

Key Points
Low-density PD-1 expression is found on resting NK cells, and scFv PD-1 sequences enhance NK cell cytolytic and cytokine production function.
PD-1 is upregulated on reconstituting NK cells after allo-HSCT.

Abstract
Expression of programmed cell death protein 1 (PD-1) on natural killer (NK) cells has been difficult to analyze on human NK cells. By testing commercial clones and novel anti-PD-1 reagents, we found expression of functional PD-1 on resting human NK cells in healthy individuals and reconstituting NK cells early after allogeneic hematopoietic stem cell transplantation (allo-HSCT). Peripheral blood samples from healthy individuals and transplant recipients were stained for PD-1 expression using the commercial anti-PD-1 clone PD1.3.1.3, fluorescein isothiocyanate (FITC)–labeled pembrolizumab, or an FITC-labeled single-chain variable fragment (scFv) reagent made from pembrolizumab. These reagents identified low yet consistent basal PD-1 expression on resting NK cells, a finding verified by finding lower PD-1 transcripts in sorted NK cells compared with those in resting or activated T cells. An increase in PD-1 expression was identified on paired resting NK cells after allo-HSCT. Blockade of PD-1 on resting NK cells from healthy donors with pembrolizumab did not enhance NK function against programmed death-ligand 1 (PD-L1)–expressing tumor lines, but blocking with its scFv derivative resulted in a twofold increase in NK cell degranulation and up to a fourfold increase in cytokine production. In support of this mechanism, PD-L1 overexpression of K562 targets suppressed NK cell function. Interleukin-15 (IL-15) activity was potent and could not be further enhanced by PD-1 blockade. A similar increase in function was observed with scFv PD-1 blockade on resting blood NK cells after allo-HSCT. We identify the functional importance of the PD-1/PD-L1 axis on human NK cells in which blockade or activation to overcome inhibition will enhance NK cell–mediated antitumor control.
Introduction
Immune checkpoint blockade has revolutionized immunotherapy for the treatment of various malignancies. Programmed cell death protein 1 (PD-1) is of high interest because 1 of its ligands, programmed death-ligand 1 (PD-L1), has been found to be upregulated on many tumors.1,2 PD-1 (CD279) is a type I transmembrane receptor with an immunoglobulin (Ig)–like extracellular domain and a cytoplasmic tail containing an immunoreceptor tyrosine-based inhibitory motif.3 PD-1 has been shown to suppress both the duration and intensity of the immune response to tumor cells, in part to limit damage to surrounding tissues.4,5 Previous studies have identified the inducible expression of PD-1 on B and T cells as a result of BCR or TCR engagement or stimulation through various γ-chain cytokines (ie, interleukin-2 [IL-2], IL-7, IL-15, and IL-21).6,7 Ligation of PD-1 on T cells triggers the phosphorylation of SHP-1/2, which prevents activation of phosphatidylinositol 3-kinase (PI3K) and Akt.3,8,9 This results in increased expression of glucose transporters and upregulation of glycolytic enzyme activity that impedes T-cell division and effector function.3 However, our understanding of PD-1 expression and function is mostly limited to T and B cells.10,11
There are several PD-1 antagonists approved for treating solid malignancies. The 2 most commonly used are Keytruda (pembrolizumab)12,13 and Opdivo (nivolumab),14 which are approved as therapeutics for treating a variety of solid and hematologic malignancies. Despite clinical efficacy, the mechanism of action during the course of treatment requires further elucidation. Data regarding PD-1 checkpoint blockade support a strong role for enhanced T-cell function and a supporting role for B cells,15 but little is understood about its potential role and function on components of the innate immune response, including natural killer (NK) cells.
NK cells constitute an important component of the innate immune response to tumor-transformed cells. Unlike T and B cells, NK cells do not recognize malignant cells through antigen specificity.16 Rather, NK cells recognize cells that have downmodulated major histocompatibility complex class I (MHC-I) molecules 17,18 and upregulate cell stress ligands (eg, UL16-binding proteins and MHC-I chain-related proteins A and B).19-21 NK cells act as first responders to tumor cells by limiting tumor growth and spread until the adaptive immune response has been primed against specific tumor antigens. Because of this important role for NK cells early in tumor surveillance and with the knowledge that several tumors upregulate PD-1 ligands, it may be critically important to determine whether and to what extent PD-1 is expressed on NK cells to better understand how tumors evade the NK cell response. This information might provide insights into how to manipulate NK cells to better kill PD-L1–expressing tumors.
Previous reports on the use of various antibodies suggest that low levels of PD-1 are expressed only on specific NK cell subsets or in response to specific activation.22,23 A study of patients with multiple myeloma showed an increase of PD-1 on all peripheral blood NK cells compared with a lack of expression on NK cells from healthy individuals, but that study used a detection reagent that was ultimately determined by its licenser (CureTech Innovative Therapies) to bind the Notch ligand DLL1 and not PD-1.23 These experiments also indicated a role for the γ-chain cytokine IL-2 in the upregulation of PD-1 on healthy donor NK cells. Another study used a large cohort of cytomegalovirus–seropositive and -seronegative individuals to examine the effects of previous cytomegalovirus infection on PD-1 expression on NK cells.22 However, despite substantial data on PD-1 expression and function on T cells, PD-1 expression on NK cells remains unclear.
In this study, we evaluated PD-1 expression and function on NK cells by using commercially available anti-PD-1 antibodies and a laboratory-generated single-chain variable fragment (scFv) reagent derived from pembrolizumab that is known to bind specifically to PD-1 with high affinity. Our findings indicate a consistent basal expression of PD-1 on all circulating human NK cells and also a strong role for PD-1 suppression of resting NK cell function in response to PD-L1–expressing tumor targets.
Materials and methods
NK cell isolation
Buffy coats collected from healthy donors were obtained from the Memorial Blood Bank (Minneapolis, MN). Peripheral blood mononuclear cells (PBMCs) were isolated by density gradient centrifugation using Ficoll-Paque media (GE Healthcare). NK cells were purified from PBMCs using an NK cell enrichment magnetic bead kit (STEMCELL Technologies). The purity of NK cells was >95%.
Patients and samples
In all, 20 transplant recipients with acute myeloid leukemia (AML) (10 siblings and 10 umbilical cord blood [UCB] recipients) were selected for analysis. PBMCs were collected from transplant recipients at 28, 60, and 100 days posttransplant and were cryopreserved. Baseline samples from sibling donors were also collected. All transplantations were performed at the University of Minnesota. Samples from patients who had received a transplant were acquired after obtaining informed consent and approval from the University of Minnesota Institutional Review Board according to the Declaration of Helsinki.
Cell lines
K562 chronic myelogenous leukemia and THP-1 monocytic cell lines were cultured in RPMI-1640 with 10% fetal bovine serum (Gibco) (hereafter, RPMI-10). Cell lines were purchased from the American Type Culture Collection in 2016. K.PD-L1 cells were generated by transducing K562 cells with a lentiviral vector expressing PD-L1 and then sorting for single clones as previously described.24
Generation of pembrolizumab scFv
The sequences for the variable heavy (VH) and variable light (VL) chains were taken directly from the pembrolizumab (MK-3475) patent. The hybrid gene encoding pembrolizumab was synthesized using DNA shuffling and DNA ligation techniques. The fully assembled gene (from the 5′ end to the 3′ end) encoded an NcoI restriction site, an ATG start codon, pembrolizumab, and an NcoI restriction site. The resulting 747 base pair NcoI/NcoI fragment gene was spliced into the pET28c expression vector under the control of an isopropyl-β-D-thiogalactopyranoside–inducible T7 promoter. DNA sequencing analysis (Biomedical Genomics Center, University of Minnesota, St. Paul, MN) was used to verify that the gene sequence was correct and had been cloned in frame.
qPCR and Octet binding analyses
Cells were homogenized using QIAGEN QIAshredder cell and tissue lysate columns, and RNA was then extracted using the QIAGEN RNeasy Mini Kit according to the manufacturer’s instructions. RNA was treated with Invitrogen DNase I before being quantified using a Thermo Fisher Scientific Nanodrop spectrophotometer. Reverse transcription (RT) was performed using Invitrogen SuperScript IV Reverse Transcriptase. RT quantitative polymerase chain reaction (RT-qPCR) was performed with an Applied Biosystems QuantStudio 5 qPCR machine using TaqMan reagents and primers specific to PD-1 (Thermo Fisher Scientific catalog No. 4331182) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Data that failed 1 or more quality checks were omitted, and normalized reporter (Rn) values were calculated using Applied Biosystems Design and Analysis Software v1.5.1. Data values were then exported, and Graphpad Prism version 8 was used to perform a one-way analysis of variance with a significance cutoff of 95% in which the mean Rn for each sample was tested against the mean Rn value of a no-template control. Octet analysis of label-free pembrolizumab and pembrolizumab scFv for affinity and binding kinetics was performed by Precision Antibody (Columbia, MD). Purified recombinant human PD-1/Fc (R&D Systems, Minneapolis, MN) was used as the binding target.
pAKT activation
Purified NK cells were cultured in a 96-well v-bottom plate in the presence of THP-1 cells for up to 60 minutes with fluorescently labeled anti-CD3 and anti-CD56 antibodies. At the end of the time course, cells were washed and immediately stained for pAKT using the FOXP3 Fix/Perm buffer kit (Thermo Fisher Scientific).
Antibodies and reagents
Fluorochrome-conjugated antibodies were purchased from BioLegend (anti-CD56 BV711 [clone HCD56], CD107a fluorescein isothiocyanate [FITC; clone H4A3], interferon-γ [IFN-γ] BV650 [clone 4S.B3], CD4 BV650 [clone OKT4], CD8 AF700 [clone SK1]), BD Biosciences (anti-CD3 phycoerythrin [PE]-CF594 [clone UCHT1], pAKT (pS473) BV421 [clone M89-61], CellTrace Violet, Live/Dead Near IR), and Miltenyi Biotec (anti-PD-1 PE [clone PD1.3.1.3]. Pembrolizumab (Merck) and pembrolizumab scFv were FITC labeled using the FITC Antibody Labeling Kit (Pierce) according to the manufacturer’s instructions. Cells were analyzed on an LSRII flow cytometer (BD Biosciences), and data were analyzed with FlowJo software (Tree Star Inc.).
CyTOF
Conjugation of heavy metals to a specific antibody was conducted using the Maxpar Antibody Labeling Kit (Fluidigm). The following antibody clones and metal tags were used: CD56 clone N901 176Yb, CD3 clone UCHT1 170Er, CD14 clone M5E2 151Eu, CD4 clone RPA-T4 145Nd, CD8 clone RPA-T8 148Nd, and PD-1 clone EH12.2H7 174Yb.The protocol involves partial antibody reduction using 0.5 M Bond-Breaker TCEP Solution (Thermo Fisher Scientific Product No. 77720) and comprehensive buffer exchange using centrifugal 3 kDa and 50 kDa filter units (Millipore Product No. UFC500396 and No. UFC505096). After conjugation of the antibody, yield was measured and the final reagent was stored in antibody stabilizer (Boca Scientific Product No. 131000). The reagent was then titrated and verified against known flow cytometry antibodies. Cells were stained with Cell-ID Cisplatin (Fluidigm Product No. 201064) followed by barcoding using the Cell-ID 20-Plex Pd Barcoding Kit (Fluidigm Product No. 201060). Cells were combined into a single 5-mL polystyrene U-bottom tube and incubated in the surface marker antibody cocktail for 30 minutes at 4°C. Cells were then fixed using 2% paraformaldehyde. For intracellular staining, cells were permeabilized with Triton X 0.1% for 5 minutes at room temperature, followed by incubation with intracellular antibody cocktail for 30 minutes at 4°C. Stained cells were then incubated overnight with Cell-ID Intercalator (Fluidigm Product No. 201192A). Cells were washed the next morning using either Maxpar PBS (phosphate-buffered saline) (Fluidigm Product No. 201058), Maxpar Cell Staining Buffer (Fluidigm Product No. 201068), or Millipore water at 1600 rpm for 4 minutes. Samples were run on the CyTOF 2 mass cytometry platform (Fluidigm) and analyzed with Cytobank (Cytobank Inc.)
CD107a assay
Purified NK cells or whole PBMCs (where indicated) were cocultured with targets at an effector-to-target ratio of 2:1 in the presence of anti-CD107a. Anti-PD-1 blocking antibodies were added to NK cells for 30 minutes before the addition of target cells at a concentration of 370.5 nM. One hour after target cell addition, golgiStop and golgiPlug (BD Biosciences) were added, and the culture was incubated for another 3 hours. At the end of the 4-hour incubation, cells were stained for viability and NK markers. Cells were fixed in 2% paraformaldehyde in phosphate-buffered saline and permeabilized with 0.1% TritonX solution before intracellular staining for IFN-γ. Samples were analyzed on an LSRII flow cytometer (BD Biosciences).
CD8+ T-cell stimulation assays
CD8 T cells were positively selected from PBMCs using EasySep CD8+ Selection Kit (STEMCELL Technologies) and cultured at 3 × 106 cells per mL in RPMI-10 with 20 U/mL IL-2 and 1 μg/mL pooled peptides derived from cytomegalovirus, Epstein-Barr virus, and flu viruses (MabTech) for 4 days. On day 4, cells were washed and put back into culture for 3 more days without peptide or IL-2. At 12 hours before evaluation, CD8 cells were harvested, washed, and restimulated with 1 μg/mL of the pooled peptide in the presence of pembrolizumab, anti-PD-1, or pembrolizumab scFv. Anti-CD107a FITC was added to all cultures. At the end of the 12-hour incubation, cells were washed, fixed, permeabilized, and stained for IFN-γ. CD107a and IFN-γ expression were then assessed by flow cytometry.
Real-time killing assays
K562 and THP-1 cells were labeled with CellTrace Violet before plating into 96-well plates, and caspase-3 and caspase-7 were added at 5 μM per well. Enriched NK cells were added at an effector-to-target ratio of 2:1 along with the appropriate treatments (at 370.5 nM concentration) and cocultured for 48 hours in RPMI-10. The normalized percentage of live K562 or THP-1 cells was calculated from the number of caspase-3– and caspase-7–negative CellTrace Violet–positive target cells acquired using IncuCyte Zoom software at noted time points and normalized against targets alone and against 0 hours in coculture groups. GraphPad Prism software (GraphPad Software, La Jolla, CA) was used to generate a normalized killing count graph and to conduct one-way analysis of variance statistical tests.
Study approval
Blood from healthy donors was obtained after receipt of written informed consent at the Memorial Blood Bank. Use of PBMCs from donors was approved by the Committee on the Use of Human Subjects in Research at the University of Minnesota (IRB 9709M00134) in accordance with the Declaration of Helsinki.
Results
Peripheral blood NK cells from healthy individuals expressed PD-1
Because of the variances in the staining patterns for PD-1 on NK cells using different commercially available antibodies and fluorophores, we opted to use the clinical reagent pembrolizumab conjugated with FITC to identify PD-1 expression (Figure 1A). The PE-conjugated anti-PD-1 (clone PD1.3.1.3) was the best commercial clone tested (Figure 1B). Staining with this reagent identified a low, but consistent expression of PD-1 on healthy donor NK cells compared with control stains. When using FITC-labeled pembrolizumab, the PD-1 expression seen was similar to that in the commercial clone. However, because FITC-pembrolizumab mean fluorescence intensity was low, a high proportion of stained NK cells overlapped with the isotype control (Figure 1C). It is known that scFv binds differently than intact antibody, so we designed an scFv of the pembrolizumab monoclonal antibody (mAb) (Figure 1A) to determine whether the smaller scFv molecule has better binding and functional activity than the intact mAb. Interestingly, the scFv derivative of pembrolizumab had a higher fluorescence intensity than an equimolar amount of the clinical reagent (Figure 1D). To confirm that the scFv had an affinity for PD-1, we performed an Octet binding assay with immobilized recombinant human PD-1 (Figure 1D). qPCR for PD-1 transcripts was then performed on purified NK and T cells from 3 healthy donors. T cells from these donors either remained unstimulated or were stimulated for 7 days with anti-CD3 and anti-CD28 beads and IL-2. qPCR data for PD-1 are reflective of flow analysis for PD-1 from the same donor lymphocyte subsets (Figure 1E-F). These data indicate that PD-1 identified by the scFv of pembrolizumab is in fact expressed on resting peripheral blood NK cells in healthy donors at levels lower than those on resting T cells and markedly lower than those on activated T cells.
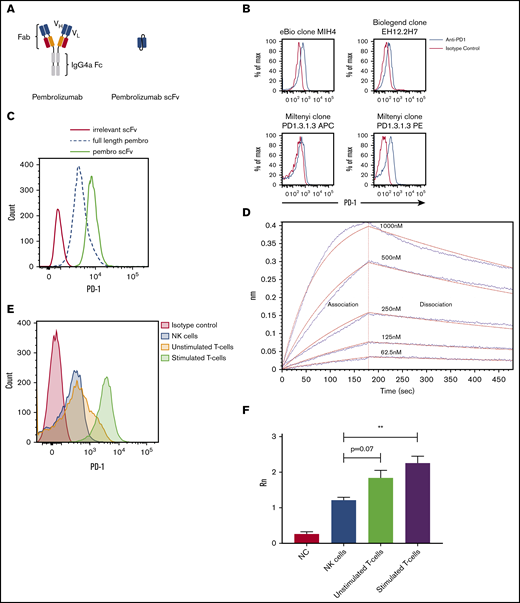
Schematic and binding of anti-PD-1 antibodies on primary NK cells. (A) Pembrolizumab is a complete mAb containing a human IgG4a Fc and 2 PD-1–specific Fab fragments compared with the scFv composed of the VH and VL chains of pembrolizumab connected by a short peptide linker. (B) PD-1 staining of healthy donor NK cells was assessed by using various commercially available antibodies. Shown is a representative example of 10 normal donors. (C) Flow cytometric analysis of PD-1 expression on peripheral blood NK cells from healthy donors is identified by the FITC-labeled pembrolizumab (pembro) or pembrolizumab scFv. (D) Confirmation of pembrolizumab scFv affinity for PD-1 is demonstrated by an Octet binding assay. (E) NK cells and T cells were isolated from healthy donor PBMCs by negative selection. T cells were left unstimulated or stimulated with anti-CD3 or anti-CD28 and IL-2 for 7 days and then stained for PD-1. (F) NK cells and resting and activated T cells were used for qPCR for PD-1 transcripts (n = 3). Error bars indicate the mean ± standard error of the mean (SEM). **P < .01. APC, fluorochrome allophycocyanin; NC, no template control.
Schematic and binding of anti-PD-1 antibodies on primary NK cells. (A) Pembrolizumab is a complete mAb containing a human IgG4a Fc and 2 PD-1–specific Fab fragments compared with the scFv composed of the VH and VL chains of pembrolizumab connected by a short peptide linker. (B) PD-1 staining of healthy donor NK cells was assessed by using various commercially available antibodies. Shown is a representative example of 10 normal donors. (C) Flow cytometric analysis of PD-1 expression on peripheral blood NK cells from healthy donors is identified by the FITC-labeled pembrolizumab (pembro) or pembrolizumab scFv. (D) Confirmation of pembrolizumab scFv affinity for PD-1 is demonstrated by an Octet binding assay. (E) NK cells and T cells were isolated from healthy donor PBMCs by negative selection. T cells were left unstimulated or stimulated with anti-CD3 or anti-CD28 and IL-2 for 7 days and then stained for PD-1. (F) NK cells and resting and activated T cells were used for qPCR for PD-1 transcripts (n = 3). Error bars indicate the mean ± standard error of the mean (SEM). **P < .01. APC, fluorochrome allophycocyanin; NC, no template control.
PD-1 inhibited NK cell cytotoxic and cytokine secreting functions when exposed to PD-L1–expressing tumor targets
To determine the functional role of low-level PD-1 on resting NK cells, we cocultured purified NK cells from healthy donors with tumor targets, displaying varying levels of PD-L1 expression in the presence or absence of pembrolizumab, pembrolizumab scFv, or an irrelevant scFv (CD19). We first tested NK cell responses against the PD-L1 low-expressing tumor cell line K562. NK cell function was evaluated by flow cytometric detection of degranulation by CD107a and cytokine production by intracellular staining for IFN-γ. Blockade of PD-1 on NK cells resulted in significant increases in both degranulation and IFN-γ production against the PD-L1 low-expressing K562 cells (Figure 2A-B). When exposed to the monocytic AML cell line THP-1, which expresses significantly higher levels of PD-L1, blockade of PD-1 resulted in a markedly greater increase in NK cell function than seen against K562 (Figure 2C-D). Furthermore, when cocultured with THP-1 cells that had been stimulated overnight with IFN-γ to further upregulate PD-L1 (supplemental Figure 1A), PD-1 blockade restored NK cell function (supplemental Figure 1B-C). Blockade enhanced NK function against the IFN-γ–stimulated THP-1 cells, but it did so to a slightly lower extent than against the unstimulated THP-1 cells. This incomplete restoration of function may be due to other effects of IFN-γ stimulation on the THP-1 cells such as upregulation of MHC-I molecules, which can suppress NK function through inhibitory receptor ligation. Interestingly, blockade of PD-L1 did not significantly increase NK activity against the target compared with PD-1 blockade with scFv (supplemental Figure 1D-E). This mirrors what we observed with the full-length pembrolizumab antibody and further highlights the benefit of using the pembrolizumab scFv to enhance function. PD-L2 blockade was not evaluated here because this ligand has been shown to be largely absent on cells from AML and myelodysplastic syndrome.25 Moreover, the enhancement in NK cell degranulation with PD-1 blockade is reflected in target cell killing. When cocultured for 48 hours in the presence of PD-L1–expressing targets, PD-1 blockade via the pembrolizumab scFv resulted in a significant increase in K562 (Figure 3A) and THP-1 (Figure 3B-E) cell killing compared with untreated NK cells or NK cells treated with the full-length pembrolizumab antibody. To determine whether PD-L1 can suppress NK function, we generated a K562 line that overexpresses PD-L1 (K.PD-L1) (Figure 3F). NK cells cultured with these cells resulted in substantially lower NK cell function compared with normal K562 cells (Figure 3G).
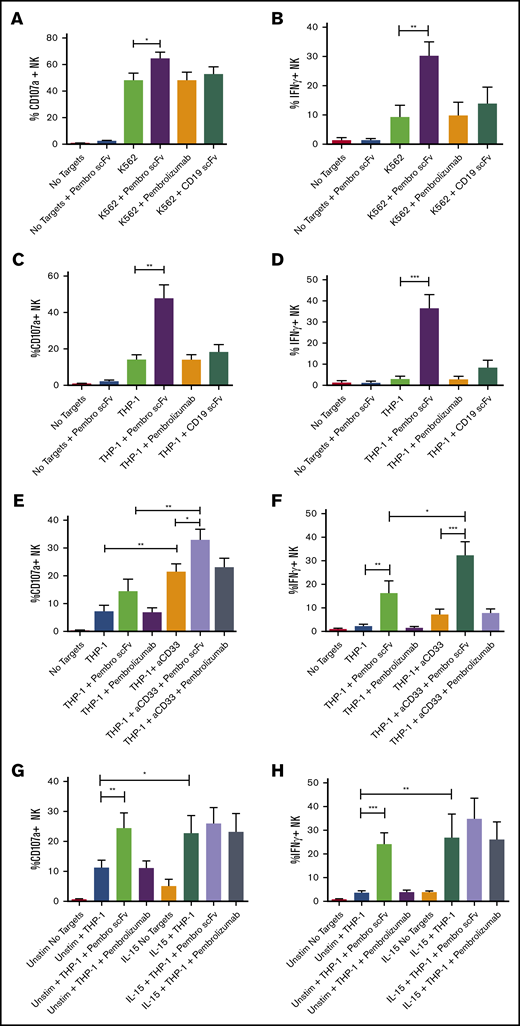
PD-1 on NK cells impairs degranulation and IFN-γ production against PD-L1–expressing targets and is restored by PD-1 blockade. (A-D) NK cells were purified from peripheral blood and co-incubated with K562 (A-B) or THP-1 (C-D) cells with or without PD-1 blocking antibodies or an irrelevant anti-CD19 scFv for 4 hours. (E-F) The ability of PD-1 blockade to enhance ADCC was also tested with an anti-CD33 (aCD33) antibody against THP-1. (G-H) PD-1 blockade was also evaluated on NK cells preactivated with 10 ng/mL IL-15 against THP-1 targets. Function was assessed via flow cytometry staining for CD107a and IFN-γ (n = 5). Error bars indicate the mean ± SEM. *P < .05; **P < .01; ***P < .001. Unstim, unstimulated.
PD-1 on NK cells impairs degranulation and IFN-γ production against PD-L1–expressing targets and is restored by PD-1 blockade. (A-D) NK cells were purified from peripheral blood and co-incubated with K562 (A-B) or THP-1 (C-D) cells with or without PD-1 blocking antibodies or an irrelevant anti-CD19 scFv for 4 hours. (E-F) The ability of PD-1 blockade to enhance ADCC was also tested with an anti-CD33 (aCD33) antibody against THP-1. (G-H) PD-1 blockade was also evaluated on NK cells preactivated with 10 ng/mL IL-15 against THP-1 targets. Function was assessed via flow cytometry staining for CD107a and IFN-γ (n = 5). Error bars indicate the mean ± SEM. *P < .05; **P < .01; ***P < .001. Unstim, unstimulated.
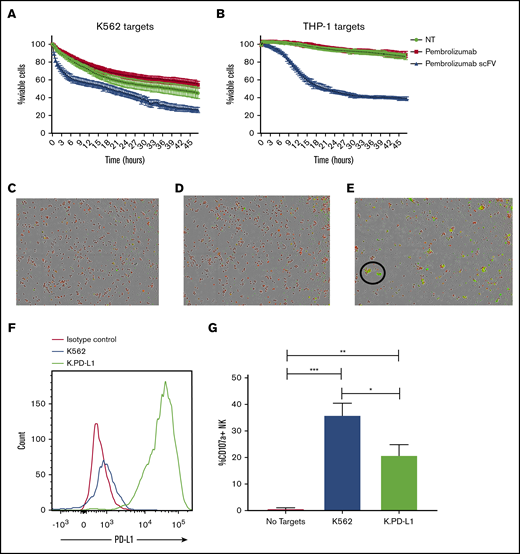
PD-1 blockade enhances NK cell killing of PD-L1–expressing targets, and PD-L1 overexpression suppresses NK function. (A-B) Purified NK cells from peripheral blood of healthy donors were cocultured with CellTrace-labeled K562 (A) or THP-1 (B) in the presence or absence of PD-1 blocking antibodies. (C-E) Visualization of killed targets is shown 24 hours after coculture with THP-1 cells without blockade (C), with pembrolizumab (D), or pembrolizumab scFv (E). Target cell killing was identified by CellTrace and activated caspase-3 and caspase-7 positivity. The killing assay was carried out in an IncuCyte Zoom (IncuCyte) for 48 hours with images taken every 30 minutes (n = 3). Magnification ×4. (F) K562 and K.PD-L1 cells were stained for PD-L1. (G) K562 and K.PD-L1 cells were cocultured for 4 hours with purified NK cells from healthy donors for 4 hours and stained for CD107a (n = 6). Error bars indicate the mean ± SEM. *P < .05; **P < .01; ***P < .001. NT, no treatment.
PD-1 blockade enhances NK cell killing of PD-L1–expressing targets, and PD-L1 overexpression suppresses NK function. (A-B) Purified NK cells from peripheral blood of healthy donors were cocultured with CellTrace-labeled K562 (A) or THP-1 (B) in the presence or absence of PD-1 blocking antibodies. (C-E) Visualization of killed targets is shown 24 hours after coculture with THP-1 cells without blockade (C), with pembrolizumab (D), or pembrolizumab scFv (E). Target cell killing was identified by CellTrace and activated caspase-3 and caspase-7 positivity. The killing assay was carried out in an IncuCyte Zoom (IncuCyte) for 48 hours with images taken every 30 minutes (n = 3). Magnification ×4. (F) K562 and K.PD-L1 cells were stained for PD-L1. (G) K562 and K.PD-L1 cells were cocultured for 4 hours with purified NK cells from healthy donors for 4 hours and stained for CD107a (n = 6). Error bars indicate the mean ± SEM. *P < .05; **P < .01; ***P < .001. NT, no treatment.
PD-1 blockade can not only enhance NK cell natural cytotoxicity, it can also improve antibody-dependent cell-mediated cytotoxicity (ADCC). This was tested against THP-1 cells (which express CD33) by using an anti-CD33 antibody capable of mediating ADCC responses. When PD-1 was blocked on NK cells before coculture with the anti-CD33 antibody, both degranulation and cytokine production were significantly enhanced compared with PD-1 blockade or anti-CD33–mediated ADCC alone (Figure 2E-F). Although PD-1 blockade of resting NK cells resulted in enhanced function against PD-L1–expressing targets and a concurrent increase in pAKT (supplemental Figure 2A), activation of NK cells with the common γ-chain cytokine IL-15 was robust. This eliminated any substantial increase in function when PD-1 was blocked (Figure 2 G-H), indicating that the inhibitory effects of PD-1 can be overcome by sufficient activation. Although the pembrolizumab scFv blocked interactions between PD-1 and PD-L1 on NK cells, the clinical reagent had no apparent effect in our in vitro 4-hour assays. We confirmed the effect of PD-1 blockade on NK cells by using an scFv derived from the clinical PD-1 antagonistic antibody tislelizumab, which was generated by BeiGene and is currently in phase 2 (NCT03777657) and phase 3 (NCT03744468) clinical trials. Similar to the pembrolizumab scFv, the tislelizumab scFv potently enhanced the function of resting NK cells (supplemental Figure 2B-C). Furthermore, PD-1 blockade using the full pembrolizumab antibody and the pembrolizumab scFv on peptide-pulsed CD8+ T cells indicated that both reagents elicited an increase in function on the T cells with the scFv reagent, which resulted in a greater than twofold increase over that of the full antibody (supplemental Figure 2D-E).
To preclude the possibility that Fc binding of the full-length pembrolizumab antibody resulted in cross-linking of PD-1 and activating PD-1 (inhibitory) signaling on NK cells, rather than acting as an antagonist, we treated pembrolizumab with the endoglycosidease Endo S to remove the N-linked oligosaccharide on IgG4a, which has been shown to be important in the binding of IgG to Fc receptors.26 In a second approach, we removed the Fc entirely by generating antigen-binding fragments (Fab) from pembrolizumab using papain digestion and subsequent Fc fraction removal by protein A purification. Purification of the Fab fragment was confirmed by enrichment of the 50-kDa fragment by western blot (supplemental Figure 3A). Unlike our pembrolizumab scFv, the use of the deglycosylated or Fab fragment versions of pembrolizumab did not result in any enhancement of NK cell function (supplemental Figure 3B-E).
PD-1, upregulated on NK cells in AML patients after allo-HSCT, suppressed NK function against PD-L1–expressing tumor targets
Considering that PD-L1 is frequently upregulated on tumors and that NK cells are the first lymphocyte population to reconstitute after allogeneic hematopoietic stem cell transplantation (allo-HSCT), we wanted to evaluate PD-1 expression on the emerging NK cell population and how it might affect the antitumor response in this setting. To test this, we selected 10 AML patients who received matched sibling allo-HSCT and evaluated PD-1 expression on the reconstituting NK cell population in comparison with the donor sample before transplantation. By using the FITC-labeled pembrolizumab scFv, we found that PD-1 expression was upregulated on NK cells at day 28 posttransplant and remains upregulated to at least day 100 posttransplant (Figure 4A). These same samples were further evaluated by mass cytometry (CyTOF) up to day 100 posttransplant. PD-1 expression was found to be upregulated on both NK cell (CD56+) and CD8+ T-cell populations during immune reconstitution (Figure 4B). These CyTOF studies used commercial reagents and detected PD-1 with greater sensitivity than by using commercial antibodies with flow cytometry. Further analysis of the NK population showed an enrichment of PD-1-positive cells in both the CD56bright and CD56dim populations, with a preferential increase in more mature CD56dim NK cells (Figure 4C). When PBMCs from these patients at day 28 posttransplant were used in a 4-hour functional assay against K562 cells in the presence or absence of PD-1 blockade, the pembrolizumab scFv significantly enhanced NK cell degranulation and cytokine production (Figure 5A-B). A similar enhancement of function was observed with day 28 posttransplant samples from recipients of UCB transplantation (Figure 5C-D).
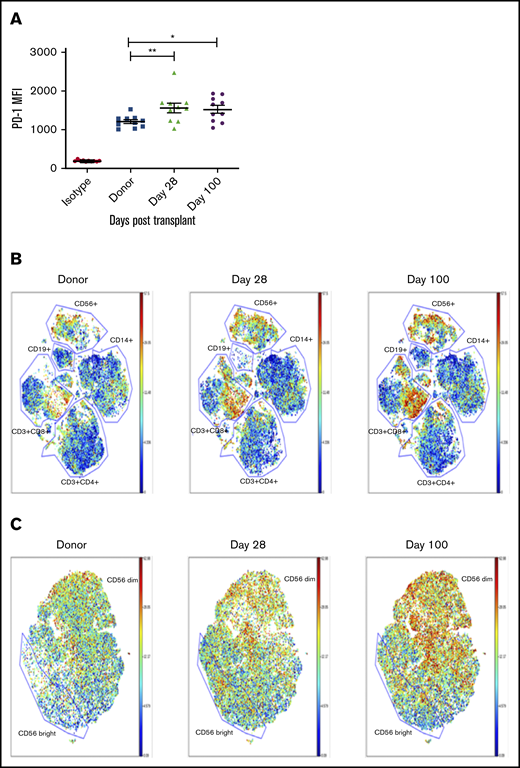
PD-1 is upregulated on NK cells in transplant recipients by day 28 posttransplant compared with donor NK cells and impairs NK cell function against PD-L1–expressing tumor targets. (A) NK cells from 10 sibling-matched donors and recipients 28 days posttransplant were stained for PD-1 expression. (B-C) PBMCs from the same 10 donors and recipients were analyzed by CyTOF up to day 100 posttransplant and visualized by bulk lymphocyte subsets (B) or focused specifically on NK cells (C) (n = 10). Horizontal bars indicate the mean ± SEM. *P < .05; **P < .01. MFI, mean fluorescence intensity.
PD-1 is upregulated on NK cells in transplant recipients by day 28 posttransplant compared with donor NK cells and impairs NK cell function against PD-L1–expressing tumor targets. (A) NK cells from 10 sibling-matched donors and recipients 28 days posttransplant were stained for PD-1 expression. (B-C) PBMCs from the same 10 donors and recipients were analyzed by CyTOF up to day 100 posttransplant and visualized by bulk lymphocyte subsets (B) or focused specifically on NK cells (C) (n = 10). Horizontal bars indicate the mean ± SEM. *P < .05; **P < .01. MFI, mean fluorescence intensity.
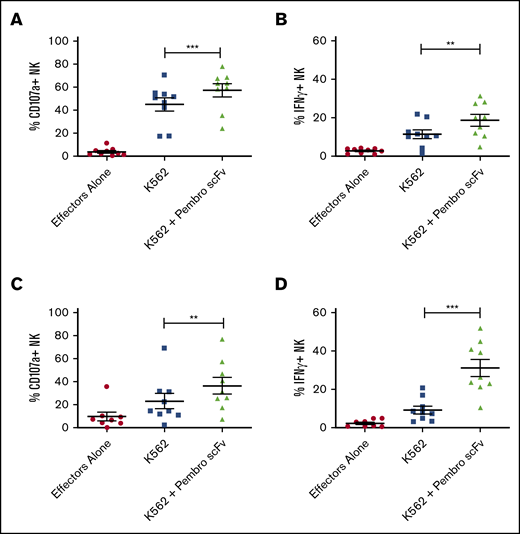
PD-1 blockade of reconstituting NK cells from sibling transplant recipients results in enhanced NK function against PD-L1–positive tumor targets. PBMCs from sibling (A-B) or UCB (C-D) recipients 28 days posttransplant were cocultured with K562 cells with or without PD-1 blockade for 4 hours and stained for CD107a and IFN-γ (n = 10). Horizontal bars indicate the mean ± SEM. **P < .01; ***P < .001.
PD-1 blockade of reconstituting NK cells from sibling transplant recipients results in enhanced NK function against PD-L1–positive tumor targets. PBMCs from sibling (A-B) or UCB (C-D) recipients 28 days posttransplant were cocultured with K562 cells with or without PD-1 blockade for 4 hours and stained for CD107a and IFN-γ (n = 10). Horizontal bars indicate the mean ± SEM. **P < .01; ***P < .001.
Discussion
Expression of PD-1 on NK cells was variable, but it was still detectable with the commercially available anti-PD-1 antibody from Miltenyi Biotec and was also detectable by using a labeled version of the clinical therapeutic mAb pembrolizumab (Merck). However, the staining intensity was low and variable between donors, and it overlapped significantly with isotype matched controls, which made definitive detection difficult. The most definitive flow cytometry staining results were seen with FITC labeling of bacterially synthesized scFv fragments from known PD-1–binding antibody sequences (derived from pembrolizumab and tislelizumab antibodies). With the better detection afforded by these novel reagents, we were able to describe a basal level of PD-1 expression on all resting peripheral blood NK cells from steady-state healthy individuals. The finding that 2 different scFv sequences enhanced functional blocking against PD-L1–positive targets supports the notion that the scFv of anti-PD1 was important for uncovering the biology of low levels of PD-1 on resting NK cells. Octet binding analysis confirmed that the scFv derived from pembrolizumab was specific for human PD-1. Importantly, staining intensity correlated well with functional activity on resting NK cells. Commercial reagents such as the intact clinical antibody pembrolizumab that gave weak flow cytometry staining did not functionally block target cell killing in vitro. In marked contrast, the 2 scFv sequences produced by Escherichia coli detected surface staining and, more importantly, resulted in potent functional inhibition of PD-1 on resting NK cells despite the low expression. Importantly, stimulation with a short-term γ-chain cytokine (IL-15) seemed to abrogate the need for PD-1 blockade in our in vitro studies, perhaps suggesting that PD-1 is predominantly a functional checkpoint on resting NK cells. However, a previous study showed that IL-2 cytokine signaling can induce PD-1 expression on NK cells,23 and common γ-chain cytokines have been shown to induce PD-1 expression on T cells and also to induce PD-L1 expression.7 Clinical testing to study PD-1 blockade in the setting of IL-15 stimulation is in progress to better understand this finding.
Blockade of PD-1 in the presence of tumor cells lines that express PD-L1 yielded no enhancement of NK cell function when treated with pembrolizumab. Yet blockade with the scFv of pembrolizumab and a different PD-1 targeting scFv resulted in a significant enhancement of NK cell degranulation as well as production of IFN-γ. This effect was seen against the PD-L1 low-expressing HLA-null chronic myelogenous leukemia K562 cell line as well as the AML THP-1 cell line, which expresses a significant amount of PD-L1.27 Higher PD-L1 expression resulted in higher differentials in functionality upon blockade. Furthermore, resting NK cells with PD-1 blockade cocultured with THP-1 cells that were treated overnight with IFN-γ to upregulate PD-L1 had an intermediate functional enhancement compared with the untreated THP-1 cells. This might be a result of upregulation of other checkpoint proteins in the cells treated with IFN-γ.28 The enhancement of function with blockade is reflected in target cell killing in which the pembrolizumab scFv, but not the intact antibody blockade, resulted in significantly greater killing of both K562 and THP-1 cells compared with no blockade. Furthermore, PD-1 seems to suppress natural cytotoxicity-mediated NK cell function and also ADCC. NK cells cocultured with THP-1 cells and an anti-CD33 antibody resulted in strong degranulation and cytokine production that was significantly enhanced by PD-1 blockade, which indicates a strong role for interactions between PD-1 and PD-L1 in the suppression of resting NK cell function without cytokine activation.
NK cells are the first lymphocytes to differentiate and homeostatically expand after allo-HSCT. Considering the importance of NK cells in antitumor responses and tumor upregulation of PD-1 ligands, we evaluated the expression of PD-1 on the reconstituting NK cells in AML patients who received either matched sibling donor or UCB transplants. PD-1 was increased on reconstituting NK cells in sibling transplant recipients by 28 days after transplantation compared with the matched steady-state donor sample. The CyTOF methodology had better sensitivity for detecting PD-1 when compared with flow cytometry using commercial antibodies. PD-1 blockade with pembrolizumab scFv from reconstituting NK cells in the presence of K562 cells resulted in enhanced NK degranulation and cytokine production, which shows that this increase may be physiologically important in this setting. A similar functional effect was seen with PD-1 blockade of NK cells from UCB transplant recipients. These data indicate that PD-1 expression increases on NK cells posttransplant, which may inhibit the capacity of the reconstituting NK compartment from mounting an effective antitumor response.
Why has there been so much controversy regarding the expression of PD-1 on NK cells? We believe this is partly a result of imperfect detection reagents with variable sensitivity for picking up low levels of PD-1. Previous publications show distinct populations of PD-1–expressing NK cells of which the vast majority are PD-10–negative. A recent study by Judge et al29 identified minimal PD-1 present on intratumoral human NK cells. Flow cytometric analysis of PD-1 in that study was performed with EH12.2H7, a clone we tested (Figure 1), and in agreement with that study, this antibody detects minimal PD-1 expression on resting NK cells. However, detection depends on the flow cytometry reagent used, and scFv PD-1 sequences have a unique ability for both detection and functional blockade. Our data indicate that on healthy NK cells, PD-1 is expressed at a low level in a broad spectrum of the population and is upregulated posttransplant.
To ensure that our staining was accurate, we tested 3 commercial clones, FITC-labeled full-length pembrolizumab, and scFv pembrolizumab, and we controlled this staining with appropriate isotype controls and irrelevant scFv’s. In support of the presence and function of PD-1 on human NK cells, another study found that a CRISPR knockout of PD-1 on primary NK cells demonstrated enhanced in vitro function of human NK cells against multiple targets and enhanced in vivo function using a xenogeneic model of ovarian cancer.30 Through the creation of scFv reagents from 2 whole antibodies (one in common clinical use), we have identified both PD-1 expression and inhibitory function on resting NK cells that had not previously been definitively detected. The clinical reagent pembrolizumab comprises 2 Fab fragments and an IgG4a Fc portion, whereas the scFv is a smaller monovalent single VH and VL chain connected by a short peptide linker. This scFv has been previously identified to efficiently bind human PD-1 during x-ray crystallographic studies that explored the interactions between PD-1 and pembrolizumab.31 Treatment of pembrolizumab with Endo S, which has a high specificity for the N-linked glycans on the heavy chain of IgG,32 did not rescue its ability to enhance NK cell function. Creating Fab fragments from pembrolizumab by using enzymes also did not reproduce the functional effect of the scFv.
A third possibility to explain these differences is that the glycosylation of sites outside the Fc domain may be active in altering function more than binding affinity.33-35 An anti-factor VIII (FVIII) antibody (LE2E9) isolated from a patient with hemophilia A neutralized FVIII by up to 90%. Removal of glycans from the complementarity-determining region 1 of the Fab fragment had no deleterious effects on FVIII binding but resulted in an antibody that was 40% less effective at inhibiting FVIII activity.36 This is important in our comparison of clinical pembrolizumab and its scFv because their posttranslational modifications are inherently different, considering the expression systems used for production. The clinical version of pembrolizumab is produced in Chinese hamster ovary cells, a mammalian system that mediates posttranslational glycosylations that are very different from those of the prokaryotic E coli system, which is how we produce our scFv’s. Differences in glycosylations of the VH or VL chains may explain differences in functionality between the intact clinical antibody and its scFv produced from bacteria.
Despite some uncertainties regarding what drives differences, our conclusions are clear but should not be overextrapolated, and they need to be interpreted with caution. Our in vitro results with PD-1 blockade with the pembrolizumab scFv suggest that the clinical PD-1 antibody pembrolizumab may not be effective on NK cells, but we cannot make this same conclusion in vivo. In fact, for practical reasons, this cannot be definitively tested because the pharmacokinetics of whole mAb is several weeks compared with hours at most with an scFv, which will be renally excreted. It is possible that the longevity of whole antibody blockade may overcome some potency differences seen in the in vitro assays used here. Future in vivo studies are planned to test the effect of clinical PD-1–blocking antibodies using human NK cells in a xenogeneic tumor-bearing model, but even these models may be difficult because they are dependent on γ-chain cytokine activation that may obscure PD-1 function. Despite the lack of blocking by the full antibody on NK cells, we did observe an enhancement of function on peptide-stimulated CD8+ T cells, with a substantially greater increase when the scFv version is used. We hypothesized that the discrepancy between the enhanced functionality of the full pembrolizumab antibody on T cells compared with NK cells may have to do with the increased density of PD-1 on activated T cells, which results in a lower threshold of signaling to activate PD-1.
Our data clearly demonstrate low levels of PD-1 on resting NK cells and the functional impact these low levels mediate, suggesting the importance of this checkpoint on NK cell biology. This adds to the growing list of NK cell checkpoints37-39 that control the antitumor function of NK cells in addition to other activating and inhibitor receptors and the importance of missing self. PD-1 is also increased on early-reconstituting resting NK cells, a physiologic setting of NK cell development. This may inhibit a graft-versus-leukemia effect in the early posttransplant setting against PD-L1 tumors. Checkpoint blockade after transplantation is not feasible because of the known complications of potentially lethal graft-versus-host disease,40,41 but targeting checkpoint blockade specifically to NK cells might be an approach for enhancing NK cell immunotherapy in the future.
To request data, please send an e-mail to Jeffrey S. Miller at mille011@umn.edu.
Acknowledgments
The authors thank the Flow Cytometry Shared Resource, Clinical Trials Office, Translational Therapy Laboratory, Cancer Research Translational Initiative, and Clinical Informatics Shared Services.
This work was supported by grants from the National Institutes of Health, National Cancer Institute (CA111412 [J.S.M.], CA65493 [B.R.B. and J.S.M.], and CA197292 [J.S.M.], and in part by 5P30CA077598-18 [funding for the University of Minnesota Masonic Cancer Center]), and by grants from the National Institute of Allergy and Infectious Diseases (R37 AI 34495 [B.R.B.] and AI149680 [J.L.R.]).
Authorship
Contribution: Z.D., M.F., and J.S.M. conceptualized the study, collected, assembled, analyzed and interpreted data, and wrote the manuscript; Z.D., T.L., S.B., J.T.W., and P.H. performed experiments and assembled the data; J.L.R. and D.A.V. helped with reagent production; and B.R.B and all the other authors helped with manuscript preparation.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jeffrey S. Miller, Blood and Marrow Transplant Program, Department of Medicine, University of Minnesota, 420 Delaware St SE, Minneapolis, MN 55455; e-mail: mille011@umn.edu.

