

Key Points
Baseline and serial molecular profiling by NGS can predict outcomes with HMAs in MDS patients.
Serial molecular profiling during therapy of patients with mutant TP53 can identify patients who may benefit from allogeneic transplantation.

Abstract
Hypomethylating agents (HMAs) are widely used in the treatment of myelodysplastic syndromes (MDSs), yet identifying those patients unlikely to benefit remains challenging. We assessed response and overall survival (OS) in 247 patients molecularly profiled by next-generation sequencing (NGS) before first-line HMA therapy, and a subset of 108 patients were sequenced serially during treatment. The most common mutations included TP53 (33.1%), ASXL1 (19%), TET2 (16.5%), DNMT3A (14.1%), and SRSF2 (12.1%). The overall response rate was 42.1%, with the composite TET2-mutant/ASXL1 wild-type genotype representing the strongest predictor of response (overall response rate, 62.1%; complete remission rate, 34.5%). The median OS for the cohort was 15 months, and the number of mutations detected by NGS (hazard ratio [HR], 1.22; P = .02), as well as mutations in TP53 (HR, 2.33; P = .001) and EZH2 (HR, 2.41; P = .04) were identified as independent covariates associated with inferior OS in multivariable analysis. Serial molecular profiling revealed that clearance of TP53 mutations during HMA therapy was associated with superior OS (HR, 0.28; P = .001) and improved outcome in patients proceeding to allogeneic hematopoietic cell transplantation. These data support baseline molecular profiling by NGS in MDS patients treated with HMAs and provide novel observations of sequential profiling during therapy that provide particular value in TP53-mutated disease.
Introduction
Myelodysplastic syndromes (MDSs) display hematologic and prognostic heterogeneity, which illustrates the need for accurate risk stratification and individualized therapy. In patients with higher-risk disease, the hypomethylating agents (HMAs) azacitidine and decitabine are the standard first-line treatment owing to their clinical activity and the potential to extend overall survival (OS).1-3 However, despite their widespread adoption, fewer than 50% of patients respond with poor outcomes observed after treatment failure, which highlights the need for a reliable strategy to identify patients most likely to benefit from therapy.4,5
In up to 90% of MDS patients, recent advances in next-generation sequencing (NGS) have identified recurrent somatic mutations in genes that function in RNA splicing, epigenetic regulation, gene transcription, and cell signaling.6-11 These mutations underlie key pathogenic features of MDS and, in addition to classic clinical and cytogenetic features, independently impact OS.6,7,12 It has been hypothesized that somatic mutations may also serve as biomarkers predictive for response to HMA therapy, but studies to date have been inconsistent. Mutations in the TET2 gene, a dioxygenase involved in DNA demethylation, were linked to a higher rate of response to HMAs in several studies,13-15 but in other studies, they had no impact.16-18 One such study showed that improved response rates in patients with a TET2 mutation were largely dependent on concurrent ASXL1 genotype.13 Mutations in the vital tumor suppressor gene TP53 are consistently associated with inferior survival; however, the impact of TP53 mutations on response to HMA therapy has been inconsistent.12,13,18-20 The adverse effect of TP53 mutations on treatment outcome also extends to allogeneic hematopoietic cell transplantation (allo-HCT), in which only a small subset of patients achieve long-term remissions.21-23
Sequential molecular profiling of disease during active treatment has been investigated as a means to characterize clonal evolution.11 Falconi et al18 found that although allelic frequencies in the majority of genes do not change, TP53-mutant clone size was reduced with HMA treatment, whereas clones remained detectable in all patients evaluated. Nonetheless, the value of serial molecular profiling of patients to help direct treatment strategies remains largely unexplored.24 Notably, we recently reported that achievement of NGS negativity was a powerful independent predictor of OS in MDS and in patients with secondary acute myeloid leukemia (AML).25
Heterogeneity and sample size have been limitations in previous studies that aimed to identify genetic covariates predictive for outcome with HMA therapy. Because of the widespread commercial availability of NGS, molecular profiling is increasingly used, so its utility in the clinical setting requires further delineation. Herein, we report on molecular predictors of outcome in one of the largest reported cohorts to date of predominantly high-risk MDS patients treated with HMA therapy. Baseline molecular profiling before treatment was required for study entry, with sequential profiling during treatment performed in a subset of patients.
Patients and methods
Patient selection and inclusion
Patients treated at the H. Lee Moffitt Cancer Center and Research Institute between 2010 and 2019 were retrospectively analyzed. Patients with a World Health Organization (WHO)–defined diagnosis of MDS, myelodysplastic syndrome/myeloproliferative neoplasm (MDS/MPN) overlap syndrome, or AML (with 20% to 30% bone marrow myeloblasts) were included. All patients received first-line therapy with an HMA, which included azacitidine, decitabine, or azacitidine in combination with an additional agent. NGS was performed within 6 months before the first cycle of HMA therapy (median time from NGS to day 1 of therapy was 17 days; maximum time was 170 days). Clinical characteristics were abstracted from multiple timepoints, including the date of diagnosis, at initiation of HMA therapy (timepoint used for analyses involving baseline predictors of response and OS), and at the time of response to HMA. Patients proceeding to allo-HCT after HMA therapy were included. This study was approved by the Institutional Review Board of the H. Lee Moffitt Cancer Center and Research Institute.
Mutational profiling
All patients underwent molecular profiling by NGS with sequencing performed on DNA extracted from peripheral blood or bone marrow mononuclear cells. The NGS panels targeted up to 406 genes, and each gene that was included in statistical analyses (supplemental Table 1) was sequenced in a minimum of 92% of patients. A minimum variant allele frequency (VAF) of 5% was used to call single nucleotide variants, and a cutoff of 10% was used for insertions or deletions. Each reported mutation was further evaluated by the study team and referenced in databases including Catalogue Of Somatic Mutations In Cancer (COSMIC) and dbSNP to ensure that reported mutations were both somatic and pathogenic. Sequential molecular profiling was evaluated in a subset of patients who had NGS testing performed during or at the completion of HMA therapy (before any subsequent therapy or allo-HCT).
Definitions of response and survival
Response to HMA therapy was assessed by using the International Working Group 2006 response criteria for MDS.26 Comparisons for evaluating predictors of response to HMA therapy assessed both the overall response rate (ORR) and the complete remission (CR) rate. ORR was defined as patients achieving CR, marrow CR (mCR), partial remission (PR), or hematologic improvement (HI). OS was defined as the duration from first day of HMA therapy until death, with surviving patients censored at time of last patient contact.
Statistical methods
Categorical variables were compared using Fisher’s exact and χ2 tests, and quantitative data were compared using the Mann-Whitney U test and Wilcoxon rank sum tests. The Kaplan-Meier method was used to estimate OS and the log-rank test was used to compare OS between groups in univariable analyses. Patients were not censored at the time of allo-HCT in reported analyses. A binary logistic regression model was used in multivariable analyses to determine covariates for response. Multivariable analysis of OS used the Cox proportional hazards model, with clinical and molecular variables having a P value < .10 in univariable analysis included in the model. The backward elimination method was used to select variables for the final multivariable model, and variables with a P value > .05 were excluded. Patients sequenced on targeted panels that did not include a specific gene of interest were excluded from all analyses evaluating that gene. Statistical analyses were performed using GraphPad Prism version 8.3 and IBM SPSS statistics version 27.
Results
Baseline characteristics of the cohort
A total of 247 patients were included in the cohort, and baseline characteristics are summarized in Table 1. The median age at diagnosis was 69 years (range, 24-89 years) and 64.7% of the patients were male. The majority of patients (79.1%) were classified at diagnosis as higher-risk (intermediate, high, or very high risk) by using the Revised International Prognostic Scoring System (IPSS-R). Among 51 patients classified as lower risk at diagnosis, 31 (60.8%) progressed to higher-risk disease before treatment with HMA was initiated; 90.3% of patients were classified as higher risk by IPSS-R at the time of HMA therapy. The most common subtypes by WHO classification at diagnosis were MDS with excess blasts-2 (25.5%) and MDS with excess blasts-1 (23.9%), and 184 patients (74.5%) had ≥5% blasts at treatment initiation. The diagnosis was classified as therapy-related in 24.3% of patients.
Baseline characteristics of the cohort at time of diagnosis
| Baseline characteristic | Total cohort (N = 247) |
|---|---|
| Median age (range), y | 69 (24-89) |
| Percent male | 64.7 |
| IPSS-R risk stratification | |
| Very low | 15 (6.1) |
| Low | 36 (14.8) |
| Intermediate | 57 (23.4) |
| High | 51 (20.9) |
| Very high | 85 (34.8) |
| WHO classification | |
| MDS-SLD | 11 (4.4) |
| MDS-MLD | 52 (21.1) |
| MDS-RS-SLD | 2 (0.8) |
| MDS-RS-MLD | 14 (5.7) |
| MDS with isolated del(5q) | 2 (0.8) |
| MDS-EB1 | 59 (23.9) |
| MDS-EB2 | 63 (25.5) |
| MDS-U | 7 (2.8) |
| MDS/MPN | 16 (6.5) |
| AML (20%-30% blasts) | 21 (8.5) |
| Therapy-related myeloid neoplasm | 60 (24.3) |
| AML transformation | 81 (35.8) |
| First-line HMA therapy | |
| Azacitidine | 200 (81) |
| Decitabine | 29 (11.7) |
| HMA plus additional agent | 18 (7.3) |
| Proceeded to allo-HCT | 61 (24.7) |
| Patients with detectable mutation on NGS | 213 (86.2) |
| Median no. of mutated genes (range) | 2 (0-8) |
| Median absolute neutrophil count (range), × 109/L | 1.16 (0.06-28.8) |
| Median hemoglobin (range), g/dL | 9 (6.8-13.6) |
| Median platelet count (range), × 109/L | 64.5 (8-1073) |
| Median bone marrow blast percentage (range) | 8 (0-30) |
| Baseline characteristic | Total cohort (N = 247) |
|---|---|
| Median age (range), y | 69 (24-89) |
| Percent male | 64.7 |
| IPSS-R risk stratification | |
| Very low | 15 (6.1) |
| Low | 36 (14.8) |
| Intermediate | 57 (23.4) |
| High | 51 (20.9) |
| Very high | 85 (34.8) |
| WHO classification | |
| MDS-SLD | 11 (4.4) |
| MDS-MLD | 52 (21.1) |
| MDS-RS-SLD | 2 (0.8) |
| MDS-RS-MLD | 14 (5.7) |
| MDS with isolated del(5q) | 2 (0.8) |
| MDS-EB1 | 59 (23.9) |
| MDS-EB2 | 63 (25.5) |
| MDS-U | 7 (2.8) |
| MDS/MPN | 16 (6.5) |
| AML (20%-30% blasts) | 21 (8.5) |
| Therapy-related myeloid neoplasm | 60 (24.3) |
| AML transformation | 81 (35.8) |
| First-line HMA therapy | |
| Azacitidine | 200 (81) |
| Decitabine | 29 (11.7) |
| HMA plus additional agent | 18 (7.3) |
| Proceeded to allo-HCT | 61 (24.7) |
| Patients with detectable mutation on NGS | 213 (86.2) |
| Median no. of mutated genes (range) | 2 (0-8) |
| Median absolute neutrophil count (range), × 109/L | 1.16 (0.06-28.8) |
| Median hemoglobin (range), g/dL | 9 (6.8-13.6) |
| Median platelet count (range), × 109/L | 64.5 (8-1073) |
| Median bone marrow blast percentage (range) | 8 (0-30) |
Data are presented as n (%) unless otherwise specified.
MDS/MPN, myelodysplastic syndrome/myeloproliferative neoplasm; MDS-EB1, MDS with excess blasts-1; MDS-EB2, MDS with excess blasts-2; MDS-MLD, MDS with multilineage dysplasia; MDS-RS-MLD, MDS with ring sideroblasts and multilineage dysplasia; MDS-RS-SLD, MDS with ring sideroblasts and single lineage dysplasia; MDS-SLD, MDS with single lineage dysplasia; MDS-U, MDS unclassifiable.
All 247 patients were treated with an HMA in the first-line setting, and azacitidine was the treatment used for 81% of the patients. A total of 29 patients received decitabine as first-line HMA therapy: 6 received a 10-day regimen, and 18 (7.3%) received azacitidine in combination with an additional agent. Those agents included investigational therapies (n = 9; no patients treated with eprenetapopt [APR-246] or magrolimab were included), lenalidomide (n = 5), checkpoint inhibitors (n = 3), and venetoclax (n = 1). The median number of cycles of HMA therapy was 4, and the median time from diagnosis to therapy initiation was 1.6 months.
Spectrum of somatic mutations
A somatic mutation was identified in at least 1 gene in 213 patients (86.2%), with a median of 2 mutated genes (range, 0-8 mutated genes). The observed frequency of mutations is detailed in Figure 1; TP53 (33.1%), ASXL1 (19%), TET2 (16.5%), DNMT3A (14.1%), and SRSF2 (12.1%) were the most commonly mutated genes.
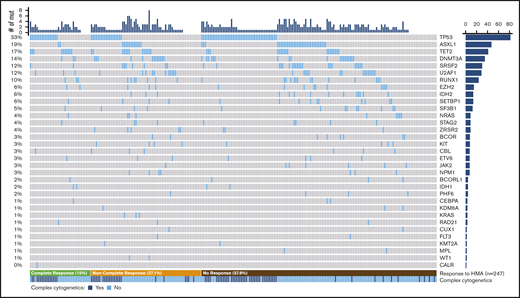
Spectrum of mutations identified in the cohort. Co-mutation plot showing mutations (mut) observed in the cohort; each column represents an individual patient. Mutations are listed in descending order by frequency observed, with the frequency of each mutation on the left and the total number of patients on the right. Patients are organized by response to HMA therapy (bottom), and the presence of complex cytogenetics is listed.
Spectrum of mutations identified in the cohort. Co-mutation plot showing mutations (mut) observed in the cohort; each column represents an individual patient. Mutations are listed in descending order by frequency observed, with the frequency of each mutation on the left and the total number of patients on the right. Patients are organized by response to HMA therapy (bottom), and the presence of complex cytogenetics is listed.
A complex karyotype was observed in 87 patients, and 71 (81.6%) harbored a mutation in TP53. Likewise, 86.6% of all TP53-mutated patients had a complex karyotype. TP53 was the sole mutation identified in 46 patients (56.1%) who had a TP53 mutation. In 63 patients (76.8%), there was a single mutation in TP53, 18 (22%) carried 2 mutations, and 1 (1.2%) harbored 3 mutations. Biallelic loss of TP53 (defined by patients with >1 TP53 mutation, TP53 mutation and a chromosome 17 abnormality, or a TP53 VAF >70%) was seen in 51 patients (62.2%). Among 47 ASXL1-mutated patients, 36 (76.6%) harbored frameshift or nonsense mutations.
Covariates for response to HMA therapy
In the total cohort, 37 patients achieved CR as best response (15%), 34 mCR, 4 PR, and 29 HI for an ORR (CR + mCR + PR + HI) of 42.1%. In univariable analysis, no clinical variables evaluated at the time of HMA initiation were predictive of ORR; however, an absolute neutrophil count <1000 cells per μL (P = .002), number of cytopenias (P = .03), and increased marrow blast percentage (P = .02) were associated with a higher CR frequency. No clinical variables were predictive of ORR or CR in multivariable analyses.
Molecular covariates for response are summarized in Table 2. In univariable analysis, the TET2-mutant/ASXL1-wild-type (WT) genotype was predictive of both CR (34.5% vs 12.1%; P = .004) and ORR (62.1% vs 39.8%; P = .03), and ASXL1 mutations (4.3% vs WT, 17.5%; P = .02) and spliceosome mutations (7.8% vs WT, 18.4%; P = .048) were associated with inferior rates of CR. In multivariable analyses (accounting for absolute neutrophil count, number of cytopenias, bone marrow blast percentage, WHO classification, age, sex, and genotype of TET2, ASXL1, TP53, and spliceosome genes), TET2 mutations (P = .03) and the TET2-mutant/ASXL1-WT genotype (P = .02) remained statistically significant covariates for CR. No specific somatic mutations or molecular patterns remained statistically significant for ORR in multivariable analysis; however, a trend was observed for the TET2-mutant/ASXL1-WT genotype (P = .10), TET2 mutations (P = .07), and a lower ORR with ASXL1 mutations (P = .08). On the basis of these findings, response rates were stratified and compared across combinations of TET2 and ASXL1 genotypes as depicted in Figure 2.
Molecular predictors of response
| Genotype | No. of patients | CR rate, % | Univariable | Multivariable | ORR rate, % | Univariable | Multivariable | ||
|---|---|---|---|---|---|---|---|---|---|
| P | OR (95% CI) | P | P | OR (95% CI) | P | ||||
| Total cohort | 247 | 15 | 42.1 | ||||||
| NGS result | .4 | .22 | |||||||
| No mutation | 28 | 21.4 | 53.6 | ||||||
| ≥1 mutation | 213 | 14.6 | 40.8 | ||||||
| TET2 | .09 | 2.80 (1.09-7.24) | .03* | .08 | 1.92 (0.93-3.94) | .07 | |||
| Mut | 41 | 24.3 | 56.1 | ||||||
| WT | 188 | 12.8 | 40.4 | ||||||
| ASXL1 | .02* | 0.13 (0.02-1.08) | .06 | .07 | 0.49 (0.22-1.08) | .08 | |||
| Mut | 47 | 4.3 | 29.8 | ||||||
| WT | 200 | 17.5 | 45.0 | ||||||
| TET2-mut/ASXL1-WT | .004* | 3.15 (1.20-8.30) | .02* | .03* | 1.99 (0.86-4.59) | .10 | |||
| Present | 29 | 34.5 | 62.1 | ||||||
| Other | 206 | 12.1 | 39.8 | ||||||
| DNMT3A | .31 | .85 | |||||||
| Mut | 35 | 8.6 | 45.7 | ||||||
| WT | 193 | 16.1 | 43.0 | ||||||
| EZH2 | >.99 | .59 | |||||||
| Mut | 15 | 13.3 | 33.3 | ||||||
| WT | 232 | 15.1 | 42.7 | ||||||
| DNA methylation mutation | .7 | .33 | |||||||
| Mut | 79 | 16.5 | 48.1 | ||||||
| WT | 148 | 14.2 | 41.2 | ||||||
| Epigenetic regulation | .47 | .36 | |||||||
| Mut | 120 | 13.3 | 40 | ||||||
| WT | 115 | 17.4 | 46.1 | ||||||
| SF3B1 | .7 | .4 | |||||||
| Mut | 13 | 7.7 | 30.8 | ||||||
| WT | 217 | 15.7 | 44.2 | ||||||
| Any spliceosome | .048* | 0.46 (0.16-1.33) | .15 | .57 | 0.86 (0.46-1.63) | .65 | |||
| Mut | 77 | 7.8 | 40.3 | ||||||
| WT | 152 | 18.4 | 44.7 | ||||||
| RUNX1 | >.99 | >.99 | |||||||
| Mut | 24 | 12.5 | 41.7 | ||||||
| WT | 223 | 15.2 | 42.2 | ||||||
| Signaling pathway | .18 | .57 | |||||||
| Mut | 32 | 6.3 | 37.5 | ||||||
| WT | 196 | 16.3 | 44.4 | ||||||
| TP53 | .08 | 1.29 (0.56-3.0) | .55 | .79 | 1.01 (0.54-1.89) | .96 | |||
| Mut | 82 | 20.7 | 43.9 | ||||||
| WT | 165 | 12.1 | 41.2 | ||||||
| Genotype | No. of patients | CR rate, % | Univariable | Multivariable | ORR rate, % | Univariable | Multivariable | ||
|---|---|---|---|---|---|---|---|---|---|
| P | OR (95% CI) | P | P | OR (95% CI) | P | ||||
| Total cohort | 247 | 15 | 42.1 | ||||||
| NGS result | .4 | .22 | |||||||
| No mutation | 28 | 21.4 | 53.6 | ||||||
| ≥1 mutation | 213 | 14.6 | 40.8 | ||||||
| TET2 | .09 | 2.80 (1.09-7.24) | .03* | .08 | 1.92 (0.93-3.94) | .07 | |||
| Mut | 41 | 24.3 | 56.1 | ||||||
| WT | 188 | 12.8 | 40.4 | ||||||
| ASXL1 | .02* | 0.13 (0.02-1.08) | .06 | .07 | 0.49 (0.22-1.08) | .08 | |||
| Mut | 47 | 4.3 | 29.8 | ||||||
| WT | 200 | 17.5 | 45.0 | ||||||
| TET2-mut/ASXL1-WT | .004* | 3.15 (1.20-8.30) | .02* | .03* | 1.99 (0.86-4.59) | .10 | |||
| Present | 29 | 34.5 | 62.1 | ||||||
| Other | 206 | 12.1 | 39.8 | ||||||
| DNMT3A | .31 | .85 | |||||||
| Mut | 35 | 8.6 | 45.7 | ||||||
| WT | 193 | 16.1 | 43.0 | ||||||
| EZH2 | >.99 | .59 | |||||||
| Mut | 15 | 13.3 | 33.3 | ||||||
| WT | 232 | 15.1 | 42.7 | ||||||
| DNA methylation mutation | .7 | .33 | |||||||
| Mut | 79 | 16.5 | 48.1 | ||||||
| WT | 148 | 14.2 | 41.2 | ||||||
| Epigenetic regulation | .47 | .36 | |||||||
| Mut | 120 | 13.3 | 40 | ||||||
| WT | 115 | 17.4 | 46.1 | ||||||
| SF3B1 | .7 | .4 | |||||||
| Mut | 13 | 7.7 | 30.8 | ||||||
| WT | 217 | 15.7 | 44.2 | ||||||
| Any spliceosome | .048* | 0.46 (0.16-1.33) | .15 | .57 | 0.86 (0.46-1.63) | .65 | |||
| Mut | 77 | 7.8 | 40.3 | ||||||
| WT | 152 | 18.4 | 44.7 | ||||||
| RUNX1 | >.99 | >.99 | |||||||
| Mut | 24 | 12.5 | 41.7 | ||||||
| WT | 223 | 15.2 | 42.2 | ||||||
| Signaling pathway | .18 | .57 | |||||||
| Mut | 32 | 6.3 | 37.5 | ||||||
| WT | 196 | 16.3 | 44.4 | ||||||
| TP53 | .08 | 1.29 (0.56-3.0) | .55 | .79 | 1.01 (0.54-1.89) | .96 | |||
| Mut | 82 | 20.7 | 43.9 | ||||||
| WT | 165 | 12.1 | 41.2 | ||||||
Response rates among the total cohort stratified by genotype; multivariable analysis was performed using a binary logistic-regression model (ORR defined as CR + mCR + PR + HI).
CI, confidence interval; OR, odds ratio.
Denotes statistical significance (P < .05).
![Response stratified by TET2 and ASXL1 genotype. Comparison of overall response rate (A) and complete remission rate (B) among combinations of TET2 and ASXL1 genotypes (TET2-mutated [mut]/ASXL1-wt [wild-type], n = 29; TET2-wt/ASXL1-wt, n = 159; ASXL1-mut, n = 47). The vertical axis represents the frequency of response and comparisons between specific groups are annotated. *Denotes statistical significance (P < .05).](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/5/4/10.1182_bloodadvances.2020003508/2/m_advancesadv2020003508f2.png?Expires=1769864991&Signature=qhS1y9lw6brNrHWda1hhoDb0JoZyD7GvO-5xX45bBIWcUMnanmSmkBsrKtqzkOpJINTKNoClro-KqzMJgDxXeyCs35A8NVvIURmS3A1xUoJ6y~2ng4DH0AGqqWVB-2tPmoB2cDhnV4mDDsb6IsQr8yF8NRWTeXJfdRBGIKWQADLfEozf3HHF4jR8yANxV1leRJvvXLgBQhxPbGTqtfXctaRw2K4p0h1Tv8aUeZ-mRvvcYTV9CxeYhcRI-ydU6kCPvM1FYDnO~x21nXF9J5FTGDnxDHuosKljOapXPfyTFTBC6gbccdNMlynEXc8tHV-5tdiuBEvzDmH-KBREHjfB3Q__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Response stratified by TET2 and ASXL1 genotype. Comparison of overall response rate (A) and complete remission rate (B) among combinations of TET2 and ASXL1 genotypes (TET2-mutated [mut]/ASXL1-wt [wild-type], n = 29; TET2-wt/ASXL1-wt, n = 159; ASXL1-mut, n = 47). The vertical axis represents the frequency of response and comparisons between specific groups are annotated. *Denotes statistical significance (P < .05).
Response stratified by TET2 and ASXL1 genotype. Comparison of overall response rate (A) and complete remission rate (B) among combinations of TET2 and ASXL1 genotypes (TET2-mutated [mut]/ASXL1-wt [wild-type], n = 29; TET2-wt/ASXL1-wt, n = 159; ASXL1-mut, n = 47). The vertical axis represents the frequency of response and comparisons between specific groups are annotated. *Denotes statistical significance (P < .05).
Responses were compared between therapeutic agents with an ORR of 40.4% for azacitidine regimens compared with 55.2% for decitabine (P = .16) and CR rates of 13.3% and 27.6% (P = .05), respectively. Identical comparisons were made in TP53-mutated patients with an ORR of 40.6% vs 55.6% (P = .29) and CR rates of 17.2% vs 33.3% (P = .19) with azacitidine compared with decitabine, respectively.
Patterns of HMA failure were investigated in a subset of patients (n = 106) that identified TP53 as the most common somatic mutation in patients who progressed to AML during or at completion of therapy (13 of 24 patients), and 37.1% of TP53-mutated patients exhibited progression. In contrast, progression to AML was rare in patients with spliceosome mutations: 1 instance occurred in an SRSF2-mutated patient (n = 12), and no instances were observed among SF3B1- or U2AF1-mutated patients.
Covariates for OS
The median OS of HMA-treated patients was 15 months (95% confidence interval, 12.4-17.5 months). OS was significantly longer in patients responding to HMA therapy (18.5 vs 12.8 months in nonresponders; HR, 0.60; P = .009). Among responders, there was no difference in OS observed in patients achieving CR vs HI (15.6 vs 21.7 months; HR, 0.74; P = .46), whereas patients with mCR exhibited an OS comparable to that of nonresponders (12.2 vs 12.8 months; HR, 0.93; P = .78). Additional clinical variables and their impact on OS are summarized in supplemental Table 2. Only IPSS-R category (P < .001), response to HMA (HR, 0.30; P < .001), and allo-HCT (HR, 0.28; P < .001) retained independent significance in the final multivariable model.
Analysis of somatic mutations and their impact on survival is summarized in Table 3. In univariable analysis, mutations in EZH2 (Figure 3A), TP53 (Figure 3B), the absence of detectable mutations by NGS (Figure 3C), and the number of mutated genes were associated with OS. Stratifying patients into groups based upon the total number of mutated genes was highly predictive in TP53-WT patients, with cut points of 0 vs 1 to 3 vs 4+ mutations most informative (Figure 3D). In multivariable analysis accounting for molecular (Table 3) and clinical (supplemental Table 2) variables, the number of somatic mutations (P = .02) and mutations in TP53 (HR, 2.33; P = .001) and EZH2 (HR, 2.41; P = .04) retained significance. Complex karyotype was also associated with inferior OS (10.2 vs 25.8 months in those without a complex karyotype; HR, 3.26; P < .001), which remained significant in multivariable analysis when replacing TP53 mutations (P = .001). Among TP53-mutated patients, there was no significant difference in OS between patients with monoallelic vs biallelic loss (10.2 vs 9.6 months; HR, 0.82; P = .5).
Molecular predictors of OS
| Genotype | No. of patients | Median OS, mo | Univariable analysis | Multivariable analysis | |||
|---|---|---|---|---|---|---|---|
| HR | P | HR | 95% CI | P | |||
| Total cohort | 247 | 15 | |||||
| NGS result | 0.46 | .02* | 1.34 | 0.54-3.33 | .52 | ||
| No mutation | 28 | 24 | |||||
| ≥1 mutation | 213 | 14.1 | |||||
| No. of mutations | .005* | 1.22 | 1.03-1.44 | .02* | |||
| TET2 | 0.89 | .67 | |||||
| Mut | 41 | 16.1 | |||||
| WT | 188 | 14.4 | |||||
| ASXL1 | 0.84 | .49 | |||||
| Mut | 47 | 18.6 | |||||
| WT | 200 | 14.4 | |||||
| TET2-mut/ASXL1-wt and TP53-wt | 0.51 | .13 | |||||
| Present | 21 | 16.1 | |||||
| Other | 218 | 14.4 | |||||
| DNMT3A | 1.37 | .24 | |||||
| Mut | 35 | 11.4 | |||||
| WT | 193 | 15.5 | |||||
| EZH2 | 2.83 | <.001* | 2.41 | 1.03-5.64 | .04* | ||
| Mut | 15 | 9.9 | |||||
| WT | 232 | 16.1 | |||||
| DNA methylation mutation | 0.96 | .83 | |||||
| Mut | 79 | 14.2 | |||||
| WT | 148 | 14.4 | |||||
| Epigenetic regulation mutation | 1.04 | .82 | |||||
| Mut | 120 | 14.2 | |||||
| WT | 115 | 15.5 | |||||
| SF3B1 | 0.77 | .57 | |||||
| Mut | 13 | 18.5 | |||||
| WT | 217 | 14.4 | |||||
| Any spliceosome | 0.68 | .07 | 1.06 | 0.58-1.95 | .84 | ||
| Mut | 77 | 19.1 | |||||
| WT | 152 | 13.1 | |||||
| RUNX1 | 1.09 | .78† | |||||
| Mut | 24 | 12.4 | |||||
| WT | 223 | 15.5 | |||||
| Signaling pathway | 1.15 | .61 | |||||
| Mut | 32 | 14.4 | |||||
| WT | 196 | 14.5 | |||||
| TP53 | 2.82 | <.001* | 2.33 | 1.41-3.85 | .001* | ||
| Mut | 82 | 9.7 | |||||
| WT | 165 | 21.7 | |||||
| Genotype | No. of patients | Median OS, mo | Univariable analysis | Multivariable analysis | |||
|---|---|---|---|---|---|---|---|
| HR | P | HR | 95% CI | P | |||
| Total cohort | 247 | 15 | |||||
| NGS result | 0.46 | .02* | 1.34 | 0.54-3.33 | .52 | ||
| No mutation | 28 | 24 | |||||
| ≥1 mutation | 213 | 14.1 | |||||
| No. of mutations | .005* | 1.22 | 1.03-1.44 | .02* | |||
| TET2 | 0.89 | .67 | |||||
| Mut | 41 | 16.1 | |||||
| WT | 188 | 14.4 | |||||
| ASXL1 | 0.84 | .49 | |||||
| Mut | 47 | 18.6 | |||||
| WT | 200 | 14.4 | |||||
| TET2-mut/ASXL1-wt and TP53-wt | 0.51 | .13 | |||||
| Present | 21 | 16.1 | |||||
| Other | 218 | 14.4 | |||||
| DNMT3A | 1.37 | .24 | |||||
| Mut | 35 | 11.4 | |||||
| WT | 193 | 15.5 | |||||
| EZH2 | 2.83 | <.001* | 2.41 | 1.03-5.64 | .04* | ||
| Mut | 15 | 9.9 | |||||
| WT | 232 | 16.1 | |||||
| DNA methylation mutation | 0.96 | .83 | |||||
| Mut | 79 | 14.2 | |||||
| WT | 148 | 14.4 | |||||
| Epigenetic regulation mutation | 1.04 | .82 | |||||
| Mut | 120 | 14.2 | |||||
| WT | 115 | 15.5 | |||||
| SF3B1 | 0.77 | .57 | |||||
| Mut | 13 | 18.5 | |||||
| WT | 217 | 14.4 | |||||
| Any spliceosome | 0.68 | .07 | 1.06 | 0.58-1.95 | .84 | ||
| Mut | 77 | 19.1 | |||||
| WT | 152 | 13.1 | |||||
| RUNX1 | 1.09 | .78† | |||||
| Mut | 24 | 12.4 | |||||
| WT | 223 | 15.5 | |||||
| Signaling pathway | 1.15 | .61 | |||||
| Mut | 32 | 14.4 | |||||
| WT | 196 | 14.5 | |||||
| TP53 | 2.82 | <.001* | 2.33 | 1.41-3.85 | .001* | ||
| Mut | 82 | 9.7 | |||||
| WT | 165 | 21.7 | |||||
HR, hazard ratio.
Denotes statistical significance (P < .05).
Associated with a statistically significant (P < .001) impact on OS when stratifying by both RUNX1 and TP53 genotype (see supplemental Figure 2).
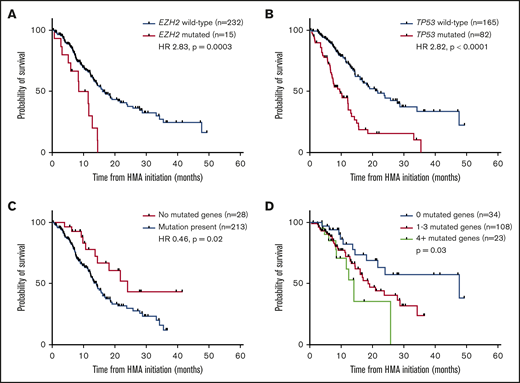
OS of specific molecular predictors. Survival impact of EZH2 mutations (A), TP53 mutations (B), and the absence of detectable mutations (C) on NGS. (D) Survival stratified by number of mutations in TP53-wt patients.
OS of specific molecular predictors. Survival impact of EZH2 mutations (A), TP53 mutations (B), and the absence of detectable mutations (C) on NGS. (D) Survival stratified by number of mutations in TP53-wt patients.
Analysis of OS according to specific HMA (ie, azacitidine vs decitabine) showed similar outcomes within the entire cohort (15.5 vs 12.3 months; HR, 0.72; P = .30) and among TP53-mutated patients (7.8 vs 12.3 months; HR, 1.21; P = .62). Similarly, there was no difference in OS among TP53-mutated patients treated with 5-day (n = 12) vs 10-day (n = 6) decitabine regimens (12.3 vs 7 months; HR, 0.33; P = .08).
Sequential molecular profiling
Sequential molecular profiling was performed in 108 patients (43.7%) either during or at completion of HMA therapy, among whom 12 had no mutation detectable at baseline. Sequential NGS was performed on bone marrow samples in 89 patients (82.4%) and peripheral blood samples in 19 patients (17.6%), with a median time to sequential NGS of 4.5 months. Among 96 patients with a detectable mutation at baseline, 25 (26%) had clearance of all mutations with HMA therapy; these patients exhibited a median OS of 15.6 months compared with 14.2 months in those with mutation persistence (HR, 0.62; P = .27). Findings were similar when not accounting for clearance of the clonal hematopoiesis-associated genes DNMT3A, TET2, and ASXL1 (HR, 0.76; P = .48), and clearance was observed in 29 patients (30.5%). Fifty patients (52.1%) had clearance of at least 1 mutation (median OS, 15.6 vs 14.2 months in those without clearance of at least 1 mutation; HR, 0.77; P = .45), and 33 patients (30.6%) acquired at least 1 new mutation (median OS, 21.7 vs 14.2 months in those who did not acquire at least 1 new mutation; HR, 0.93; P = .83) during therapy. At completion of HMA therapy, acquisition of new mutations was identified in 26.7% of HMA nonresponders, 43.2% of HMA responders who subsequently progressed, and 47.1% of patients who progressed to AML.
Similar analyses were performed among patients harboring TP53 mutations (n = 47), with 15 patients (31.9%) clearing all baseline mutations (median OS, 13.3 vs 10.3 months with persistent mutations; HR, 0.46; P = .06). When solely analyzing clearance of TP53, this was predictive of an improvement in median OS (15.6 vs 7.7 months with TP53 persistence; HR, 0.28; P = .001; Figure 4A) among the 22 patients (46.8%) exhibiting TP53 clearance with HMA therapy. Of these 22 patients, 12 (54.5%) achieved CR, 3 (13.6%) mCR, 3 (13.6%) HI, and 4 (18.2%) stable disease, with no difference in OS between patients who achieved CR and those who did not (median OS, 14.5 vs 13.1 months; HR, 0.68; P = .61). Patients with clearance of TP53 mutations demonstrated a lower median VAF (12% vs 33.3%; P = .02) but no difference in frequency of biallelic loss (P > .99), associated chromosome 17 abnormalities (P > .99), class of mutation (P = .36), or complex cytogenetics (P = .47). In addition, there was no difference in time to sequential NGS analysis (3.7 vs 4.1 months; P = .68).
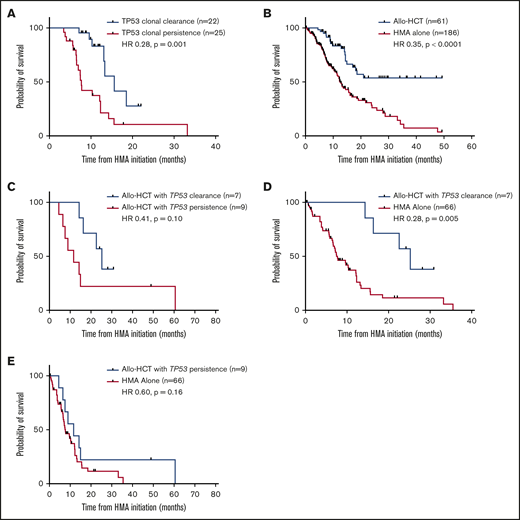
Impact of TP53 clonal clearance on OS with HMA therapy and allo-HCT. (A) Impact of TP53 clonal clearance on OS in all HMA-treated patients undergoing sequential molecular profiling (median OS, 15.6 months with TP53 clearance and 7.7 months with TP53 persistence). (B) OS of all patients within the cohort proceeding to allo-HCT after first-line therapy with an HMA. (C) Impact of TP53 clonal clearance on OS among patients proceeding to allo-HCT (median OS, 25.2 vs 11.7 months, respectively). Comparison of OS among patients proceeding to allo-HCT vs patients receiving HMA therapy alone in those with TP53 clonal clearance before transplant (25.2 vs 7.7 months) (D) vs those with TP53 clonal persistence (11.7 vs 7.7 months) (E).
Impact of TP53 clonal clearance on OS with HMA therapy and allo-HCT. (A) Impact of TP53 clonal clearance on OS in all HMA-treated patients undergoing sequential molecular profiling (median OS, 15.6 months with TP53 clearance and 7.7 months with TP53 persistence). (B) OS of all patients within the cohort proceeding to allo-HCT after first-line therapy with an HMA. (C) Impact of TP53 clonal clearance on OS among patients proceeding to allo-HCT (median OS, 25.2 vs 11.7 months, respectively). Comparison of OS among patients proceeding to allo-HCT vs patients receiving HMA therapy alone in those with TP53 clonal clearance before transplant (25.2 vs 7.7 months) (D) vs those with TP53 clonal persistence (11.7 vs 7.7 months) (E).
Within the total cohort, patients who proceeded to allo-HCT (n = 61) after first-line treatment demonstrated improved OS compared with those treated with HMA alone (not reached vs 12.4 months; HR, 0.35; P < .0001; Figure 4B). Among these patients, 27 underwent serial molecular analysis before allo-HCT; 10 patients cleared all detectable mutations (median OS not reached vs 14.4 months with detectable mutations; HR, 0.47; P = .47; supplemental Figure 1). Among 16 TP53-mutated patients proceeding to allo-HCT, 7 achieved a complete molecular remission before transplantation with a trend toward improved OS compared with patients with clonal persistence (median OS, 25.2 vs 11.7 months; HR, 0.41; P = .10; Figure 4C). Furthermore, patients with TP53 clearance before allo-HCT had a survival benefit compared with patients treated solely with an HMA (25.2 vs 7.7 months; HR, 0.28; P = .005; Figure 4D), but patients with TP53 persistence did not (11.7 vs 7.7 months; HR, 0.60; P = .16; Figure 4E). Patients demonstrating TP53 clearance before allo-HCT were younger than those with TP53 persistence (median, 53 vs 67 years old, respectively; P = .09), but time to serial molecular analysis was similar (median, 4.1 vs 4.0 months; P = .56).
Discussion
Treatment of higher-risk MDS with HMAs remains the standard of care, but the rate of nonresponse is high, and putative biomarkers for treatment benefit show inconsistent results across studies. Furthermore, the clinical impact of monitoring mutations longitudinally during therapy remains largely unexplored. To this end, we present results from the largest single-institution cohort to date of higher-risk MDS patients treated with HMA therapy after baseline molecular profiling by NGS. We identify several NGS-based molecular covariates for response and survival with novel observations from a large subset analyzed serially during therapy.
Mutations in TET2 and ASXL1 genes emerged as the most informative variables associated with response to HMA therapy in multivariable analysis that accounted for both clinical and molecular variables. Although TET2-mutated patients as a whole exhibited ORR and CR rates higher than those of the overall cohort, this benefit was largely driven by patients harboring the TET2-mutant/ASXL1-WT genotype, confirming previous observations by Bejar et al13 from a smaller cohort. Notably, ASXL1 mutations were the strongest molecular covariate for inferior response, in particular CR, and they largely negated the benefit of TET2 mutations. Similar outcomes were observed in patients with dual TET2 and ASXL1 mutations compared with TET2-WT/ASXL1-mutated patients.
Mutation of a spliceosome component gene was the only other molecular covariate associated with response in univariable analysis, which demonstrated a lower rate of CR but no difference in ORR. Nonetheless, multivariable analysis by logistic regression revealed no impact of spliceosome mutations and identified no baseline clinical variables that retained significance. Although DNMT3A mutations were initially hypothesized to predict for response to HMAs, this study aligns with more recent reports that have not identified such a benefit, which provides further definitive evidence that no such association exists.13,15,16,18,27,28 Thus, stratifying by composite TET2/ASXL1 genotype seems to be the strongest molecular biomarker for response to HMA therapy, outperforming all other clinical and molecular variables evaluated. As in previous studies, improved response rates in TET2-mutated patients did not correlate with a survival advantage, although a trend toward improved OS was found in TET2-mutated patients who had both ASXL1 and TP53 WT.13,14
The median OS of the total cohort was 15 months, considerably lower than that observed in the AZA-001 study but comparable to that reported in similar real-world post-marketing data for higher-risk MDS patients treated with HMA.1,29-31 Patients achieving a response to an HMA experienced an improvement in OS, with benefit largely driven by patients achieving CR or HI. Patients who achieved mCR as best response had no evidence of a survival benefit and their outcomes were nearly identical to those of nonresponders, thus raising the question of the value of including mCR in response assessments in future studies.
An improvement in OS was observed in patients who proceeded to allo-HCT after first-line HMA therapy. This benefit was restricted to patients with ≥5% blasts at treatment initiation, but it was independent of response to HMA. Importantly, the survival advantage with allo-HCT was maintained in multivariable analysis that accounted for both clinical and molecular variables, which supports the use of HMA in the pretransplant setting.
Molecular covariates for inferior OS in univariable analysis included the total number of mutations and mutations in the TP53 and EZH2 genes, whereas the absence of mutations was associated with improved OS. Each of the molecular categories predictive of inferior OS retained independent significance in the final multivariable model. Previous work has yielded conflicting survival data on the impact of ASXL1 mutations and HMA therapy.12,32,33 In this study, ASXL1 mutations had no impact on OS despite inferior response rates, whereas EZH2 mutations were associated with a poor OS, which is contrary to previous observations.
Interestingly, genotypes that were associated with inferior OS (ie, TP53- and EZH2-mutated) were not associated with primary resistance to HMAs; TP53-mutated patients actually had a trend toward higher CR rates. Likewise, genotypes associated with primary resistance to HMAs (ie, TET2-WT/ASXL1-mutated) had no direct impact on OS and were largely mutually exclusive with those that did, suggesting distinct mechanisms of primary vs secondary resistance.
Our findings are in agreement with previous reports that HMA therapy does not overcome the adverse prognostic impact of TP53 mutations, which remain perhaps the strongest predictor of inferior outcomes.13,19,32 TP53 was the most commonly mutated gene in this study (33%), which is a considerably higher frequency than that observed in larger genomic studies of MDS.6,7 However, a higher frequency has been reported in cohorts from tertiary cancer centers, which likely reflects referral bias.13,19,34,35 Importantly, this represents the largest cohort to date of TP53-mutated patients treated with first-line HMA and provides important historical data for ongoing trials evaluating novel agents in combination with HMA, including eprenetapopt and magrolimab, in this molecular subgroup.
To account for a potential adverse prognostic impact related to the high frequency of TP53 mutations, patients with TP53-WT were analyzed as a separate cohort for both response rate (supplemental Table 3) and OS (supplemental Table 4). A trend toward improved OS was seen in spliceosome-mutated patients overall, but this seemed to be driven by the majority of TP53-mutated patients who fall within the spliceosome WT group. No survival impact was seen in the TP53-WT cohort or in multivariable analysis of the total cohort. On the contrary, no difference was observed when solely evaluating RUNX1 genotypes, but stratifying by both RUNX1 and TP53 status did delineate 3 groups (supplemental Figure 2); RUNX1-mutated patients had an intermediate OS closer to that of TP53-mutated patients (median OS, 12.4 vs 9.7 months for TP53; HR, 0.59; P = .10). In further evaluating the impact of mutational burden, stratifying patients into specific groups based on the number of mutations was less predictive in the total cohort. This seemed to be largely a result of the inferior impact of TP53 mutations which were associated with a lower number of coexisting mutations, thus impacting the survival of patients with 1 or 2 mutations. When isolating the TP53-WT cohort, however, patients with 1 to 3 mutations demonstrated similar outcomes, and grouping patients into categories of those with 0, 1 to 3, or 4 or more mutations was predictive of OS.
In addition to assessing baseline somatic mutations before initiation of therapy, 108 patients had serial sequencing performed during or at the completion of therapy, primarily from bone marrow samples. The number of each individual mutation was relatively low in this subgroup, so analyses instead focused on gain or clearance of any mutation. Of particular interest, complete clearance of all mutations, clearance of at least 1 mutation, or the gain of 1 or more mutations had no impact on survival. These findings are in line with investigations in AML showing that preleukemic mutations commonly observed in clonal hematopoiesis and MDS (including DNMT3A, TET2, and ASXL1) are significantly less likely to clear with therapy, with persistence having no impact on outcome.36,37
Falconi et al18 previously reported that while the VAF of the majority of somatic mutations remained stable during HMA therapy, TP53 mutations were the exception, and a reduction in mutational burden was routinely observed. In this study, nearly half the evaluable patients achieved TP53 clearance by NGS (VAF <5%), and these patients demonstrated a significant improvement in OS. Interestingly, mutation clearance was seen in the majority of patients with CR (12 of 15 patients), but nearly half of such patients had a response less than CR, including 4 patients with stable disease. Stratifying by quality of response had no further impact on OS, suggesting that TP53 clearance supersedes clinical response in prognostic importance in TP53-mutated patients.
Similarly, clearance of TP53 mutations also portended for improved survival after allo-HCT, albeit in a small sample size. These findings align with those reported by Welch et al20 in TP53-mutated MDS and AML patients treated with a 10-day regimen of decitabine. Although further validation in larger cohorts is necessary, these findings support a novel strategy for selecting TP53-mutated patients as allo-HCT candidates. Despite improved outcomes, however, just 3 of the 16 TP53-mutated patients undergoing allo-HCT experienced a relapse-free survival lasting longer than 24 months, which supports the need for new therapeutic strategies in this group.
Patient heterogeneity is a potential limitation of the study. The majority of the cohort consisted of patients with higher-risk MDS, but small numbers of patients with lower-risk MDS, MDS/MPN, and AML (20% to 30% blasts) were included. However, this is consistent with typical populations treated with HMAs in clinical practice. To account for any bias related to diagnosis, WHO category was included in multivariable analyses and demonstrated no impact on outcome. Similarly, variability in the timing of sequential NGS analysis has the potential to bias these results, although the median time to sequential NGS was 4.5 months, consistent with expected timing of response assessments in patients treated with HMA.
In conclusion, this study expands on previous work that evaluated molecular biomarkers for outcome with HMA therapy and supports the incorporation of baseline molecular profiling before treatment. It also provides further insight into the application of serial sequencing during treatment, which seems particularly valuable in TP53-mutated patients. These findings suggest that somatic mutations and classic clinical variables do not fully account for variability in outcomes, with novel biomarkers yet to be discovered.
To request data, please contact David A. Sallman by e-mail at david.sallman@moffitt.org.
Acknowledgments
This work was supported in part by the S Foundation Young Investigator Grant, the Early Career Award of the Dresner Foundation, and the Edward P. Evans Foundation Award (all to D.A.S.).
Authorship
Contribution: A.M.H. and D.A.S. conceived of and designed the study; A.M.H., R.S.K., N.A.A., O.C., D.A.S., J.S., and M.H. collected and assembled the data; A.M.H., R.S.K., and D.A.S. analyzed and interpreted the data; and A.M.H., R.S.K., N.A.A., O.C., S.Y., E.P., J.S., M.H., C.T., K.L.S., J.E.L., A.F.L, and D.A.S. wrote the manuscript.
Conflict-of-interest disclosure: D.A.S., E.P., and A.F.L received research funding from Celgene. The remaining authors declare no competing financial interests.
Correspondence: David A. Sallman, Malignant Hematology Department, H. Lee Moffit Cancer Center, 12902 USF Magnolia Dr, Tampa, FL 33612; e-mail: david.sallman@moffitt.org.

