

Abstract
Tyrosine kinase inhibitors (TKIs) are used to target dysregulated signaling pathways in virtually all hematologic malignancies. Many of the targeted signaling pathways are also essential in nonmalignant immune cells. The current coronavirus severe acute respiratory syndrome coronavirus 2 pandemic catalyzed clinical exploration of TKIs in the treatment of the various stages of COVID-19, which are characterized by distinct immune-related complications. Most of the reported effects of TKIs on immune regulation have been explored in vitro, with different class-specific drugs having nonoverlapping target affinities. Moreover, many of the reported in vivo effects are based on artificial animal models or on observations made in symptomatic patients with a hematologic malignancy who often already suffer from disturbed immune regulation. Based on in vitro and clinical observations, we attempt to decipher the impact of the main TKIs approved or in late-stage development for the treatment of hematological malignancies, including inhibitors of Bruton’s tyrosine kinase, spleen tyrosine kinase, BCR-Abl, phosphatidylinositol 3-kinase/ mammalian target of rapamycin, JAK/STAT, and FMS-like tyrosine kinase 3, to provide a rationale for how such inhibitors could modify clinical courses of diseases, such as COVID-19.
Introduction
Over the last decades, dysregulation of signaling pathways involved in proliferation, activation, and survival have been identified in virtually all hematologic malignancies. This has led to the development and application of a diverse set of tyrosine kinase inhibitors (TKIs) that have drastically improved the clinical outcomes of these diseases.1
In addition to their role in tumorigenesis, many of the targeted signaling pathways are essential in nonmalignant immune cells. Furthermore, many TKIs have significant off-target effects.1
Their immunomodulatory effects led to an increased interest in TKIs developed for hematologic malignancies in applications other than cancer. The current coronavirus severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pandemic catalyzed clinical exploration of TKIs in the treatment of the various stages of COVID-19.2 Because a standard treatment for COVID-19 is still lacking, drug repurposing strategies for TKIs are being considered in the management of this potentially lethal disease. Despite the vast number of publications on TKIs, an up-to-date systematic review on the presently exploited immunomodulatory effects of TKIs is lacking. We focus on the main TKIs approved or in late-stage development for the treatment of hematological malignancies: Bruton’s tyrosine kinase (BTK), spleen tyrosine kinase (SYK), BCR-Abl, phosphatidylinositol 3-kinase (PI3K)/mammalian target of rapamycin (mTOR), JAK/STAT, and FMS-like tyrosine kinase 3 (FLT3). First, we summarize the putative roles of these kinases in innate (neutrophils, dendritic cells [DCs], monocytes, and natural killer [NK] cells) and adaptive (B cells and T cells) immune cells and then systematically discuss in vitro and in vivo evidence for their effects in these cells to provide a rationale for how such inhibitors could modify clinical courses of diseases, such as COVID-19. COVID-19 has major global impacts on health and economy, and our understanding of its underlying pathophysiological mechanisms has progressed with unprecedented speed. The pandemic has also triggered a storm of studies on treatment options and vaccine development. Amid this ongoing research, any review has the obvious limitation that current knowledge may quickly be outdated. Nevertheless, current and future research on COVID-19 and other diseases characterized by immune-related complications could benefit from an overview of the immunomodulatory effects of TKIs provided in this review.
The main TKIs and their targets
TKIs, their indications, and the clinically relevant (off-target) kinases that they inhibit, will be discussed are summarized in Table 1. The available data on their immunomodulatory effects are summarized by TKI class in supplemental Tables 1 through 6.
TKIs, their indications, and the (off-target) kinases they inhibit
| Class/drugs | Indication (T)rial/(R)egistration/(O)ff-label | Relevant immunomodulatory off-target molecules |
|---|---|---|
| BTK* | ||
| Ibrutinib | CLL/SLL(R), WM(R), MCL(R), MZL(R), BCL(R), cGvHD(R)103 | ITK, EGFR, JAK3103 |
| Acalabrutinib | CLL/SLL(R), NHL(R), MCL(R)104 | |
| Zanubrutinib | MZL(R), CLL/SLL(T), WM(T), MCL(T)105 | |
| Tirabrutinib | CLL(T), BCL(T), WM(T), PCNSL(T)104 | |
| SYK† | ||
| Fostamatinib | ITP(R), RA(T), CLL(T), BCL(T)106,107 | FLT3, JAK263,108 |
| Entospletinib | AML(T), CLL(T), BCL(T), GvHD(T), MCL(T), NHL(T)106 | JAK2106 |
| Cerdulatinib | DLBCL(T), CLL(T), NHL(T)106 | JAK1/JAK3, AKT, NF-κB106,107 |
| BCR-Abl‡ | ||
| Imatinib | Ph+CML(R)109 | |
| Nilotinib | Ph+CML(R)110 | |
| Dasatinib | Ph+CML(R)58 | BTK58 |
| Bosutinib | Ph+CML(R), Ph+ALL(R)111 | |
| Ponatinib | Ph+CML(R), Ph+ALL(R)111 | Abl, FLT3111 |
| Radotinib | Ph+CML(R)112 | |
| PI3K/mTOR§ | ||
| Idelalisib (p110δ) | ALL(R), CLL/SLL(R), FL(R)113 | |
| Copanlisib (PI3Kδ & PI3Kδ) | FL(R), CLL/SLL(R)114,115 | |
| Duvelisib | FL(R), CLL/SLL(R)115 | |
| Umbralisib (PI3Kδ) | MZL(T), CLL(T), FL(T), DLBCL(T)116 | |
| Temsirolimus | NHL(R), MCL(R).117 | |
| JAK¶ | ||
| Ruxolitinib (JAK1/2) | PV(R), MF(R), GvHD(R)118 | |
| Fedratinib (JAK2) | MF(R)119 | FLT3, BCR-Abl120 |
| Momelotinib (JAK1/2) | MPN(T)121 | |
| Pacritinib (JAK2) | MPN(T)122 | FLT3122 |
| FLT3|| | ||
| Midostaurin | FLT3-mutated AML(R), ASM(R), MC(R)123 | SYK123 |
| Sunitinib | RCC(R), GI(R), FLT3-mutated AML(O)124 | |
| Sorafenib | HCC(R), RCC(R), FLT3-mutated AML(O)125 | |
| Gilteritinib | FLT3-mutated AML(R)125 | |
| Crenolanib | FLT3-mutated AML(T)125 | |
| Quizartinib | FLT3-mutated AML(T)125 |
| Class/drugs | Indication (T)rial/(R)egistration/(O)ff-label | Relevant immunomodulatory off-target molecules |
|---|---|---|
| BTK* | ||
| Ibrutinib | CLL/SLL(R), WM(R), MCL(R), MZL(R), BCL(R), cGvHD(R)103 | ITK, EGFR, JAK3103 |
| Acalabrutinib | CLL/SLL(R), NHL(R), MCL(R)104 | |
| Zanubrutinib | MZL(R), CLL/SLL(T), WM(T), MCL(T)105 | |
| Tirabrutinib | CLL(T), BCL(T), WM(T), PCNSL(T)104 | |
| SYK† | ||
| Fostamatinib | ITP(R), RA(T), CLL(T), BCL(T)106,107 | FLT3, JAK263,108 |
| Entospletinib | AML(T), CLL(T), BCL(T), GvHD(T), MCL(T), NHL(T)106 | JAK2106 |
| Cerdulatinib | DLBCL(T), CLL(T), NHL(T)106 | JAK1/JAK3, AKT, NF-κB106,107 |
| BCR-Abl‡ | ||
| Imatinib | Ph+CML(R)109 | |
| Nilotinib | Ph+CML(R)110 | |
| Dasatinib | Ph+CML(R)58 | BTK58 |
| Bosutinib | Ph+CML(R), Ph+ALL(R)111 | |
| Ponatinib | Ph+CML(R), Ph+ALL(R)111 | Abl, FLT3111 |
| Radotinib | Ph+CML(R)112 | |
| PI3K/mTOR§ | ||
| Idelalisib (p110δ) | ALL(R), CLL/SLL(R), FL(R)113 | |
| Copanlisib (PI3Kδ & PI3Kδ) | FL(R), CLL/SLL(R)114,115 | |
| Duvelisib | FL(R), CLL/SLL(R)115 | |
| Umbralisib (PI3Kδ) | MZL(T), CLL(T), FL(T), DLBCL(T)116 | |
| Temsirolimus | NHL(R), MCL(R).117 | |
| JAK¶ | ||
| Ruxolitinib (JAK1/2) | PV(R), MF(R), GvHD(R)118 | |
| Fedratinib (JAK2) | MF(R)119 | FLT3, BCR-Abl120 |
| Momelotinib (JAK1/2) | MPN(T)121 | |
| Pacritinib (JAK2) | MPN(T)122 | FLT3122 |
| FLT3|| | ||
| Midostaurin | FLT3-mutated AML(R), ASM(R), MC(R)123 | SYK123 |
| Sunitinib | RCC(R), GI(R), FLT3-mutated AML(O)124 | |
| Sorafenib | HCC(R), RCC(R), FLT3-mutated AML(O)125 | |
| Gilteritinib | FLT3-mutated AML(R)125 | |
| Crenolanib | FLT3-mutated AML(T)125 | |
| Quizartinib | FLT3-mutated AML(T)125 |
AKT, protein kinase B; ALL, acute lymphoid leukemia; AML, acute myeloid leukemia; ASM, aggressive systemic mastocytosis; BCL, B-cell lymphoma; cGvHD, chronic graft-versus-host disease; CLL, chronic lymphocytic leukemia; CML, chronic myeloid leukemia; DLBCL, diffuse large B-cell lymphoma; EGFR, epidermal growth factor receptor; FL, follicular non-Hodgkin lymphoma; GI, gastrointestinal tumors; GvHD, graft-versus-host disease; HCC, hepatocellular carcinoma; ITP, idiopathic thrombocytopenic purpura; ITK, interleukin-2–inducible T-cell kinase; MC, mast cell leukemia; MCL, mantle cell lymphoma; MF, myelofibrosis; MPN, myeloproliferative neoplasm; MZL, marginal zone lymphoma; NHL, non-Hodgkin lymphoma; PCSNL, primary central nervous system lymphoma; Ph+, Philadelphia chromosome positive; PV, polycythemia vera; RA, rheumatoid arthritis; RCC, renal cell carcinoma; SLL, small lymphocytic leukemia; WM, Waldenström macroglobulinemia.
Summarized in supplemental Table 1.
Summarized in supplemental Table 2.
Summarized in supplemental Table 3.
Summarized in supplemental Table 4.
Summarized in supplemental Table 5.
Summarized in supplemental Table 6.
Role of the target kinases BTK, SYK, BCR-Abl, PI3K/mTOR, JAK/STAT, and FLT3 in normal immune function
The innate immune system
Cells of the innate immune system act as first responders for the detection and clearance of (viral) infections.3 They secrete proinflammatory cytokines that inhibit viral replication, stimulate the adaptive immune response, and recruit other immune cells to the site of infection.3 A schematic overview of a normal immune response during a viral infection can be found in Figure 1.
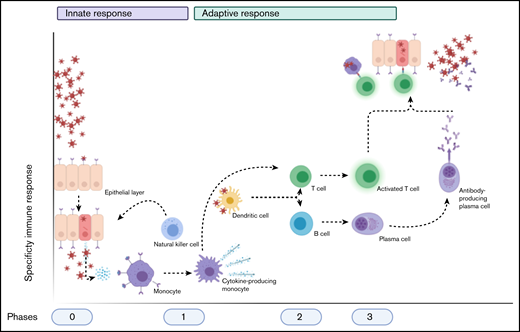
Schematic overview of the normal immune after a viral infection. Phase 0 consists of epithelial infection with the virus, after which monocytes detect the pathogen and subsequent cytokine production and differentiate into cytokine-producing macrophages and other cell types. Phase 1 entails the attraction and activation of other innate immune cells, such as NK cells, which kill infected cells, and DCs, which then travel to naive T cells and lymph nodes. Phase 2 consists of the subsequent activation of T cells and B cells by DCs (and the cytokines produced) into activated and differentiated T cells and antibody-producing plasma cells, respectively. This results in phase 3, in which antibodies and T cells attack the virus and virus-infected cells, respectively. Image created with Biorender.com.
Schematic overview of the normal immune after a viral infection. Phase 0 consists of epithelial infection with the virus, after which monocytes detect the pathogen and subsequent cytokine production and differentiate into cytokine-producing macrophages and other cell types. Phase 1 entails the attraction and activation of other innate immune cells, such as NK cells, which kill infected cells, and DCs, which then travel to naive T cells and lymph nodes. Phase 2 consists of the subsequent activation of T cells and B cells by DCs (and the cytokines produced) into activated and differentiated T cells and antibody-producing plasma cells, respectively. This results in phase 3, in which antibodies and T cells attack the virus and virus-infected cells, respectively. Image created with Biorender.com.
Granulocytes and monocytes detect extracellular pathogens via Toll-like receptors (TLRs).4 Granulocytes will release enzymes and toxic proteins at the site of infection, whereas monocytes traffic to inflamed tissues and differentiate into (cytokine-producing) macrophages and monocyte-derived DCs.3 Macrophages and neutrophils phagocytose and destroy pathogens, as well as infected cells. These sequential processes within neutrophils and monocytes require complex signaling cascades that involve BTK, SYK, Abl, PI3K/mTOR, and JAK/STAT.3,5-8 BTK is involved in granulocyte maturation, and SYK signaling is required for leukocyte adhesion, pathogen recognition, and phagocytosis.8-10 Abl is critical in podosome formation and the function of macrophages and is required for neutrophil recruitment and migration.6,7 PI3K/mTOR modulate oxidative bursts in neutrophils and limit TLR signaling in macrophages.11 JAK/STAT is involved in cytokine production of macrophages, as well as in the differentiation, survival, and activation of neutrophils.8,12
Activated DCs present pathogen-derived antigens to naive helper T (Th) cells to initiate the adaptive immune response.3 BTK mediates maturation and antigen presentation, whereas SYK is involved in the production of reactive oxygen species and recognition of tissue damage, and PI3K negatively regulates TLR signaling in DCs.5,10,11 JAK/STAT signaling is involved in their maturation, antigen presentation, and cytokine production.11,12
NK cells kill infected cells via degranulation, receptor-mediated apoptosis, and antibody-dependent cell-mediated cytotoxicity (ADCC). Their maturation and activation are mediated by BTK and PI3K/mTOR, whereas PI3K/mTOR and JAK/STAT are involved in cytokine production.11,13,14 Their cytolytic function is supported by PI3K/mTOR and (indirectly) by FLT3.11,15
The adaptive immune system
Responses of the adaptive immune system are based on antigen-specific receptors expressed on T- and B-cell surfaces. T-cell receptors recognize peptide fragments when presented by antigen-presenting cells, such as DCs.3 The (cytotoxic) CD8+ subset primarily kills infected cells, whereas the CD4+ subset regulates cellular and humoral responses and contains the immunosuppressive regulatory T-cell (Treg) subset.3,11 CD4+ cells differentiate into Th1, Th2, and Th17 cells. Th1 cells lead to an increased cell-mediated response, and Th2 cells lead to a humoral immune response. Th17 cells are developmentally distinct from Th1 and Th2 lineages and specifically produce proinflammatory interleukin-17 (IL-17).3 SYK and Abl are involved in early T-cell development and maturation, whereas PI3K signaling is involved in the proliferative responses of all T cells and the survival of Tregs.10,16 JAK/STAT signaling plays a key role in T-cell responses, specifically with respect to differentiation and cytokine production.12
B cells express (and produce) different classes of immunoglobulins based on their maturation and activation status.3 After activation by binding of antigen to the B-cell receptor, B cells undergo affinity-based differentiation into plasma cells that produce specific antibodies, or into memory cells that can rapidly respond following reencounter of their specific antigen.3 BTK and PI3K/mTOR form fundamental parts of the B-cell receptor–signaling cascade in which SYK acts as upstream signal transducer. Activation of this cascade leads to key processes that are involved in proliferation, differentiation, and survival.10,17,18 JAK/STAT signaling is involved in the process of class switching.
Impact of the main TKIs on the immune response
Based on above-described roles of these kinases in innate and adaptive immunity, it may seem predictable how TKIs will affect immune responses. Be that as it may, effects of TKIs on immune regulation have primarily been explored in vitro with different class-specific drugs having nonoverlapping target affinities. Moreover, many of the reported in vivo effects are based on artificial animal models or observed in symptomatic patients with a hematologic malignancy who often already suffer from disturbed immune regulation. Furthermore, because TKIs have effects on innate and adaptive immune functions, the timing of administration within the infectious cycle and development could greatly influence the response. Effects of a specific inhibitor within a TKI class can also differ greatly, because these inhibitors display distinct drug-protein interactions and off-target kinase-inhibition profiles, also resulting in inconsistent immunomodulatory effects.19 Bearing in mind these caveats, we attempt to delineate the impact of the major TKIs on the frequency and function of each cell type of the innate and the adaptive immune response.
Innate immune cells
Monocytes/neutrophils.
The SYK and JAK inhibitors, fostamatinib and ruxolitinib, respectively, are associated with acquired neutropenia, without neutrophil dysfunctions.20,21 TKI-induced functional alterations of neutrophils and monocyte-derived cells have been widely reported. Duvelisib improves macrophage function by polarizing them to an M1 phenotype, in contrast to the immunosuppressive effects seen with other PI3K inhibitors.22 Most BCR-Abl inhibitors improve monocyte chemotaxis.23 mTOR inhibition by temsirolimus stimulates neutrophil and macrophages infiltration into lung epithelium, possibly explaining the interstitial lung disease seen in temsirolimus-treated patients.24 Impairment of activation and cytotoxicity/phagocytosis of neutrophils and monocytes occur in patients treated with ibrutinib or idelalisib.25-27 Proliferation and cytokine production is reduced by the SYK and JAK inhibitors fostamatinib, ruxolitinib and fedratinib.28-30
In summary, granulocyte and monocyte function is impaired by inhibitors of BTK, SYK, and JAK and improved by Abl and mTOR inhibitors. PI3K inhibitors seem to have stimulating and suppressive effects in distinct cell types.
DCs.
DC maturation and activation are improved by ibrutinib in vitro.31 However, SYK and JAK inhibition with fostamatinib and ruxolitinib, respectively, decreases signaling, activation, and differentiation.32,33 Because ruxolitinib’s effect on DCs is expected to be due to on-target JAK inhibition, it is expected that all JAK inhibitors impair DC function.32 Midostaurin decreases DC differentiation and activation, and sorafenib limits their migration and T-cell activation, both seen in vitro.34,35 Because sunitinib does not have any effects on the functionality of DCs, it remains questionable whether the aforementioned effects are the actual results of FLT3 inhibition.35 Treatment with BCR-Abl inhibitors can result in suppression and improvement of DC expansion and function (including antitumor effects), depending on the timing and duration of drug exposure.36 This might be explained by the negative impact on DC maturation and differentiation (eg, lower levels of costimulatory molecules) observed with long-term imatinib cultures of progenitor cells in vitro but the increased chemotaxis seen during short-term cultures of mature DCs.37,38
Thus, DC function is decreased by SYK, JAK, and FLT3 inhibition and increased by the BTK inhibitor ibrutinib, whereas the effects of BCR-Abl inhibitors depend on the timing and duration of drug exposure.
NK cells.
Alterations in the frequency of NK cells occur during treatment with BCR-Abl and JAK inhibitors. An increased NK cell/lymphocyte ratio is observed in chronic myeloid leukemia (CML) patients treated with dasatinib, imatinib, and nilotinib.39 Ruxolitinib treatment leads to an absolute decrease in NK cells in patients with myeloproliferative neoplasms, likely caused by an increased ratio of immature/mature subsets.40,41 At the functional level, sorafenib increases degranulation and interferon-γ (IFN-γ) secretion, possibly leading to more antitumor NK cell activity.42 However, granule exocytosis and the cytotoxic function of NK cells are reduced in patients treated with ibrutinib, as well as in vitro by idelalisib.43-45 Data on the suppression of NK cell activity by the more selective BTK inhibitors are conflicting, with less pronounced effects with acalabrutinib and zanubrutinib but equal suppression of ADCC with tirabrutinib and ibrutinib.45 Whether BTK inhibitors potentiate or suppress anti-CD20–induced ADCC is still a matter of debate, as reviewed recently.46 Because many of the necessary cytokines in NK cell development and maturation exert their impact through JAK/STAT signaling, blocking the effects of IL-2 and IL-15 by ruxolitinib, fedratinib, and momelotinib prevents (in different degrees) activation and therefore IFN-γ and tumor necrosis factor-α (TNF-α) secretion of NK cells in vitro, hampering their killing capacity.40,41,45 NK cell–related effects of BCR-Abl inhibitors are not uniform. Although nilotinib suppresses NK cell reactivity in CML patients, dasatinib treatment results in enhanced reactivity through an unknown mechanism.23
In summary, NK cell cytotoxicity is impaired by BTK inhibitors, to the greatest extent by ibrutinib. PI3K and JAK/STAT inhibition lower the killing capacity and cytokine production, whereas BCR-Abl inhibition has variable effects. On the other hand, FLT3 inhibition increases NK cell function.
Adaptive immune cells
T cells.
T cells (subsets) are frequently altered during TKI treatment. Ibrutinib increases total CD4+ and CD8+ T cells in chronic lymphocytic leukemia (CLL) patients without lymphocytosis; however, in patients with significant tumor burden, ibrutinib decreases total T-cell numbers, which could be viewed as pseudonormalization.47 Ibrutinib and (the more selective) zanubrutinib increase the frequency of Th17 cells and reduce the number of Tregs in CLL patients, possibly resulting in a more immune-supportive phenotype.47,48 Treg frequency is also reduced by idelalisib, which correlates with autoimmune-related toxicities, as reviewed by Greenwell et al.49 Tregs are probably less affected by umbralisib, because it leads to less severe T-cell–mediated toxicity than seen with idelalisib.50 Imatinib, ruxolitinib, and certain FLT3 inhibitors also reduce Treg numbers.35,39,51-53
Functional T-cell alterations occur with most TKIs, although they vary in effect and degree. Ibrutinib reverses the polarization of CD4+ Th2 cells to a Th1 cell phenotype through off-target ITK inhibition.54 Ibrutinib, as well as the more selective BTK inhibitors, reduces the (over)activated T-cell state of CLL patients, as measured by lower plasma cytokine levels and decreased exhaustion.47,55,56 These observations are probably the indirect result of on-target (BTK) effects on leukemia cells, resulting in decreased suppressive interactions of the malignant B cells with T cells.57 Sorafenib and sunitinib treatment also results in lower PD-1 expression on CD4+ and CD8+ T cells in vivo and in vitro, respectively.53,58
Low-dose sorafenib also increases activation, whereas high-dose sorafenib decreases T-cell proliferation and augments PD-1 expression.59 Furthermore, low-dose sorafenib increases tumor-infiltrating T cells compared with high-dose sorafenib treatment of hepatocellular carcinoma patients in vivo, consistent with the dose-dependent immunomodulatory effects seen in vitro.58,59 The increased T-effector activation observed during idelalisib and duvelisib treatment is caused, at least in part, by Treg inhibition.49 Furthermore, mTOR inhibition by temsirolimus increases differentiation and enhances the function of Tregs.60 Sunitinib and sorafenib improve the Th1-type response in vivo, most likely through off-target effects.58,61,62
On the other hand, fostamatinib, entospletinib, and cerdulatinib reduce activation, proliferation, and cytokine production of T cells in vitro.63 All JAK inhibitors lower T-cell proliferation and cytokine secretion, resulting in less proinflammatory signals.64 Th1 cytokine production is inhibited by midostaurin in vitro.65
BCR-Abl inhibitors again produce various effects; although nilotinib reduces T-cell activation in vivo, dasatinib treatment leads to enhanced proliferation of T cells in leukemia patients, which correlates with improved disease responses.39,58 The effects of imatinib on T cells are conflicting, as reviewed in detail.38,66
Altogether, BTK inhibitors lead to changes in absolute T-cell numbers (depending on the clinical status of the patient) and skewing of specific subsets within the T-cell population. They also lead to reduced T-cell exhaustion. BTK, BCR-Abl, PI3K, JAK, and FLT3 inhibitors lower Treg numbers. SYK and PI3K inhibition reduces T-cell function, whereas mTOR inhibition promotes it. Effects of BCR-Abl inhibition are inconsistent between available drugs. JAK/STAT inhibition results in diminished T-cell function over multiple aspects. The effect of FLT3 inhibitors is dose dependent, with low doses leading to improved T-cell functions.
B cells.
Because many TKIs are targeted toward hematological malignancies occurring in B cells, they also have various effects on healthy B cells. These are significantly less sensitive to ibrutinib-, momelotinib-, or idelalisib-induced apoptosis than their malignant counterparts, resulting in stable B-cell numbers.45,55 However, B-cell numbers and immunoglobulin levels decrease in CML patients treated with any of the BCR-Abl inhibitors, without changes in B-cell subsets.67 Although fostamatinib clearly reduces the number of class-switched germinal center B cells, contradictory results have been reported with regard to fostamatinib’s effect on the total B-cell number.28,33
In terms of functional changes, decreased immunoglobulin G (IgG) levels are observed in CLL patients after treatment with ibrutinib.55,68 Likewise, IgM levels are decreased by ibrutinib in the graft-versus-host disease setting.69 However, humoral immunity seems to be positively affected by ibrutinib. Increased IgA has been observed across clinical trials of ibrutinib, and patients with greater improvements in IgA developed fewer infections.70 Similarly, although IgM and IgG remain unaffected, acalabrutinib treatment also augments IgA levels.70 CLL patients on ibrutinib also respond better to influenza vaccination than do untreated patients, but titers are diminished compared with healthy individuals.71 Effects on B cells in CML patients differ among BCR-Abl inhibitors. Although imatinib reduces all immunoglobulin levels, dasatinib only lowers IgM, bosutinib only lowers IgG, and nilotinib does not incite any immunoglobulin decrease.36,67,72
Fostamatinib and cerdulatinib lower B-cell activation and BCR-mediated antigen presentation in vitro and attenuate autoantibody production by B cells in vivo, most likely through on-target SYK inhibition.33,73 The effect of FLT3 inhibitors on B cells is modest, with only midostaurin showing an in vitro negative effect on the viability of B cells.74 Although proliferation and IgG production in healthy B cells are decreased by idelalisib in vitro, this is not seen upon idelalisib treatment in vivo.45
In summary, antibody changes are seen after BTK, BCR-Abl, and, possibly, PI3K inhibition. SYK inhibitors lower B-cell (auto)activity.
Immunomodulation in relation to SARS-CoV-2
The high morbidity and mortality of the current COVID-19 pandemic and the lack of effective treatment has driven research toward application of TKIs outside of the malignant spectrum. Immune-related complications by SARS-CoV-2 infection and other inflammatory diseases led to the consideration of these inhibitors as treatment options. The initial “innate” phase of SARS-CoV-2 infection is set in motion following entry in nasal and bronchial epithelial cells through the viral structural spike protein binding to the angiotensin-converting enzyme 2 receptor.75 Like most viral infections, tissue-resident macrophages in close proximity to infected cells will incite a proinflammatory state, resulting in recruitment of innate immune cells and cytokine production, leading to subsequent activation of adaptive immune responses.75 However, in the case of (severe) COVID-19, massive invasion of innate immune cells into the infected tissues leads to hyperproduction of such cytokines, eventually resulting in a so-called “cytokine storm.”76 As a consequence, recruited T cells reach a state of exhaustion, resulting in T-cell dysfunction and depletion that cause lymphopenia.76 These detrimental inflammatory effects can potentially result in acute respiratory distress syndrome (ARDS), secondary infections, multiple organ failure, and death.76 This chain of events might be interrupted by blocking specific kinases at different phases of the disease. Based on the data and considerations summarized above, we attempt to hypothetically assess the immunomodulatory potential of the TKIs in the pathophysiological process of COVID-19. Ongoing trials with these TKIs are summarized in Table 2. A schematic summary of the hypothetical application and timing of TKIs in COVID-19 is shown in Figure 2.
Ongoing trials with TKIs in treatment of COVID-19
| Class/agent | Goal | Comments |
|---|---|---|
| BTK | ||
| Ibrutinib | Efficacy in reducing respiratory failure | Quadruple-blind RCT, NCT04375397 |
| Efficacy in reducing respiratory failure and preventing death | Double-blind RCT, NCT04439006 | |
| Acalabrutinib | Efficacy in preventing deterioration, mortality, and respiratory failure or speeding up recovery of hospitalized patients with COVID-19 | Multiarm RCT, 2020-001736-95 (ACCORD 2) |
| Zanubrutinib | Efficacy in increasing proportion of respiratory failure-free patients | Double-blind RCT, NCT04382586 |
| SYK | ||
| Fostamatinib | Efficacy in reducing mortality, SAEs, inflammatory biomarkers, and hospital admission | RCT, 2020-001750-22 |
| Efficacy in preventing progression to severe/critical disease | Double-blind RCT, NCT04629703 | |
| BCR-Abl | ||
| Imatinib | Efficacy in preventing vascular leak | Single blind-RCT, 2020-001236-10 |
| Efficacy of baricitinib, imatinib, or lopinavir in combination with hydroxychloroquine | RCT, 2020-001321-31 | |
| Efficacy in preventing need for invasive mechanical ventilation | RCT, NCT04422678 | |
| Safety and efficacy in hospitalized adults | Double-blind RCT, NCT04394416 | |
| Time to clinical improvement with treatment with hydroxychloroquine in combination with baricitinib, imatinib, or lopinavir/ritonavir | RCT, 2020-001321-31 | |
| Time to clinical improvement | RCT, NCT04346147 | |
| Efficacy in preventing severe COVID-19 in hospitalized aged patients | RCT, NCT04357613 | |
| PI3K/mTOR | ||
| Duvelisib | Overall survival, length of hospital/ICU stay | Triple-blind RCT, NCT04372602 |
| Efficacy in requirement for mechanical ventilation or dying | Double-blind RCT, NCT04487886 | |
| Temsirolimus | Efficacy in preventing respiratory failure | Double-blind RCT, NCT04482712 |
| Time to clinical recovery and viral clearance | Single-blind RCT, NCT04461340 | |
| Proportion of patients alive and free from advanced respiratory support measures | Quadruple-blind RCT, NCT04341675 | |
| JAK | ||
| Ruxolitinib | Efficacy in reducing mortality, SAEs, inflammatory biomarkers, and hospital admission | RCT, 2020-001750-22 |
| Incidence of SAEs and severe disease, safety | Single arm, NCT04348071 | |
| Efficacy in preventing clinical worsening of respiratory status | Single arm, NCT04414098 | |
| Treatment of cytokine storm | Open label, NCT04355793 | |
| Efficacy in preventing death and influencing SAEs/ICU-free days | Double-blind RCT, NCT04377620 | |
| Recovery from pneumonia | Open label, NCT04334044 | |
| Efficacy in reducing respiratory failure and ICU care and preventing death | Double-blind RCT, NCT04362137, NCT04337359 | |
| Reduction of cytokine load by single plasma volume exchanges, with or without ruxolitinib | Open label, NCT04374149 | |
| Efficacy in reversing hyperinflammation (CIS) | Open label, NCT04338958 | |
| Efficacy in preventing death, ICU admission, or mechanical ventilation | Triple-blind RCT, NCT04477993 | |
| Efficacy in preventing SAEs and COVID-19 patients from becoming critically ill | Single arm, NCT04331665 | |
| Overall survival | Open label, NCT04359290 | |
| Efficacy in preventing severe respiratory failure and reducing ICU/hospital stay and survival, in combination with simvastatin | Open label, NCT04348695 | |
| Ability to prevent all-cause mortality, noninvasive or invasive ventilation | Double-blind RCT, NCT04581954 | |
| Ability to achieve a change from baseline in the clinical assessment score COVID-19 with colchicine, ruxolitinib, secukinumab, or standard treatment | Open label, NCT04403243 | |
| Safety and efficacy of ruxolitinib combined with mesenchymal stem cells | Single-blind RCT, ChiCTR2000029580 | |
| Efficacy and safety in patients on respiratory support or approaching need for respiratory support | Open label, 2020-001459-42 | |
| Efficacy of anakinra/tocilizumab, alone or in combination with ruxolitinib, in increasing ventilation-free days | Open-label RCT, 2020-001754-21, NCT04424056 | |
| Clinical and biological efficacy of anakinra, alone or in combination with ruxolitinib, in reducing oxygen dependency, need for invasive ventilation, and systemic inflammation | Open-label RCT, 2020-001963-10 | |
| Efficacy in preventing deterioration requiring mechanical ventilation | Open label, 2020-001777-71 | |
| Overall survival after 28 d | Single arm, 2020-001732-10 | |
| Pacritinib | Efficacy in preventing mechanical ventilation or ECMO | Double-blind RCT, NCT04404361 |
| Class/agent | Goal | Comments |
|---|---|---|
| BTK | ||
| Ibrutinib | Efficacy in reducing respiratory failure | Quadruple-blind RCT, NCT04375397 |
| Efficacy in reducing respiratory failure and preventing death | Double-blind RCT, NCT04439006 | |
| Acalabrutinib | Efficacy in preventing deterioration, mortality, and respiratory failure or speeding up recovery of hospitalized patients with COVID-19 | Multiarm RCT, 2020-001736-95 (ACCORD 2) |
| Zanubrutinib | Efficacy in increasing proportion of respiratory failure-free patients | Double-blind RCT, NCT04382586 |
| SYK | ||
| Fostamatinib | Efficacy in reducing mortality, SAEs, inflammatory biomarkers, and hospital admission | RCT, 2020-001750-22 |
| Efficacy in preventing progression to severe/critical disease | Double-blind RCT, NCT04629703 | |
| BCR-Abl | ||
| Imatinib | Efficacy in preventing vascular leak | Single blind-RCT, 2020-001236-10 |
| Efficacy of baricitinib, imatinib, or lopinavir in combination with hydroxychloroquine | RCT, 2020-001321-31 | |
| Efficacy in preventing need for invasive mechanical ventilation | RCT, NCT04422678 | |
| Safety and efficacy in hospitalized adults | Double-blind RCT, NCT04394416 | |
| Time to clinical improvement with treatment with hydroxychloroquine in combination with baricitinib, imatinib, or lopinavir/ritonavir | RCT, 2020-001321-31 | |
| Time to clinical improvement | RCT, NCT04346147 | |
| Efficacy in preventing severe COVID-19 in hospitalized aged patients | RCT, NCT04357613 | |
| PI3K/mTOR | ||
| Duvelisib | Overall survival, length of hospital/ICU stay | Triple-blind RCT, NCT04372602 |
| Efficacy in requirement for mechanical ventilation or dying | Double-blind RCT, NCT04487886 | |
| Temsirolimus | Efficacy in preventing respiratory failure | Double-blind RCT, NCT04482712 |
| Time to clinical recovery and viral clearance | Single-blind RCT, NCT04461340 | |
| Proportion of patients alive and free from advanced respiratory support measures | Quadruple-blind RCT, NCT04341675 | |
| JAK | ||
| Ruxolitinib | Efficacy in reducing mortality, SAEs, inflammatory biomarkers, and hospital admission | RCT, 2020-001750-22 |
| Incidence of SAEs and severe disease, safety | Single arm, NCT04348071 | |
| Efficacy in preventing clinical worsening of respiratory status | Single arm, NCT04414098 | |
| Treatment of cytokine storm | Open label, NCT04355793 | |
| Efficacy in preventing death and influencing SAEs/ICU-free days | Double-blind RCT, NCT04377620 | |
| Recovery from pneumonia | Open label, NCT04334044 | |
| Efficacy in reducing respiratory failure and ICU care and preventing death | Double-blind RCT, NCT04362137, NCT04337359 | |
| Reduction of cytokine load by single plasma volume exchanges, with or without ruxolitinib | Open label, NCT04374149 | |
| Efficacy in reversing hyperinflammation (CIS) | Open label, NCT04338958 | |
| Efficacy in preventing death, ICU admission, or mechanical ventilation | Triple-blind RCT, NCT04477993 | |
| Efficacy in preventing SAEs and COVID-19 patients from becoming critically ill | Single arm, NCT04331665 | |
| Overall survival | Open label, NCT04359290 | |
| Efficacy in preventing severe respiratory failure and reducing ICU/hospital stay and survival, in combination with simvastatin | Open label, NCT04348695 | |
| Ability to prevent all-cause mortality, noninvasive or invasive ventilation | Double-blind RCT, NCT04581954 | |
| Ability to achieve a change from baseline in the clinical assessment score COVID-19 with colchicine, ruxolitinib, secukinumab, or standard treatment | Open label, NCT04403243 | |
| Safety and efficacy of ruxolitinib combined with mesenchymal stem cells | Single-blind RCT, ChiCTR2000029580 | |
| Efficacy and safety in patients on respiratory support or approaching need for respiratory support | Open label, 2020-001459-42 | |
| Efficacy of anakinra/tocilizumab, alone or in combination with ruxolitinib, in increasing ventilation-free days | Open-label RCT, 2020-001754-21, NCT04424056 | |
| Clinical and biological efficacy of anakinra, alone or in combination with ruxolitinib, in reducing oxygen dependency, need for invasive ventilation, and systemic inflammation | Open-label RCT, 2020-001963-10 | |
| Efficacy in preventing deterioration requiring mechanical ventilation | Open label, 2020-001777-71 | |
| Overall survival after 28 d | Single arm, 2020-001732-10 | |
| Pacritinib | Efficacy in preventing mechanical ventilation or ECMO | Double-blind RCT, NCT04404361 |
CIS, chronic inflammation score; ECMO, extracorporeal membrane oxygenation; ICU, intensive care unit; RCT, randomized controlled trial; SAE, serious adverse event.
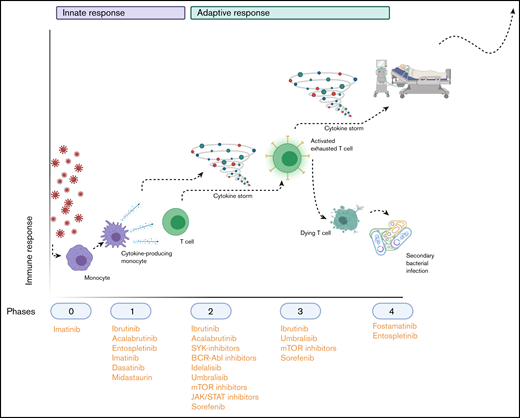
Hypothetical application of TKIs during the SARS-COV-2–induced immunopathology. Phase 0 consists of infection with the virus; phase 1 entails the polarization of macrophages to an M1 phenotype (and their subsequent activation) and their production of cytokines to attract other immune cells. Phase 2 is the recruitment and activation of cytokine-producing innate immune cells and T cells and B cells, which (together with the cytokine storm that results from phase 1) results in phase 3: depletion and exhaustion of lymphocytes and, subsequently, secondary bacterial infections. In phase 4, acute respiratory distress syndrome, secondary infections and multiorgan failure arise, leading to respiratory insufficiency and life-threatening situations. The applicable TKIs are indicated below the phases during which their immunomodulatory effects would be the most beneficial. Image created with Biorender.com.
Hypothetical application of TKIs during the SARS-COV-2–induced immunopathology. Phase 0 consists of infection with the virus; phase 1 entails the polarization of macrophages to an M1 phenotype (and their subsequent activation) and their production of cytokines to attract other immune cells. Phase 2 is the recruitment and activation of cytokine-producing innate immune cells and T cells and B cells, which (together with the cytokine storm that results from phase 1) results in phase 3: depletion and exhaustion of lymphocytes and, subsequently, secondary bacterial infections. In phase 4, acute respiratory distress syndrome, secondary infections and multiorgan failure arise, leading to respiratory insufficiency and life-threatening situations. The applicable TKIs are indicated below the phases during which their immunomodulatory effects would be the most beneficial. Image created with Biorender.com.
BTK inhibitors
Ibrutinib reduces macrophage numbers, activation, infiltration, and degranulation.77,78 Moreover, ibrutinib treatment decreases serum levels of inflammatory cytokines associated with cytokine release syndrome, such as IL-1β, IL-6, IL-10, and TNF-α.79 In addition, ibrutinib polarizes the T-cell population toward effector cells and reduces their exhaustion marker expression, which possibly enables the adaptive immune system to a more capable immune response.56,80 B-cell activation and a subsequent increase in IgG levels, which are both inhibited by ibrutinib, are specifically increased in patients with severe COVID-19.45,68,76 Because of these immunomodulatory effects, ibrutinib, and perhaps the more selective BTK inhibitors as well, might be beneficial in the early and late stages of COVID-19.81 However, zanubrutinib also lowers Tregs in CLL patients and could potentially ameliorate immune responses.48 First results of a clinical trial with acalabrutinib in COVID-19 (the CALAVI trial) showed decreased IL-6 levels, reflecting the inhibition of monocytes and attenuation of their cytokine production.82 However, it was recently reported that the trial did not meet its primary end point of increasing the patient proportion remaining alive and free of respiratory failure. It remains to be seen whether specific subgroups of patients benefit from acalabrutinib treatment.83
SYK inhibitors
The SYK inhibitors affect some of the essential players in the cytokine storm seen in severe COVID-19 patients: IL-6, IL-10, and TNF-α.28,63,84 Entospletinib also reduces pulmonary inflammation by macrophages and increases Treg numbers, which might mitigate the lung damage seen in the later stages of severe COVID-19.63,85,86 In a high-content screen for mucin-1–reducing compounds, a biochemical marker predicting ARDS, fostamatinib reduced mucin-1 in vitro and in vivo.87 However, all SYK inhibitors reduce T- and B-cell frequency and activation, potentially impairing an adequate adaptive immune response.28,33,63,73,88 Therefore, SYK inhibitors are expected to be of therapeutic benefit, primarily in the initial phase of the disease.
BCR-Abl inhibitors
Prior to injection of viral RNA into the host cytoplasm for replication, all enveloped viruses, including coronaviruses, must fuse with cellular membranes or with endosomes, which is mediated by Abl proteins.89 Indeed, in the early phases of infection, imatinib prevents viral entry via Abl-mediated cytoskeletal rearrangement, whereas in later infection phases, imatinib and dasatinib inhibit SARS-CoV and Middle East respiratory syndrome coronavirus (MERS-CoV) replication through blocking of the Abl2 protein.89 In addition to its effects on viral replication, BCR-Abl inhibitor’s immunomodulatory effects include attenuation of proinflammatory cytokine release (among them IL-1β and TNF-α) and reduction of macrophage development and M1 polarization, which means that their application could result in a decrease in, or perhaps even prevention of, severe cases of COVID-19.23,39,90,91 Nevertheless, BCR-Abl inhibitors also cause a decrease in B-cell numbers, antibody production, and Treg viability.39,67,72 Therefore, application of BCR-Abl inhibitors could be tested at early stages of the disease, but not at later stages, to preserve long-term memory and adequate antibody formation.39,67,72 The wide variation in the immunomodulatory effects within this class of TKIs must always be considered carefully when choosing a specific inhibitor.
PI3K/mTOR inhibitors
PI3K/mTOR signaling was reported to be overactivated in MERS-CoV–infected cell lines, and treatment with the nonselective PI3K inhibitor wortmannin and the mTOR inhibitor rapamycin led to inhibited MERS-CoV infection.92 The PI3K inhibitors described in this review decrease multiple inflammatory cytokines, including IL-1, TNF-α, IL-10, and IFN-γ.43,93 However, idelalisib reduces cytokine production in macrophages, whereas duvelisib polarizes them to an M1 phenotype, leading to a more proinflammatory state.22,50,55 Idelalisib and umbralisib reduce exhaustion marker expression on CD8+ T cells, which may assist in preventing lymphocyte dysfunction and depletion.93 Therefore, PI3K inhibitors could be useful in innate response modulation by inhibiting viral replication, improving early macrophage activation, and ameliorating the T-cell dysfunction seen in later stages. However, the last should be approached cautiously because hyperinflammatory side effects (due to a decrease in Tregs) are relatively common.94 Because mTOR inhibitors are known to cause interstitial lung disease, possibly caused by increased monocyte infiltration in lung epithelium, their application in this ARDS-prone disease seems counterintuitive.24 However, multiple trials of rapamycin are underway to assess the possibility that it might decrease cytokine release and T-cell dysfunction (Table 2).
JAK inhibitors
The key role of JAK-STAT–mediated cytokines, such as IL-6, IL-10, and TNF-α, in the severity of COVID-19 suggests a role for JAK-STAT inhibitors in ameliorating disease severity.95 However, blocking the JAK-STAT pathway strongly inhibits IFN signaling and decreases T-cell proliferation and response.45,96 Because IFN signaling is instrumental in initiating the innate immune response and in preventing viral replication, JAK-STAT inhibitors should be avoided during the very early stages of the disease.97 Because preventing the (IFN-triggered) cytokine storm would be highly desirable, attenuating JAK-STAT signaling could very well improve disease outcome when such symptoms are occurring. Indeed, preliminary data from COVID-19 patients treated with the JAK inhibitor baricitinib in the early phases of their disease demonstrated improvements in inflammatory symptoms and in pulmonary function tests.95 Ruxolitinib treatment was also proven to reduce severe systemic hyperinflammation in a pilot study, with more extensive phase 2 testing underway.98 Because JAK inhibitors affect T-cell skewing and function, inhibition of an adequate T-cell response to secondary infections might be expected upon prolonged treatment.45 The JAK inhibitors baricitinib, ruxolitinib, and tofacitinib are being tested in clinical trials (Table 2).
FLT3 inhibitors
Although FLT3 signaling seems to have a less distinctive role in cytokine production compared with the other pathways mentioned, possible helpful immunomodulatory effects could be expected upon administration of FLT3 inhibitors to COVID-19 patients. Midostaurin treatment leads to macrophage apoptosis and lowers IL-6, IL-10, and TNF-α.52,65,99 Quizartinib also lowers IL-6 and TNF-α, suggesting that these TKIs might be useful in attenuating the cytokine storm.100 However, midostaurin also decreases the proliferation and activation of T and B cells. On the other hand, sorafenib and sunitinib increase IFN-γ levels, lower PD-1 and CTLA4 expression on T cells, and enhance T-cell responses (primarily Th1 cells) in general, which could hypothetically strengthen the primary immune response against the virus.42,53,58,62
Concluding remarks
Because most kinases play a role in >1 cell type, and because most inhibitors have promiscuous effects, the use of these drugs will affect immune responses at multiple levels. However, depending on the timing, method, and state of cellular development in which cells are exposed to TKIs, specific skewing could be feasible. Use of kinase inhibitors to mediate specific immune alterations is becoming more widespread, as seen in the treatment of graft-versus-host disease, rheumatoid arthritis, and other immune-mediated diseases, although the specific way in which these TKIs perturb the pathophysiology is often not completely understood.
The recent widespread adoption of dexamethasone treatment in patients with severe COVID-19, based on the RECOVERY trial, will change the need for some of the immunomodulatory schemes described here.101 Specific subgroups of COVID-19 patients could very well benefit from immunosuppression through the downregulation of proinflammatory cytokine production achieved by low doses of corticosteroids.102 Because corticosteroids could also reduce the initial immune responses needed to induce pathogen recognition and control viral replication, the timing of treatment seems to be of paramount importance.101,102 Thus, well-timed corticosteroid treatment could potentially be combined with, or followed by, regimens with some of the TKIs mentioned in this review. It could also limit the need for TKIs in the later (phase 3 and 4) stages of immunopathological developments, in COVID-19 and other inflammatory diseases.
Expectations are high for the application of TKIs in the treatment of various diseases that display imbalances in immune functions. Investigating their effects in clinical trials necessitates careful timing of treatment initiation and treatment duration, as well as close immune monitoring using biomarkers of the innate and adaptive immune response. The severity of the current pandemic will no doubt accelerate clinical testing, which can provide the requisite insight, hopefully in the nearby future.
Send data sharing request via email to the corresponding author, Arnon P. Kater, a.p.kater@amsterdamumc.nl.
Authorship
Contribution: C.F.J., E.E., and A.P.K. selected and collected data and wrote the manuscript.
Conflict-of-interest disclosure: A.P.K. receives research funding from Janssen, AbbVie, Roche/Genentech, Astra Zeneca, and Celgene and is a member of advisory boards for Janssen, AbbVie, Roche/Genentech, and Juno. E.E. receives research funding from Janssen, AbbVie, Roche/Genentech, Astra Zeneca, and Celgene. C.F.J. declares no competing financial interests.
Correspondence: Arnon P. Kater, Department of Hematology, D3-221.1, Amsterdam University Medical Center, Location AMC, Meibergdreef 9, 1105 AZ Amsterdam, The Netherlands; e-mail: a.p.kater@amsterdamumc.nl.

