

Key Points
ALK– ALCL represents 15% of the global mature T-cell lymphomas, with significant geographic variation.
In patients with ALK– ALCL, multiagent chemotherapy results in 49% and 43% rates of 5-year OS and PFS, respectively.

Abstract
Anaplastic lymphoma kinase–negative anaplastic large cell lymphoma (ALK– ALCL) is an aggressive neoplasm of T-cell/null-cell lineage. The T-Cell Project is a global prospective cohort study that consecutively enrolled patients newly diagnosed with peripheral T-cell lymphoma, registered through a centralized computer database between September 2006 and February 2018. Of 1553 validated cases from 74 sites in 13 countries worldwide, 235 were reported as ALK– ALCL. The median age at diagnosis was 54 years (range, 18-89 years), with a male predominance (62%). Stage III to IV disease was identified in 71% of patients, bulky disease and bone marrow involvement were uncommon, and 66% of patients presented with a low (0-1) International Prognostic Index score. Of all treated patients, 85% received multiagent initial chemotherapy, and 8% were consolidated with autologous hematopoietic cell transplantation. The initial overall and complete response rates were 77% and 63%, respectively. After a median follow-up of 52 months (95% confidence interval [CI], 41-63), the median progression-free survival (PFS) and overall survival (OS) were 41 months (95% CI, 17-62) and 55 months (95% CI, 36-75), respectively. The 3- and 5-year PFS rates were 52% and 43%, and the 3- and 5-year OS rates were 60% and 49%. Treatments containing both anthracycline and etoposide were associated with superior OS (P = .05) but not PFS (P = .18). In this large prospective cohort study, outcomes comparable to those previously reported in the retrospective International Peripheral T-Cell Lymphoma Project were observed. The study underscores the need for introducing novel platforms for ALK– ALCL and establishes a benchmark for future clinical trials. This trial was registered at www.clinicaltrials.gov as #NCT01142674.
Introduction
Anaplastic large cell lymphoma (ALCL) was identified by Stein and colleagues in 1985 based on a cohesive growth pattern with frequent invasion of lymph node sinuses and uniform strong expression of CD30 by the malignant cells. Subsequent studies restricted the use of the term ALCL to lymphomas of T-cell or null-cell phenotype.1 Furthermore, identification of systemic ALCL with expression of anaplastic lymphoma kinase (ALK) protein as a distinct clinico-pathologic entity divided ALCL into ALK-positive (ALK+) and ALK-negative (ALK–) subtypes.2 The World Health Organization (WHO) classification of hematologic malignancies 2016 update finally defined ALK– ALCL as an established diagnostic entity.3
According to the previously published International Peripheral T-cell Lymphoma Project report, ALK– ALCL comprises ∼5.5% of peripheral T-cell lymphoma (PTCL) diagnoses globally, with some geographic predilection.4 In addition, a report using the Surveillance, Epidemiology, and End Results registry suggested an impact of race and ethnicity on ALK– ALCL incidence and prevalence in the United States, with higher incidence rates in Black and non-Hispanic White subjects and very low rates in Asian and Pacific Islander subjects.5 The true reasons for these variations are unknown. In both reports, the median age at diagnosis was similar (58 years and 56 years, respectively).
Anthracycline-containing multiagent protocols are associated with high response rates in ALK– ALCL; however, relapses are common, and reported 5-year progression-free survival (PFS) rates ranged from 36% to 60% in various studies.6-8 The addition of etoposide was suggested to improve outcome in a meta-analysis of patients with PTCL, including ALCL, enrolled in several prospective studies by the German Non-Hodgkin Lymphoma Study Group (DSHNHL). However, the difference was statistically significant (P = .003) only in younger patients with normal lactate dehydrogenase (LDH) values at diagnosis.9 In a recent prospective, randomized phase 3 trial, the addition of the CD30-targeting immunoconjugate brentuximab vedotin to cyclophosphamide, doxorubicin, and prednisone (CHP) chemotherapy resulted in improved PFS compared with standard cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) therapy in patients with CD30+ PTCL, of whom 70% carried the diagnosis of ALCL.10 The highest benefit was observed for ALK+ ALCL. It is unclear whether the new combination would remain superior if compared with anthracycline/etoposide–containing protocols, as historically such protocols would seem to be superior to CHOP. Furthermore, an infusional dose-adjusted etoposide, prednisone, vincristine, cyclophosphamide, and doxorubicin (EPOCH) regimen produced high rates of PFS in a mix of ALK+ and ALK– ALCL patients in a single-institution, prospective phase 2 clinical trial (12-year PFS, >70%).11
The recent advances in gene sequencing technology have informed about genomic heterogeneity of ALK– ALCL and also suggested an impact of specific chromosomal aberrations on clinical outcomes. Two of the described recurrent genomic rearrangements define subgroups of ALCL with vastly different outcomes. DUSP22-IRF4 chromosomal rearrangement t(6;7) portends an excellent prognosis with current therapies, whereas TP63 overexpression that results from either chromosomal aberration, inv(3), or cryptic gene mutation predicts dismal outcomes.12 These genetic abnormalities are found in ∼30% and 8% of patients, respectively, and are mutually exclusive. The prognostic impact of these genetic markers remained significant in multivariate analysis when adjusted for International Prognostic Index (IPI) scores.13 Unknown proportions of these genetic variants in past clinical trials and registry studies make comparisons vs historical data difficult.
The T-Cell Project (#NCT01142674) was a global prospective cohort study that accrued 1553 patients newly diagnosed with PTCL between 2006 and 2018. It was initiated to overcome limitations of a prior retrospective study and to collect more accurate demographic, clinical, and outcome data in the era of new WHO classifications of lymphoid neoplasms and the emergence of novel therapies with unique mechanisms of action and better understanding of the genomic diversity of PTCL subtypes. The current report focuses on the characterization and evaluation of outcomes in 235 (15%) cases of ALK– ALCL.
Methods
The T-Cell Project was initiated in 2006. Patients with newly diagnosed aggressive, mature, nodal, and extranodal PTCL subtypes according to the WHO 2001 or WHO 2008 classifications were registered in the T-Cell Project electronic database at initial diagnosis. The study is devised as a prospective collection of information potentially relevant to better define prognosis for the more frequent subtypes of PTCL (ie, PTCL not otherwise specified and angioimmunoblastic T-cell lymphoma) and to better outline clinical characteristics and outcome of the more uncommon PTCL subtypes (extranodal natural killer/T-cell lymphoma; enteropathy-type T-cell lymphoma; hepatosplenic T-cell lymphoma; peripheral gamma/delta T-cell lymphoma; subcutaneous panniculitis-like T-cell lymphoma; and anaplastic large-cell lymphoma, T cell/null cell, primary systemic type). Additional eligibility criteria included: age ≥18 years; tissue biopsy specimens adequate for diagnosis and classification and available for centralized review; and clinical data, including baseline information on disease localization and laboratory parameters at staging. Where reported, disease-related discomfort had to be related to the lymphoma diagnosis, as determined by investigator assessment, and included fatigue, dyspnea, cough, hemoptysis, chest pain, anorexia, abdominal pain, new-onset headaches, sensory and motor neurologic symptoms, painful adenopathy, rash, pruritus, and documented infection at the time of diagnosis. Data were collected on front-line treatment, response evaluation at the end of treatment, and updated follow-up for at least 5 years for living patients. Participating institutions were asked to provide information about a consecutive series of cases, without any selection, so that patients who did not receive any kind of treatment could also be registered in the study.
Data collection was performed with a Web-based platform via electronic case report forms at a dedicated Web site (www.tcellproject.org), with the adoption of proper technology assuring protection in Web communications of each subject’s clinical data. Data access and management were regulated by the use of passwords with different levels of admittance, providing that subject confidentiality was respected. Data management and study management were performed at the study trial office in Modena, Italy. Registration was based on locally established histologic diagnosis; a panel of expert hematopathologists was planned to review diagnosis of all patients entered into the study. Such review has not been performed after study initiation due to insurmountable challenges that arose with the logistics of human subject tissue shipments from around the globe. DUSP22 and TP63 genomic alterations that have recently emerged as important prognostic markers in ALCL were not considered at the time of study design and inception. Ad hoc analysis will be attempted to address this deficiency as part of a follow-up study.
The T-Cell Project was conducted in compliance with the Declaration of Helsinki. It was approved by the appropriate research ethics committees or institutional review boards at each participating institution. The project required each patient to provide written informed consent before registration.
End point definitions
The main end point of the study, overall survival (OS), was measured from the date of diagnosis until death from any cause or the date of the last known contact for living patients. The secondary end point was PFS; it was measured from the time of diagnosis to progressive disease, death from any cause, or the date of the last known contact for living patients. Additional secondary end points included demographic, clinical, and epidemiologic characteristics of the study subjects; descriptive statistics were used for analysis. Response assessments after the first treatment were adapted from the Standardized Response Criteria for Non-Hodgkin’s Lymphoma and from Recommendations for revised Response Criteria for Malignant Lymphoma.
Statistical analysis
Standard descriptive analyses were conducted for clinical and demographic end points. For a crude association analysis, categorical data were analyzed by using the χ2 or Fisher’s exact test (two-sided) for data analysis. Survival estimates were obtained by using the Kaplan-Meier method, and comparisons between categories were performed by using the log-rank test and Cox proportional hazards regression.
Continuous biological covariates were dichotomized according to usual clinical thresholds. Stata software version 14.0 or greater was used for data analysis.
Results
Between September 2006 and February 2018, a total of 1695 patients with newly diagnosed PTCLs were registered in the T-Cell Project database by 74 institutions in 13 countries; of these patients, 1553 were confirmed eligible for enrollment and included in the analyses. A diagnosis of ALK– ALCL was reported in 235 cases (15%), with significant differences in geographical distribution: ALK– ALCL comprised 26% (71 of 271), 14% (50 of 368), 14% (89 of 644), and 4.5% (8 of 176) of cases registered in South America, the United States, Europe, and Asia, respectively.
Patient characteristics and treatment
Table 1 summarizes the main demographic and clinical characteristics of this cohort of patients. The median age at diagnosis was 54 years (range, 18-89 years), with a male predominance (62%). More than 27% of patients were aged >65 years, and 16% were aged >70 years. Stage III to IV disease was identified in 71% of patients, and disease-related discomfort was present in 70%; bulky disease and bone marrow involvement were uncommon (6% and 8%, respectively). Elevations of β2-microglobulin and C-reactive protein were reported in 57% and 69% of patients. IPI and Prognostic Index for T-cell Lymphoma (PIT) scores were assessed in 150 cases. According to IPI, 99 patients (66%) had a low/intermediate score (0-2), and 51 patients (34%) had an intermediate/high score (3-5). Similar distribution of patients with higher prevalence of low/intermediate scores (0-1) compared with intermediate/high scores (2-4) was found in the PIT score analysis, with 95 (63%) vs 55 (37%) patients.
Patient clinical and demographic characteristics
| Parameter | n | Value |
|---|---|---|
| Age, median (range), y | 235 | 54 (18-89) |
| Age ≥60 y | 235 | 92 (39) |
| Male sex | 235 | 146 (62) |
| ECOG-PS >1 | 217 | 53 (24) |
| B symptoms | 221 | 99 (45) |
| Discomfort disease-related | 195 | 136 (70) |
| Stage III to IV | 187 | 133 (71) |
| Nodal only disease | 163 | 53 (32) |
| Extranodal involvement | 235 | 112 (48) |
| Bulky disease (≥10 cm) | 235 | 13 (6) |
| No. of extranodal sites >1 | 163 | 42 (26) |
| BM involvement | 194 | 16 (8) |
| LDH >ULN | 195 | 92 (47) |
| Hemoglobin <12 g/dL | 212 | 89 (42) |
| Platelets <150 × 103/μL | 211 | 21 (10) |
| Monocytes ≥0.8 × 103/μL | 197 | 53 (27) |
| ANC >6.5 × 103/μL | 209 | 76 (36) |
| β2-microglobulin >ULN | 88 | 50 (57) |
| CRP >ULN | 89 | 61 (69) |
| Parameter | n | Value |
|---|---|---|
| Age, median (range), y | 235 | 54 (18-89) |
| Age ≥60 y | 235 | 92 (39) |
| Male sex | 235 | 146 (62) |
| ECOG-PS >1 | 217 | 53 (24) |
| B symptoms | 221 | 99 (45) |
| Discomfort disease-related | 195 | 136 (70) |
| Stage III to IV | 187 | 133 (71) |
| Nodal only disease | 163 | 53 (32) |
| Extranodal involvement | 235 | 112 (48) |
| Bulky disease (≥10 cm) | 235 | 13 (6) |
| No. of extranodal sites >1 | 163 | 42 (26) |
| BM involvement | 194 | 16 (8) |
| LDH >ULN | 195 | 92 (47) |
| Hemoglobin <12 g/dL | 212 | 89 (42) |
| Platelets <150 × 103/μL | 211 | 21 (10) |
| Monocytes ≥0.8 × 103/μL | 197 | 53 (27) |
| ANC >6.5 × 103/μL | 209 | 76 (36) |
| β2-microglobulin >ULN | 88 | 50 (57) |
| CRP >ULN | 89 | 61 (69) |
Values are n (%) except as noted.
ANC, absolute neutrophil count; BM, bone marrow; CRP, C-reactive protein; ULN, upper limit of normal.
Treatment details were available in 220 patients, of whom 15 (6.8%) received only best supportive care. Of the remaining 205 patients, 168 (82%) were treated with anthracycline-containing regimens, 31 (15%) with anthracycline/etoposide–containing regimens, and 6 (3%) with other regimens. Sixteen patients (8%) underwent high-dose chemotherapy with autologous stem cell support as consolidation of first-line therapy. Finally, 4 patients were treated with radiotherapy alone (2%).
Response to treatment and survival
Of 205 patients who were treated with curative intent, 129 (63%) achieved a complete response, and 29 (14%) had a partial response, with an overall response rate of 77%. In the remaining 47 patients (23%), the response was recorded as stable or progressive disease.
The median follow-up for the entire cohort of 235 patients was 52 months (95% confidence interval [CI], 36-75). Seven patients (3%) were lost to follow-up after a median time of 21 months. The minimum and median follow-up times for surviving patients were 48 months and 70.3 months, respectively. The median OS was 55 months. The 3- and 5-year OS rates were 60% (95% CI, 48-72) and 49% (95% CI, 35-59). The 3- and 5-year PFS rates were 52% (95% CI, 43-69) and 43% (95% CI, 20-69) (Figure 1).
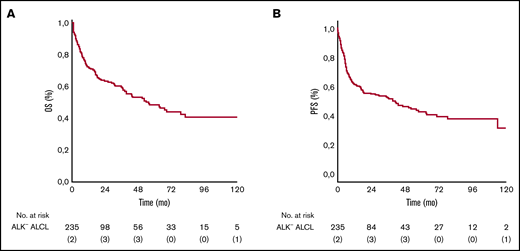
OS and PFS. Kaplan-Meier analysis of OS and PFS in ALK– ALCL patients. (A) Five-year rate of OS was 49% (95% CI, 35-59). (B) Five-year rate of PFS was 43% (95% CI, 20-69).
OS and PFS. Kaplan-Meier analysis of OS and PFS in ALK– ALCL patients. (A) Five-year rate of OS was 49% (95% CI, 35-59). (B) Five-year rate of PFS was 43% (95% CI, 20-69).
At the time of data lock, 102 deaths were recorded: 72 (70%) due to lymphoma, 9 (9%) due to infection, 2 (2%) due to organ failure, 2 (2%) due to treatment-related toxicity, and 4 (4%) due to second malignancies. For 13 patients (13%), the cause of death was unknown. Overall, 103 patients (50%) experienced progression or relapse.
Treatment with anthracycline and etoposide was associated with a superior outcome: 3-year and 5-year OS rates were 56% and 44% in the anthracycline-based treatment group, and 76% and 69% in the anthracycline/etoposide–based treatment group, and the differences were statistically significant (P = .05). Similar results were also observed in PFS. The 3-year and 5-year PFS rates were 47% (95% CI, 32-59) and 39% (95% CI, 29-48) for those treated with anthracycline-only regimens vs 65% (95% CI, 32-87) and 50% (95% CI, 36-89) for those treated with anthracycline/etoposide–containing regimens; the differences were not statistically significant (P = .186) (Figure 2).
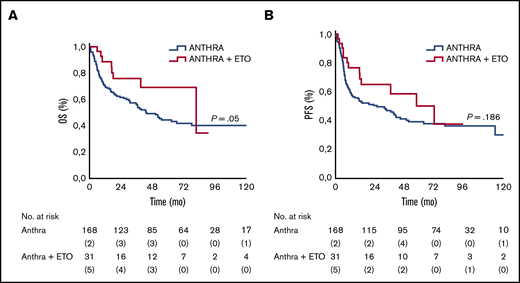
OS and PFS according to anthracycline (Anthra) and Anthra + etoposide (ETO) treatments. Kaplan-Meier analysis of OS and PFS in patients with ALK– ALCL stratified according to use of ETO in combination with Anthra. (A) Five-year rates of OS in the ETO cohort vs no ETO cohort were 69% vs 44%, respectively. (B) Five-year rates of PFS in the ETO cohort vs the no ETO cohort were 50% vs 39%, respectively.
OS and PFS according to anthracycline (Anthra) and Anthra + etoposide (ETO) treatments. Kaplan-Meier analysis of OS and PFS in patients with ALK– ALCL stratified according to use of ETO in combination with Anthra. (A) Five-year rates of OS in the ETO cohort vs no ETO cohort were 69% vs 44%, respectively. (B) Five-year rates of PFS in the ETO cohort vs the no ETO cohort were 50% vs 39%, respectively.
In 150 patients, sufficient data allowed for IPI and PIT score calculations. As expected, patients with high IPI and PIT scores had a worse prognosis, as summarized in Table 2 and Figure 3.
OS and PFS rates for ALK– ALCL stratified by the IPI and PIT scores
| Variable | 5-y OS, % | 5-y PFS, % | Median OS, mo | Median PFS, mo | P |
|---|---|---|---|---|---|
| IPI | |||||
| Low (0-2) | 62 | 53 | Not reached | 64 (95% CI, 37-90) | <.001 |
| High (2-4) | 27 | 21 | 9 (95% CI, 5-13) | 7 (95% CI, 3-11) | |
| PIT | |||||
| Low (0-2) | 60 | 51 | Not reached | 64 (95% CI, 34-93) | <.001 |
| High (2-4) | 24 | 21 | 9 (95% CI, 6-13) | 7 (95% CI, 2-11) |
| Variable | 5-y OS, % | 5-y PFS, % | Median OS, mo | Median PFS, mo | P |
|---|---|---|---|---|---|
| IPI | |||||
| Low (0-2) | 62 | 53 | Not reached | 64 (95% CI, 37-90) | <.001 |
| High (2-4) | 27 | 21 | 9 (95% CI, 5-13) | 7 (95% CI, 3-11) | |
| PIT | |||||
| Low (0-2) | 60 | 51 | Not reached | 64 (95% CI, 34-93) | <.001 |
| High (2-4) | 24 | 21 | 9 (95% CI, 6-13) | 7 (95% CI, 2-11) |
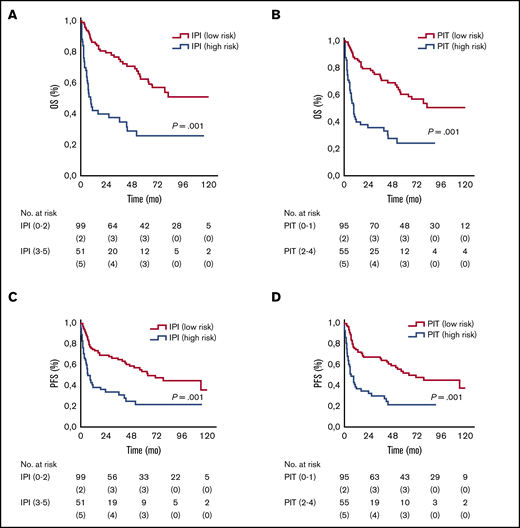
OS and PFS according to IPI and PIT. Kaplan-Meier analysis of OS (A) and PFS (C) stratified according to IPI-defined risk groups. Kaplan-Meier analysis of OS (B) and PFS (D) stratified according to PIT-defined risk groups.
OS and PFS according to IPI and PIT. Kaplan-Meier analysis of OS (A) and PFS (C) stratified according to IPI-defined risk groups. Kaplan-Meier analysis of OS (B) and PFS (D) stratified according to PIT-defined risk groups.
In a univariate analysis, several clinical and laboratory features had a significant negative impact on OS, including age >60 years (P = .025), Eastern Cooperative Group performance status (ECOG-PS) ≥2 (P = .007), presence of B symptoms (P = .002), elevated LDH levels (P = .001), and a platelet count <150 × 109 (P = .014). Moreover, stage III to IV disease (P = .01), ECOG-PS ≥2 (P = .001), presence of B symptoms (P = .001), and an elevated LDH level (P = .001) were also predictive of inferior PFS (Table 3). In the multivariate analysis, the presence of B symptoms (P = .008), elevated LDH level (P = .001), ECOG-PS ≥2 (P = .001), and a platelet count <150 × 109 (P = .05) carried significance for OS (Table 4). The presence of B symptoms (P = .02), elevated LDH level (P = .001), and ECOG-PS ≥2 (P = .001) maintained their prognostic significance for PFS.
Univariate analysis of prespecified parameters for OS and PFS
| Covariate | Group | OS | PFS | ||
|---|---|---|---|---|---|
| P | HR (95% CI) | P | HR (95% CI) | ||
| Age | <60 y | 1.00 | 1.00 | ||
| >60 y | .025 | 1.41 (0.94-2.12) | .120 | 1.28 (0.87-1.87) | |
| Disease stage | I-II | 1.00 | 1.00 | ||
| III-IV | .005 | 1.85 (1.18-2.91) | .01 | 1.72 (1.13-2.63) | |
| B symptoms | No | 1.00 | 1.00 | ||
| Yes | .002 | 1.94 (1.30-2.89) | .001 | 2.07 (1.41-3.08) | |
| Bone marrow involvement | No | 1.00 | 1.00 | ||
| Yes | .282 | 1.44 (0.72-2.87) | .11 | 1.75 (0.91-3.37) | |
| CRP | <ULN | 1.00 | 1.00 | ||
| >ULN | .113 | 1.69 (0.90-3.19) | .05 | 1.36 (0.99-1.54) | |
| Platelet count | <150 × 109 | 1.00 | 1.00 | ||
| >150 × 109 | .014 | 2.29 (1.29-4.05) | .063 | 1.99 (1.13-3.51) | |
| LDH | <ULN | 1.00 | 1.00 | ||
| >ULN | .001 | 2.73 (1.77-4.19) | .001 | 2.34 (1.57-3.49) | |
| ECOG-PS | ≥2 | 1.00 | 1.00 | ||
| ≤2 | .001 | 3.23 (2.14-4.89) | .001 | 2.71 (1.82-4.04) | |
| Covariate | Group | OS | PFS | ||
|---|---|---|---|---|---|
| P | HR (95% CI) | P | HR (95% CI) | ||
| Age | <60 y | 1.00 | 1.00 | ||
| >60 y | .025 | 1.41 (0.94-2.12) | .120 | 1.28 (0.87-1.87) | |
| Disease stage | I-II | 1.00 | 1.00 | ||
| III-IV | .005 | 1.85 (1.18-2.91) | .01 | 1.72 (1.13-2.63) | |
| B symptoms | No | 1.00 | 1.00 | ||
| Yes | .002 | 1.94 (1.30-2.89) | .001 | 2.07 (1.41-3.08) | |
| Bone marrow involvement | No | 1.00 | 1.00 | ||
| Yes | .282 | 1.44 (0.72-2.87) | .11 | 1.75 (0.91-3.37) | |
| CRP | <ULN | 1.00 | 1.00 | ||
| >ULN | .113 | 1.69 (0.90-3.19) | .05 | 1.36 (0.99-1.54) | |
| Platelet count | <150 × 109 | 1.00 | 1.00 | ||
| >150 × 109 | .014 | 2.29 (1.29-4.05) | .063 | 1.99 (1.13-3.51) | |
| LDH | <ULN | 1.00 | 1.00 | ||
| >ULN | .001 | 2.73 (1.77-4.19) | .001 | 2.34 (1.57-3.49) | |
| ECOG-PS | ≥2 | 1.00 | 1.00 | ||
| ≤2 | .001 | 3.23 (2.14-4.89) | .001 | 2.71 (1.82-4.04) | |
HR, hazard ratio.
Multivariate analysis of prespecified parameters for OS and PFS
| Covariate | Group | OS | PFS | ||
|---|---|---|---|---|---|
| P | HR (95% CI) | P | HR (95% CI) | ||
| Disease stage | I-II | 1.00 | 1.00 | ||
| III-IV | .58 | 0.491 (0.23-1.02) | .205 | 0.63 (0.32-1.28) | |
| B symptoms | No | 1.00 | 1.00 | ||
| Yes | .008 | 2.01 (1.01-3.93) | .023 | 1.58 (0.81-3.08) | |
| CRP | <ULN | 1.00 | 1.00 | ||
| >ULN | .24 | 1.31 (0.63-2.76) | .09 | 1.35 (0.65-2.81) | |
| LDH | <ULN | 1.00 | 1.00 | ||
| >ULN | .001 | 3.77 (1.61-8.77) | .001 | 2.36 (1.01-5.36) | |
| ECOG-PS | ≥2 | 1.00 | 1.00 | ||
| ≤2 | .001 | 4.04 (2.07-7.84) | .001 | 3.69 (1.67-7.50) | |
| Covariate | Group | OS | PFS | ||
|---|---|---|---|---|---|
| P | HR (95% CI) | P | HR (95% CI) | ||
| Disease stage | I-II | 1.00 | 1.00 | ||
| III-IV | .58 | 0.491 (0.23-1.02) | .205 | 0.63 (0.32-1.28) | |
| B symptoms | No | 1.00 | 1.00 | ||
| Yes | .008 | 2.01 (1.01-3.93) | .023 | 1.58 (0.81-3.08) | |
| CRP | <ULN | 1.00 | 1.00 | ||
| >ULN | .24 | 1.31 (0.63-2.76) | .09 | 1.35 (0.65-2.81) | |
| LDH | <ULN | 1.00 | 1.00 | ||
| >ULN | .001 | 3.77 (1.61-8.77) | .001 | 2.36 (1.01-5.36) | |
| ECOG-PS | ≥2 | 1.00 | 1.00 | ||
| ≤2 | .001 | 4.04 (2.07-7.84) | .001 | 3.69 (1.67-7.50) | |
Discussion
The T-Cell Project is the largest, to date, prospective cohort study with a centralized computer database allowing for uniform analysis of PTCL patients enrolled at numerous independent global sites. Collection of the large number of cases created a unique opportunity to analyze the rare subtypes of these non-Hodgkin lymphomas with sufficient statistical power and without interference from other biologically distinct entities. Although the absence of a particular defined treatment protocol might seem as a limitation of the study, it also has the benefit of presenting a real-life scenario and outcomes in patients with a particular malignancy.
ALK– ALCL comprised 15% of the diagnoses reported to the T-Cell Project by the investigators. This frequency among PTCLs is higher than the 5.5% that was previously recorded in the retrospective International Peripheral T-cell Lymphoma Project.4 The reason for such a discrepancy is not entirely clear but might involve the demographic differences in the 2 study populations. It might also be supported by a report from the Surveillance, Epidemiology, and End Results registry showing demographic predilection of this entity, with a higher incidence in Black and non-Hispanic White subjects and a very low incidence in Asian, Hispanic, American Native, and Pacific Islander subjects.5
We also admit to a limitation of our study in the lack of central pathology review and diagnostic validation. Despite the planned expert review in the study design, investigators encountered unsurmountable challenges related to secure shipments of hundreds of tissue specimens from 74 countries with varying regulatory requirements. However, with rare exceptions, the patients in the study were enrolled by academic centers around the globe with recognized expertise in hematopathology, which partially negates this limitation in the opinion of study investigators. In addition, the authors acknowledge the missing data for several patient and disease characteristics, as well as treatment and outcome details, as another weakness of the study. Despite the colossal effort by the study executive team and investigators, the hurdles of conducting a global prospective data collection of this magnitude imposed these limitations.
Anthracycline-containing multiagent chemotherapy has long been a standard initial therapy for patients with ALCL approached with curative intent.6,14 In our study, 97% of patients were treated with CHOP(-like) therapy, including 15% who received both an anthracycline and etoposide. This is consistent with findings in the retrospective international project in which 93% of ALK– ALCL patients received multiagent chemotherapy with curative intent. The very high rate of curative intent multiagent chemotherapy in both registries despite the patients’ advanced median age (ie, >60 years) underscores the curative potential of aggressive front-line therapies in ALK– ALCL. In both studies, the rate of consolidative high-dose therapy and autologous stem cell transplantation was low at 8% and 7%, respectively. The latter finding might reflect the lack of randomized trial data showing clear benefit with a consolidative approach or access to advanced-level oncologic facilities equipped to perform the transplant procedure. Furthermore, our results recapitulate the findings from the retrospective study of superior outcomes in ALK– ALCL compared with PTCL not otherwise specified or in most other histologic subtypes.
The proximity in the range of major clinical end points (ie, OS, PFS) between previously published retrospective and current prospective registries is important in that it solidifies the benchmark against which future clinical trials of novel combinations and agents should be compared. It should also be noted that our results represent outcomes in real-life environments as opposed to highly standardized clinical trials conducted at expert academic institutions. As such, our results might provide an appropriate lens through which to extrapolate the results of either successful or failed academic trials. To validate our point, in the recently reported randomized, double-blind controlled clinical trial that compared novel brentuximab vedotin plus CHP combination with standard CHOP therapy, the 5-year rate of OS in the control group was ∼65%, whereas in our study and in the retrospective registry study, the 5-year OS rates were both 49%.10 If we assume that this sizable difference represents the inherent selection bias in academic clinical trials toward lower risk patients (eg, able to travel, younger, higher socioeconomic status, less rapidly progressive or lower burden disease to afford delay of care for screening periods), then we should also be cautious about overinterpreting an unusually high OS rate of ∼80% in the experimental arm that was not previously seen in PTCL or ALCL after the front-line therapy when applying these results to real-life oncology practice. We would therefore argue that prospective registry studies provide a unique perspective of disease outcomes that is compromised by the scrutiny of therapeutic academic and/or registrational studies.
It should be noted that since the data cutoff date in our study, the standard of care for ALCL patients has changed with the report of ECHELON-2 (Brentuximab Vedotin With Chemotherapy for CD30-Positive Peripheral T-Cell Lymphoma) clinical trial results, confirming clinical benefit of brentuximab vedotin and CHP combination over standard CHOP for CD30+ PTCL in which the majority of the study patients had an ALK+ and ALK– ALCL diagnosis.10 It is noteworthy that the superiority of the novel combination has not been shown over anthracycline/etoposide–containing regimens (ie, etoposide phosphate, prednisone, vincristine sulfate, cyclophosphamide, and doxorubicin hydrochloride [CHOEP]), and the addition of etoposide to CHOP-like regimens has shown a trend toward better survival outcomes (both, OS and PFS), at least in a retrospective meta-analysis of patients enrolled in clinical trials of the DSHNHL.9 It is also hard to dismiss the favorable results of the prospective single-institution phase 2 study in which 12-year PFS in high-risk ALCL patients was >70%.11 Resonating with these results, our study also indicates superior OS (P = .05) and a trend toward higher PFS (P = .186) in patients who received treatments that contained both anthracycline and etoposide. However, the authors acknowledge that there are significant limitations to this analysis, given the lack of randomization to a particular therapy; there is an inherent bias by the treating physicians to use more intensive therapies in younger fit patients with fewer comorbidities or disease-associated sequelae, potentially selecting for a better prognosis cohort. It should also be considered that such patients would be more likely to use aggressive curative intent therapies upon relapse that would further affect OS.
In addition, while brentuximab vedotin was approved by the US Food and Drug Administration in the midst of study enrollment, it has not been available and therefore not used in several participating countries, further confounding the analysis. It is therefore impossible to make strong recommendations regarding the benefits of etoposide when added to CHOP based on these data. Conversely, our results still raise the question of whether standard CHOP is an adequate control arm in indicating superiority of new treatment regimens over a standard-of-care approach. Finally, if we consider a 5-year failure rate of 60% by the current multidrug regimens to cure ALK– ALCL and sizable treatment-related morbidities, especially in light of numerous emerging novel biologic agents, we should then ask the provocative question of whether traditional “add-on” strategies in clinical trials should give way to developing new therapeutic platforms consisting entirely or mostly of rationally developed PTCL-specific drugs.
We also observed that the rates of OS and PFS in our study are in close proximity at both the 3- and 5-year cutoffs, indicating low rescue rates with current salvage strategies and underscoring the need for novel first-line platforms. We should bear in mind that brentuximab vedotin was available only through the second half of the study enrollment, and for many study locations only for small fraction of the enrollment period. Therefore, the full impact of this novel and highly effective agent was not captured. Recent studies of composite epigenetic-targeted therapy,15 combinations of phosphatidylinositol 3–kinase inhibitors + proteasome inhibitors, phosphatidylinositol 3–kinase inhibitors + histone deacetylase inhibitors, antifolate + histone deacetylase inhibitor,16 and others reported very promising levels of clinical responses comparable to the rates observed in front-line therapies with multiagent cytotoxic combinations. Combined with better toxicity profiles of novel agents compared with traditional cytotoxic drugs, it is likely only a matter of time that we will see a major shift from established CHOP-based platforms in ALK– ALCL and other PTCL subtypes.
Recent advances in genomic characterization of hematologic malignancies challenged traditional clinical prognostic scales (eg, IPI, PIT, PINK-E). Two specific recurrent genetic alterations have defined biologically distinct ALCL subtypes. The DUSP22-IRF4 translocation is found in ∼30% of patients with ALCL and portends excellent prognosis, with 5-year OS rates of >80% with traditional therapies.12 Mutations or cryptic translocations in TP63 can be identified in 5% to 6% of ALCLs and are predictive of the early treatment failures and a dismal overall prognosis, with <10% of patients being cured of their lymphoma.12,17 Based on some reports, even early application of high-dose therapy and autologous stem cell transplantation does not overcome the negative impact of these genomic lesions.13 These new molecular markers seem to be independent of clinical risk models and will most likely replace traditional models in disease stratification. Uneven distribution of the genomic subtypes of ALCL might account for some discrepancies in the results of therapeutic and cohort studies. These new findings are in urgent need of validation in prospective clinical trials, including prospective registry trials. The second stage of the T-Cell Project that was recently initiated will focus on molecular characterization of PTCLs as well as on further clinical and epidemiologic studies, and it will hopefully better define the role of new genomic markers in driving future therapies and risk stratification.
Requests for data sharing may be submitted to the corresponding author (Andrei Shustov; e-mail: ashustov@uw.edu).
Acknowledgments
This study was supported by grants from the Fondazione Cassa di Risparmio di Modena, the Associazione Angela Serra per la Ricerca sul Cancro, the Fondazione Italiana Linfomi, Allos Therapeutics, Inc., and Spectrum Pharmaceuticals, Inc.
Authorship
Contribution: A.S. and M.F. designed the research, interpreted the data, and wrote the manuscript; M.E.C., M.C., M.B., M.M., and T.S. collected and analyzed the data; Y.H.K., S.M.H., C.A.D., J.A.R., S.B., M.V.P., A.J.M.F., C.C., M.S., J.M.V., A.C., D.L., D.M., and C.S. provided patients and study materials; and all authors reviewed and approved the manuscript.
Conflict-of-interest disclosure: S.M.H. has consulted, received honorarium from, or participated in advisory boards for ADC Therapeutics, C4 Therapeutics, Celgene, Janssen, Kura Oncology, Kyowa Hakko Kirin, Myeloid Therapeutics, Seattle Genetics, Takeda, Trillium Therapeutics, Verastem, and Vividion Therapeutics; and received research support for clinical trials from ADC Therapeutics, Affimed, Aileron, Celgene, Daiichi Sankyo, Forty Seven, Inc., Kyowa Hakko Kirin, Millennium/Takeda, Portola Pharmaceuticals, Seattle Genetics, Trillium Therapeutics, and Verastem. The remaining authors declare no competing financial interests.
A complete list of participating institutions/investigators of the T-cell Lymphoma Project appears in “Appendix.”
Correspondence: Andrei Shustov, University of Washington School of Medicine, Seattle Cancer Care Alliance, 617 Eastlake Ave E, Mail Box CE3-300, Seattle, WA 98109; e-mail: ashustov@uw.edu.
Appendix
The participating institutions/investigators of the T-cell Lymphoma Project are: British Columbia Cancer Agency, Vancouver, BC, Canada (Kerry Savage, Joseph Connors, Randy Gascoyne, Mukesh Chhanabhai); National Cancer Institute, Bethesda, MD (Wyndham Wilson, Elaine S. Jaffe); University of Nebraska Medical Center, Omaha, NE (James O. Armitage, Julie M. Vose); Dennis D. Weisenburger, James Anderson, Fred Ullrich, Martin Bast); Massachusetts General Hospital, Boston, MA (Ephraim Hochberg, Nancy Harris, Agota Smogorzewska); Los Angeles County Hospital–University of Southern California, Los Angeles, CA (Alexandra Levine, Bharat N. Nathwani); Arizona Cancer Center, Tucson, AZ (Thomas Miller, Lisa Rimsza); University of Barcelona Hospital, Barcelona, Spain (Emili Montserrat, Armando Lopez-Guillermo, Elias Campo); Spanish National Cancer Center, Madrid, Spain (Marta Cuadros, Javier Alvarez Ferreira, Beatriz Martinez Delgado); Norwegian Radium Hospital, Oslo, Norway (Harold Holte, Jan Delabie); University of Würzburg Hospital, Würzburg, Germany (Thomas Rüdiger); Institute of Pathology, University of Würzburg, Würzburg, Germany (Konrad Müller-Hermelink, Peter Reimer); Krankenhaus Munchen-Schwabing, Munich, Germany (Patrick Adam, Martin Wilhelm, Norbert Schmitz, Christoph Nerl); Saint Bartholomew's Hospital, London, United Kingdom (Andrew Lister, Andrew Norton); St James Hospital, Leeds, United Kingdom (Kenneth A. MacLennan); University of Bologna Hospital, Bologna, Italy (Pier Luigi Zinzani, Stefano A. Pileri); Intergruppo Italiano Linfomi, Bologna, Italy (Massimo Federico, Monica Bellei, Stefano Luminari); Centre Hospitalier Lyon-Sud, Lyon, France (Bertrand Coiffier, Francoise Berger); King Chulalongkorn Hospital, Bangkok, Thailand (Intragumtornchai Tanin, Pongsak Wannakrairot); Queen Mary Hospital, Hong Kong, China (Wing Y. Au, Raymond Liang, Florence Loong); Singapore General Hospital, Singapore (Sandeep Rajan, Ivy Sng); National Cancer Center Hospital of Japan, Tokyo, Japan (Kensei Tobinai, Yoshihiro Matsuno); Aichi Cancer Center, Nagoya, Japan (Yasuo Morishima, Shigeo Nakamura, Masao Seto); Okayama University Hospital, Okayama, Japan (Mitsune Tanimoto, Tadashi Yoshino); Fukuoka University Hospital, Fukuoka, Japan (Junji Suzumiya, Koichi Ohshima); and Samsung Medical Center, Seoul, Korea (Won-Seog Kim, Young-Hyeh Ko).

