enhancing NK activity.8 Instead of 3 Fabs, another format of NKCEs includes an Fc domain, naturally binding CD16, together with an anti-NKG2A checkpoint inhibitor Fab further increasing NK activity by blocking the NKG2A/MHC class I inhibitory signal.9 Being smaller than regular mAbs, such next-generation Abs can reach a broader biodistribution.10
Multivalent Abs are also attractive next-generation weapons against pathogens and notably brought broad anti-HIV specificity and protection in a nonhuman primate (NHP) model.11
Current stage of cell therapy with retargeted T cells (CAR T cells)
CAR T-cell therapy is the latest success story in cell therapy. It is based on the forced expression of a new Ag-binding receptor able to activate transduced or transfected primary T cells against a given Ag (usually a tumor Ag).12 Additional modifications of engineered T cells were also proposed to ensure local secretion by T cell of a soluble anticancer molecule, using CAR T cells as micro-pharmacies.13
As for CAR T cells tisagenlecleucel (Kymriah; Novartis) and axicabtagene ciloleucel (Yescarta; Gilead), approved in 2017 against CD19, current CAR T cells mostly rely on retroviruses and lentiviruses. Although there is no report of oncogenic insertion in the case of CAR T cells, such vectors still carry this potential risk, calling for the development of safer gene delivery strategies. Electroporation is a common mean for introducing naked DNA, RNA, or proteins into cells, and CAR T cells were indeed also obtained after simple plasmid electroporation but only with transient expression.14,15 DNA insertion and stable expression with rarer oncogenic integration than with retroviruses were also obtained using transposase-based systems.16,17
DNA mini-circles (devoid of bacterial DNA) are also efficient vectors and have yielded functional CAR T cells persisting in vivo for more than 28 days.18 They, however, still carry a risk of random genomic insertion.
Electroporation of mRNA provides an alternative to DNA and can yield expression greater than 90%, with 80% cell viability.19 Although restricted to transient (<1 week) expression, this is hereby attractive in terms of safety, with no risk of oncogenic integration and obviating the need for any suicide safety system for eliminating transfected cells in case of side effects.20 It, however, remains to be demonstrated whether such transient CAR expression would yield cancer remission.21
Although simple electroporation of nucleic acids is costly in terms of cell viability, use of nanoparticles captured through endocytosis was recently used for improving CAR T-cell transfection.22 This strategy could also be applied for mRNA transfection.23 Altogether, CAR T-cell therapy still carries limitations related to efficiency, persistence of engineered T cells, safety, and pricing. Future protocols resolving these limitations and notably including precise genome edition will crucially help to broaden their applications.
Cell therapy in the era of precise genome editing
Although random genomic insertion carries safety issues, new genome editing tools now make it possible to induce on-purpose mutations, deletions, and insertions. These tools, especially the CRISPR/Cas9 system and its variants, are entering into therapeutic applications at least based on their ex vivo use.24,25 Precise genome edition thus begins to be developed for safer generation of CAR T cells by targeting the TCR loci, simultaneously disrupting endogenous TCR expression and bringing expression of a specific CAR.
Recent developments of gene editing tools.
After zinc finger nucleases and transcription activator-like effector nucleases first provided DNA scissors and were efficiently used for TCR gene edition,26-28 CRISPR/Cas9 has become the most efficient and versatile system for genome engineering via RNA-guided cleavage.29 The CRISPR/Cas9 toolbox now uses single guide RNA molecules, combining both RNA molecules necessary for initiating a specific cleavage,30 and the Cas9 nuclease delivered to cells by DNA or mRNA transfection or simply as a protein.31 Variants of this system such as nickases, dead Cas9 binding DNA without cleavage, or concomitant use of Cas9 inhibitors can further improve specificity or promote precise base replacements.32 Cas9 breaks can be repaired by homology-directed repair (HDR) and then promote precise integration of a template DNA flanked with adequate homology arms.33,34 These technologies are in constant development and strongly expand the possibilities of manipulating DNA, with a huge diversity of potential applications.
CRISPR edition in cell therapy.
CRISPR tools recently allowed to insert a CD19-specific CAR in the T-cell receptor alpha (TCRA) locus, improving both CAR expression, cytotoxicity, and persistence of functional CAR T cells in a preclinical model.35 Successful TCR gene replacement was then obtained both with an adeno-associated virus (AAV), which favors HDR,36 and with a simple naked DNA template.37 The possibility to simultaneously target several loci with CRISPR also opens the way for multiengineered universal off-the-shelf CAR T cells (designed for tolerance by various recipients and limited graft-versus-host disease). Ren et al38 notably succeeded in simultaneous CRISPR inactivation of TCR, B2M, and PDCD1 genes, generating universal CAR T cells without human leukocyte antigen class I expression and capable of bypassing the PD-1 checkpoint inhibition.
The current challenge of CRISPR engineering in human T cells is to increase the proportion of successfully transformed cells, which is currently below that reached after viral transduction. Multiple attempts to improve HDR efficiency during CRISPR edition are thus currently tested.39,40 Cas9 variants with higher fidelity might also reduce off-target genomic lesions.41 Such developments should more efficiently yield next-generation CAR T cells in a context where this therapy is increasingly used. Beyond the remaining challenges of mastering side effects and safety issues, it is also tempting to explore gene edition strategies for the therapeutic use of other types of human lymphoid cells and notably B cells.
Future immunotherapy strategies from the immunoglobulin/B-cell side
Gene therapy for in vivo mAb production (vectored immunotherapy)
Vectored immunotherapy with viruses.
Although retro/lentiviruses have a known risk of oncogenesis,42 AAVs currently stand as convenient vectors for gene delivery, neither integrating into the host genome nor associating with any disease, and successful therapy based on AAVs was demonstrated against hemophilia.43,44 Applications to mAb delivery, termed vectored immunoprophylaxis, emerged for various infectious pathogens such as HIV45 (Figure 3) and proved efficient in mice against Plasmodium falciparum and Ebola virus and in monkeys against Simian immunodeficiency virus.46-48 Vectored immunoprophylaxis might also treat tumors and increased survival was notably obtained with AAV-encoded trastuzumab in mice carrying HER2+ tumors.49
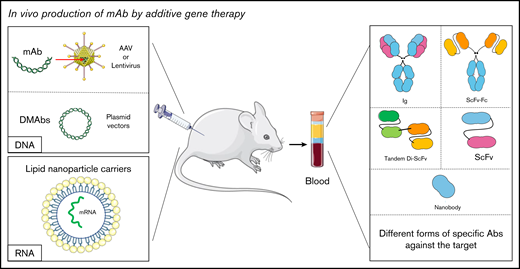
In vivo production of mAb by additive gene therapy. The mAbs can be produced by direct or indirect injection (using viruses such as lentiviruses or AAVs) of DNA encoding the desired antibody. They can also be produced by the injection of mRNA carried by lipid nanoparticles. mAbs are then detected in biological fluids. It is possible to obtain different antibody formats such as a complete immunoglobulin, an ScFv, or a nanobody depending on the original sequence.
In vivo production of mAb by additive gene therapy. The mAbs can be produced by direct or indirect injection (using viruses such as lentiviruses or AAVs) of DNA encoding the desired antibody. They can also be produced by the injection of mRNA carried by lipid nanoparticles. mAbs are then detected in biological fluids. It is possible to obtain different antibody formats such as a complete immunoglobulin, an ScFv, or a nanobody depending on the original sequence.
The limited packaging capacity of AAVs has prompted to improve the design of cassettes coding for mAb chains. Addition of a self-processing P2A peptide allowed to encode both H and L chains using a monocistronic cassette.50 Several other formats of mAb-encoding cassettes were expressed from AAVs, but the frequent anti-AAV immune response constitutes a brake for clinical applications, together with antidrug antibodies targeting the therapeutic mAb. Despite such limitations, the AAV platform remains an interesting, vectored immunotherapy option.
Naked DNA-encoded mAbs.
Naked DNA-encoded mAbs (DMAbs) stand as another platform for mAb in vivo delivery. It is relatively safe because naked DNA is neither infectious nor immunogenic by itself. Many studies thus used DMAbs to treat infection51-57 or cancer.58,59 This strategy was initially limited by low expression, but delivery and expression have now been optimized.60
Two mAb formats, either complete or restricted to the Fab, were compared by the DMAb strategy in the context of chikungunya infection, with the former providing the best immunity.53 Although this strategy can rely on a single DNA fragment linking H and L chain sequences with a P2A site, simultaneous injection of several DMAbs was also shown possible.51,52,54 In both influenza and HIV infection, combined DMAb yielded immunity.52,54 Muthumani et al53 highlighted another combination strategy by simultaneously injecting a DNA vaccine encoding the CHIKV envelope and a DMAb that neutralizes the CHIVK. Patel et al,56 for their part, successfully used DMAb to produce a bispecific mAb that targets 2 proteins essential to Pseudomonas aeruginosa pathogenicity. Preliminary experiments in NHPs with DMAb targeting the Zika virus suggested that this could be translatable to humans because it raised sufficient Ab levels for controlling viral load.56 DMAb targeting HIV also showed strong in vivo IgG expression in NHP.52
Besides infections, this strategy is pertinent to oncology and demonstrated significant antitumor activity in vivo (being as effective as a conventional mAb) for controlling tumor growth,58,59 increasing CD8 T-cell infiltration, and decreasing the infiltration of T regulatory cells into tumors.58
DMAb can thus be of interest for short- and medium-term treatment of various pathologies by rapidly and efficiently supporting the production of specific mAbs.55 Repeated DMAb injections are, however, needed when prolonged treatment is required. Safety issues also finally remain associated with the potential risk of oncogenic random genomic insertion.
RNA injection: a safer alternative?.
As for CAR induction, in vivo mAb production has also motivated safe RNA-based approaches (with no risk of genomic insertion, oncogenic hit, or vector immunogenicity). Using lipid nanoparticles as carriers, Pardi et al61 optimized the cytosolic transfer of the mRNA encoding H and L chains of an anti-HIV neutralizing mAb. A single intravenous dose of mRNA-lipid nanoparticles (LNPs) yielded in vivo mAb expression at 170 µg/mL after 24 hours, and weekly administration made it possible to maintain a high mAb concentration. This protected mice against challenge with an HIV derivative and showed that mRNA-LNP coding for mAbs could replace mAb injection. In a recent and encouraging study, Kose et al62 also showed that an mRNA encoding a chikungunya-neutralizing human mAb expressed after a single intravenous injection of mRNA-LNPs decreased the viremia of mice challenged with the virus. Besides, it has also been shown that effective RNA-based approaches can be applied to cancer immunotherapy.63,64
Contrary to DNA injection, this strategy requires repeated administration to maintain a stable mAb level in vivo. Its transient efficacy (as for regular mAb treatment) can thus be a limitation for long-term treatments of chronic diseases, and more trials are still needed to ensure the absence of side effects. Despite this limitation, this drug format is significantly cheaper than proteins because the production of synthetic mRNA therapeutics65 does not require expensive cell culture and purification systems. This is an important aspect, given the very high cost of mAb therapy.
Genome editing for in vivo mAb production
Genome editing technologies have strongly expanded the possibilities of manipulating DNA. When applied to Ab production, they allow more precise control via gene engineering of B-lineage cells.
Although in vivo cell modification therapies might be a future grail, ex vivo modification after cell sorting obviates the need for cell targeting.66 Another advantage is the possibility to analyze and characterize the modified cells before reintroducing them back into the host.67 In the context of mAb therapy, B-lineage cells isolated from peripheral blood or lymphoid organs are perfect targets, because they are the ultimate antibody-secreting cells.68 A system where B cells are isolated from peripheral blood, modified ex vivo for mAb secretion and injected back into the organism (Figure 4), would be an ideal therapeutic strategy with applications for both cancer and viral infection treatment.69
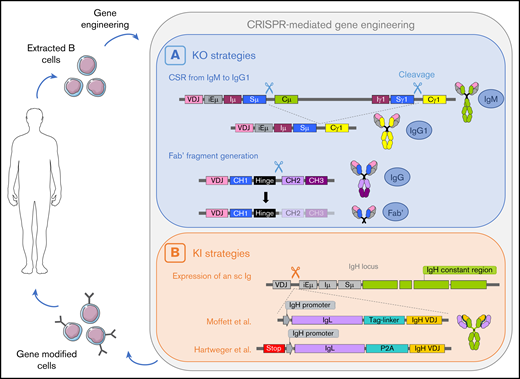
A potential cycle of B cell–mediated therapies including an ex vivo genome edition step. B cells are extracted from the patient, gene-edited using CRISPR/Cas9 technology, and infused back to the patient. (A) Two cleavages can lead to deletion of the Cµ gene and class switch recombination from IgM to IgG.77 One cleavage between the hinge region and the constant region allows the generation of a Fab′ fragment.77 (B) Insertion of a cassette encoding the VH chain and a light chain linked with a tag-linker leads to expression of an sc immunoglobulin with modified specificity.79 Hartweger et al80 described a similar strategy with an additional stop codon to disrupt expression of the endogenous immunoglobulin. iE, enhancer; I, intervening; S, switch; C, constant.
A potential cycle of B cell–mediated therapies including an ex vivo genome edition step. B cells are extracted from the patient, gene-edited using CRISPR/Cas9 technology, and infused back to the patient. (A) Two cleavages can lead to deletion of the Cµ gene and class switch recombination from IgM to IgG.77 One cleavage between the hinge region and the constant region allows the generation of a Fab′ fragment.77 (B) Insertion of a cassette encoding the VH chain and a light chain linked with a tag-linker leads to expression of an sc immunoglobulin with modified specificity.79 Hartweger et al80 described a similar strategy with an additional stop codon to disrupt expression of the endogenous immunoglobulin. iE, enhancer; I, intervening; S, switch; C, constant.
Successful B-cell modification was first obtained using lentiviral transduction methods.70-72 Primary human hematopoietic cells, including B cells, were efficiently transduced with lentiviral vectors encoding anti-HIV broadly neutralizing Abs. Edited cells engrafted and persisted in blood and lymphoid tissues in vivo in humanized mice, efficiently secreting antiviral broadly neutralizing Abs.72
However, nonspecific lentiviral insertions could target regions essential for cell viability or function, compromising clinical applications.69 CRISPR/Cas9 site-specific cleavage can by contrast result in precise insertion by HDR.67,69 Cas9, guide RNA (gRNA), and the repair template for HDR can be brought into target cells as plasmids, mRNAs, or a ribonucleoprotein (RNP).66,69 In primary human B cells, RNPs seem to be highly efficient, whereas Cas9-encoding DNA and mRNA often lead to poor or no DNA cleavage.66,69 To optimize HDR in cells at the S/G2 phases of the cell cycle,34,73,74 various B-cell expansion mixtures were tested,66,69,75 and the optimal B-cell activation cocktail included CD40L, cytosine-phosphate-guanine (CpG), IL-2, IL-10, and IL-15.75
Efficient CRISPR/Cas9-mediated knockout in human B cells was obtained in several conditions.66,69,75,76 Cheong et al77 used lentiviral vectors carrying Cas9 and gRNA sequence. By using 2 gRNAs, 1 specific for a region near Sµ and 1 near Sγ, they induced deletions and efficiently mimicked class switch recombination in both mouse and human primary B cells. They also succeeded in generating Fab′ fragment-secreting hybridomas after deleting the Fc domain-coding region, with secretion at a level comparable to the original complete immunoglobulin. This strategy would simplify the process of producing Fab′ in vitro as proteins, which is currently based mainly on protease cleavage. Recently, an integrase-defective lentiviral vector was also used to target precise insertion of an antibody cassette into the GAPDH gene and yielded efficient expression in plasma cells.78
Expressing transgenic immunoglobulins (ie, H2L2 polymers) in normal B cells in fact involves several challenges: high expression, stoichiometric expression of both chains, and, if possible, disruption of endogenous immunoglobulin genes to minimize the assembly of chimeric immunoglobulin (ie, randomly mixing transgenic and endogenous immunoglobulin chains of unpredictable specificity). It is thus desirable to design transgenic protocols also disrupting endogenous immunoglobulin production. HDR knock-in (KI) at the immunoglobulin locus was thus explored using various strategies. The first successful immunoglobulin gene KI in B cells used an RNP/AAV combination.69,75,79 Reporter cassettes were efficient inserted,69,75 and gene modification for secretion of a survival factor was achieved in human PCs.75 Moffett et al79 engineered the IgH locus to make cells secrete a mAb that bound the respiratory syncytial virus (RSV). Their cassette was introduced upstream of the Eµ enhancer and included a heavy chain promoter, the light (L) chain, a long linker, and the variable region of the H chain. Such an sc strategy both disrupted endogenous IgH chain production and forced an appropriate pairing of the transgenic heavy (H) and L chain peptides. This cassette, followed by a site for splicing of the VDJ exon to the endogenous Cµ gene, thus encoded a complete (although sc) antibody. Electroporation with RNP for cleavage was followed by incubation with AAV providing the KI cassette. This strategy led to efficient expression of engineered immunoglobulin in primary human B cells, later differentiating into PCs and secreting the engineered mAb.
Combination of RNP with double-strand or single-strand (ssDNA) DNA templates was also reported. In mice, a KI cassette for an anti-RSV mAb provided as a double-strand DNA template successfully yielded anti-RSV immunity.79 Transferred cells were then able to differentiate in vivo into both long-lived PCs and switched memory B cells. Greiner et al66 inserted ssDNA templates into the H or L chain loci to engineer B cells producing either mAbs or nanobodies that aimed at neutralizing tumor necrosis factor α. Hartweger et al80 also used an ssDNA template to produce mAbs against HIV-1 and showed them to be functional when produced either by engineered mouse or human B cells. Their strategy simultaneously disrupted the κL chain in primary B cells using RNPs and expressed a transgenic H and L chains cassette, inserted in the first IgH intron (downstream of JH). This cassette began with a stop cassette to interrupt the transcription of the endogenous VDJ, followed by a VH promoter, a sequence encoding Igκ, a P2A cleavage site, and the VDJ region of the transgenic H chain. Splicing of a KI VDJ onto the endogenous constant region (as also cited for the work of Moffett et al79 ) has the advantage to be compatible with eventual class-switch recombination and production of the engineered mAb under various classes.
For all immunoglobulin gene engineering strategies presented thus far, a common issue is the low editing efficiency, especially in primary B cells. A recent paper reported a method to increase editing yield, using nanoparticles and modifications of the HDR template.81 Truncated Cas9 target sequences were added at both ends of the repair template, allowing them to recruit Cas9. Using Cas9 variants coupled with nuclear localization sequences, as a shuttle bringing the template to the genomic DNA, was also used, together with poly-l-glutamic acid to stabilize RNP nanoparticles associated with HDR template. Altogether, these tricks enhanced editing efficiency in different cell types, especially when used jointly. In B cells, it improved editing efficiency by fivefold. Poly-l-glutamic acid even permitted stabilized RNPs to resist freeze-thaw cycles and lyophilization without losing efficiency.
Despite remaining difficulties and challenges, primary B-cell editing is obviously promising and worth efforts. Modified B cells can differentiate into memory B cells and/or Ab-secreting PCs,75,79 and successful differentiation of reinfused modified B cells was reported.82,83 Transferred PCs could eliminate the need for periodic mAb injections,69 required for some current therapies because of limited persistence in the organism.72 In some patients with immune deficiency, it could replace vaccines to protect against infections.69 Contrary to the fixed structure of mAbs, engineered B cells might also be eventually capable of evolving into variants, notably through class switching, when appropriate insertion template is used.80,83,84 Another possible evolution of an adoptive B-cell receptor in edited B lymphocytes might be the entry into new rounds of Ag binding selection and in vivo affinity maturation.79,84 Such a feature would be tremendously helpful for providing durable immunity against mutating antigens such as viruses or cancer cells (evolving during chronic infection or reinfection or cancer relapse). Such adoptive immunotherapy would then dynamically reformat humoral immunity on purpose, by redesigning B-cell specificity while preserving their ability to evolve along successive immune challenges.
Among the methodologic breaks still needed before clinical applications, increasing the amount of mAb secreted by edited B cells is first. In most published models, mAb concentration rarely reached the level needed for immunity and rapidly declined.79 Ex vivo amplification of primary B cells and conditions for their commitment into either short or long-lived survival remain ill defined. Understanding how ex vivo amplification could preserve or strengthen a long-lived commitment will need to be mastered for optimal B-cell engineering after optimized culture. Moreover, as only mouse models were used thus far, experimentation in NHP is required before clinical applications. By better mimicking the human patterns of viral infections or tumor progression, this will notably allow to estimate the number of cells required for a protective immunity and the best conditions for their successful graft.
Another major concern relates to the safety of gene edition in a lineage highly exposed to oncogenic transformation and off-target mutations driven by the activation-induced deaminase activity.79,80,84-86 Cas9 off-target mutations can be reduced by using nickase variants such as D10A85 and then needing 2 adjacent on-target cleavage sites. This strongly reduces the risk of an off-target double-strand break. Mutant Cas9 with reduced off-target activity are also reported.86 Safety issues for B-cell adoptive immunotherapy could also partly be solved by efficient schemes for clearing edited cells in case of undesired side effects. Efficient therapies are available for total B-cell or plasma cell deletion with anti-CD20 or anti-CD38 therapeutic mAbs. More specifically, suicide strategies such as inducible apoptosis by Cas1287,88 were validated in T-cell adoptive immunotherapy protocols and could be applied to edited B cells.
Overall, despite these various efficiency and safety issues, there are already strong preliminary elements showing that adoptive B-cell immunotherapy is feasible and should soon take its part in the therapeutic arsenal, solving a number of unmet needs in human health.
Acknowledgments
This work was supported by Association pour la Recherche sur le Cancer (ARC) Foundation for Cancer Research grant RF20180207070 (M. Cogné) and French National Research Agency grant 18-CE18-0022-02 (M. Cogné).
Authorship
Contribution: N.U., M. Cahen, and M. Cogné conceptualized the study; N.U., M. Cahen, Y.D., and M. Cogné prepared the original draft; N.U., M. Cahen, J.M., C.S., and M. Cogné reviewed and edited the manuscript; M. Cogné supervised the study; and C.S. and M. Cogné provided funding.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Michel Cogné, INSERM U1236, Faculté de Médecine, University of Rennes, 2 Av du Pr. Léon Bernard, 35000 Rennes, France; e-mail: michel.cogne@inserm.fr.

