

Key Points
Degradation inhibitors influence megakaryocyte differentiation in induced pluripotent stem cells and primary cells in RUNX1-FPD.
Blocking RUNX1 degradation may offer a therapeutic entry point for controlling bleeding or preventing leukemia in RUNX1-FPD.

Abstract
RUNX1 familial platelet disorder (RUNX1-FPD) is an autosomal dominant disorder caused by a monoallelic mutation of RUNX1, initially resulting in approximately half-normal RUNX1 activity. Clinical features include thrombocytopenia, platelet functional defects, and a predisposition to leukemia. RUNX1 is rapidly degraded through the ubiquitin-proteasome pathway. Moreover, it may autoregulate its expression. A predicted kinetic property of autoregulatory circuits is that transient perturbations of steady-state levels result in continued maintenance of expression at adjusted levels, even after inhibitors of degradation or inducers of transcription are withdrawn, suggesting that transient inhibition of RUNX1 degradation may have prolonged effects. We hypothesized that pharmacological inhibition of RUNX1 protein degradation could normalize RUNX1 protein levels, restore the number of platelets and their function, and potentially delay or prevent malignant transformation. In this study, we evaluated cell lines, induced pluripotent stem cells derived from patients with RUNX1-FPD, RUNX1-FPD primary bone marrow cells, and acute myeloid leukemia blood cells from patients with RUNX1 mutations. The results showed that, in some circumstances, transient expression of exogenous RUNX1 or inhibition of steps leading to RUNX1 ubiquitylation and proteasomal degradation restored RUNX1 levels, thereby advancing megakaryocytic differentiation in vitro. Thus, drugs retarding RUNX1 proteolytic degradation may represent a therapeutic avenue for treating bleeding complications and preventing leukemia in RUNX1-FPD.
Introduction
RUNX1-familial platelet disorder (FPD)1 consists of platelet granule abnormalities, thrombocytopenia, and predisposition to myelodysplastic syndrome and acute myeloid leukemia (AML).2 RUNX1-FPD is caused by heterozygous germline mutations in the transcription factor RUNX1. It is 1 of 3 runt homology DNA-binding domain family members that form heterodimers with a non–DNA binding partner, collectively known as core binding factors.3 RUNX1 is mutated in ∼10% of cases of adult AML, 30% of which are germline mutations.4,5 RUNX1-FPD mutations generate null, hypomorphic, or dominant-negative alleles.2,6-8
At conception, patients with RUNX1-FPD possess 1 wild-type allele, producing normal protein, albeit at approximately half-normal levels. Mutations involving the wild-type RUNX1 allele accompany malignant progression.7-11 Transformation may be facilitated by RUNX1 haploinsufficiency and depend on cooperating mutations, such as DNMT3A.7
The half-life of RUNX1 is ∼30 to 60 minutes,12 and phosphorylation primarily governs its stability via ubiquitin-dependent proteasomal degradation.13,14
RUNX1 autoregulates its expression by recognizing one of its promoters.15 For an autoactivating protein, steady state occurs when rate constants for synthesis and degradation are equal, independent of protein concentration. Consequently, when the rate of synthesis exceeds the rate of degradation, protein concentration rises exponentially and remains persistently elevated after the perturbation ceases and steady state is restored.16 For example, transient inhibition of proteases degrading MyoD, which similarly autoactivates its expression, causes MyoD concentration to remain elevated after withdrawal of inhibitors.16
We hypothesize that drugs retarding the proteolytic degradation of RUNX1, even transiently, may help restore steady-state levels of RUNX1 in RUNX1-FPD and normalize platelet production. To test this hypothesis, we attempted to “jump start” RUNX1 expression by transient overexpression of exogenous RUNX1 or brief treatment with small-molecule inhibitors of the ubiquitin-proteasome degradation pathway and then assess the corresponding impact on megakaryocyte differentiation in cell lines and patient-derived induced pluripotent stem cells (iPSCs) and primary cells. We found that inhibition of kinases regulating ubiquitylation or inhibition of the proteasome increased RUNX1 levels and advanced megakaryocyte differentiation, suggesting potential new therapeutic opportunities for treating bleeding and preventing leukemia in RUNX1-FPD.
Methods
Cell culture
293T cells17 were cultured in Dulbecco’s modified Eagle’s medium. MEG-0118 cells were cultured semiadherently at 2 × 105 to 4 × 105 cells per mL in RPMI-1640 medium. K56219 cells were cultured to between 2 × 105 and 2 × 106 cells per mL in Iscove’s modified Dulbecco’s modified Eagle’s medium (IMDM). The media were supplemented with 10% fetal bovine serum (FBS).
Drug treatments
Roscovitine, flavopiridol, P276-00, SCH727965, R547, bortezomib, carfilzomib, ixazomib, oprozomib, and pevonedistat (Thermo Fisher Scientific) and terameprocol (Sigma-Aldrich) were dissolved in dimethyl sulfoxide (DMSO; supplemental Table 1). For each cell line, the 50% lethal concentration was determined with the CellTiter 96 (Promega) reagent. Cells (2.5 × 106) cells were treated for 24 hours. RNA purification was performed with RNeasy Plus Mini Kits (Qiagen). Protein lysates were obtained with radioimmunoprecipitation assay buffer containing Complete Protease Inhibitor (Roche), 1 mM Na3VO4, and 1 mM phenylmethylsulfonyl fluoride (PMSF).
RUNX1-FPD patient-derived iPSCs
With written informed consent and Children’s Hospital of Philadelphia Institutional Review Board approval, Sendai virus–reprogrammed iPSCs20 were generated from peripheral blood mononuclear cells (MNCs) from an adult male patient with RUNX1-FPD without leukemia who was heterozygous for a germline RUNX1 splicing mutation (NM_00100189:c.533-1G>T).8 For isogenic controls, the mutation was corrected via CRISPR-Cas9–mediated, homology-directed repair genome editing.21 N-2.12 cells served as additional iPSC controls.22 Hematopoietic differentiation was performed as described.22 On day 8, hemopoietic stem progenitor cells in liquid culture were treated for 2 days with vehicle or drugs, dissociated with Accutase, cytometrically analyzed for CD45/CD41 expression, and plated for colony-forming unit (CFU) assays, with scoring 12 days later.
Primary samples
With written informed consent and University of Washington Institutional Review Board approval, bone marrow was aspirated from 3 females (sisters aged 11 and 20 years and their 40-year-old aunt) without leukemia, sharing the heterozygous RUNX1 mutation NM_001754 c.1242C>A, p.Y414X.23 MNCs were isolated by using lymphocyte separation medium (Corning). For patients with AML, cryopreserved MNCs from their peripheral blood or bone marrow were cultured in IMEM with 20% horse serum and 20% FBS (initial analysis depicted in supplemental Table 2; supplemental Figure 1). The patients’ characteristics, including relevant DNA sequence analysis (MyAML 194-gene panel; Invivoscribe), are summarized in supplemental Table 3.
RUNX1 expression constructs
Human RUNX1 variant-1 cDNA (NM_001754.3) with carboxyl-terminal FLAG epitope tag in pcDNA3.1 vector (pcDNA3.1-RUNX1–FLAG) was purchased from Genscript (OHu26363). Cells (2 × 106) 293T cells were transfected with 4 µg pcDNA3.1-RUNX1–FLAG plasmid with 12 µL Lipofectamine 2000 (Thermo Fisher Scientific) per 10-cm plate. One day later, the cells were passaged, pooled, and collected at the 24-hour posttransfection time point. The remaining cells were seeded into 10-cm plates at equal densities for collection at later time points.
Messenger RNA quantification
Superscript IV reverse transcriptase (Thermo Fisher) was used to generate cDNA, and quantitative reverse transcription-polymerase chain reaction (qRT-PCR) was performed with the Thermo Fisher Scientific StepOnePlus Real-Time PCR System with TaqMan probes for RUNX1 (Hs01021970_m1) and β-actin (4333762F) control, by the 2–ΔΔCT method. Semiquantitative analysis was performed by agarose gel electrophoresis.
Immunoblot analysis
Protein concentrations were determined by bicinchoninic assay (Pierce). Samples were diluted in Laemmli buffer, heated at 90°C for 10 minutes, and electrophoresed on Mini-Protean TGX 4% to 10% Tris-glycine sodium dodecyl sulfate precast gels (Bio-Rad), transferred to a polyvinylidene difluoride membrane, blocked for 30 minutes, and washed with Tris-buffered saline-Tween 20. Blots were developed on Immobilon Western Chemiluminescent HRP Substrate (EMD Millipore), SuperSignal West Pico, or Femto chemiluminescent substrate (Thermo Fisher Scientific) with Kodak BioMax XAR film or with the ChemiDoc MP Imaging System (BioRad). Antibody sources, dilutions, and buffers are listed in supplemental Table 4.
Clonogenic assays
A megakaryocyte CFU assay was performed with the MegaCult-C Complete Kit with Cytokines (Stemcell Technologies). At day 8 of hematopoietic differentiation, the hemopoietic stem progenitor cells were treated for 2 days with vehicle or drugs. After treatment, the cells were harvested, dissociated with Accutase, and cytometrically analyzed for CD41 expression. The cells (103) were mixed with MegaCult-C and plated in duplicate, then incubated at 37°C for 12 days before staining and scoring, with normalization to CD41a+ proportion at the time of plating.
Primary cell megakaryocyte differentiation
MNCs were thawed and seeded at 0.2 × 106 to 1 × 106 cells per 12-well plate with the various drugs or vehicle. StemSpan SFEM II basal medium containing Megakaryocyte Expansion Supplement (Stemcell Technologies) was used for megakaryocyte differentiation. After 24 hours of treatment (day 1), the cells were removed from the plate, and the pellets were resuspended in freshly added differentiation medium containing drug or vehicle and incubated for an additional 24 hours. On day 2 (48 hours of drug exposure), the cells were removed, washed with phosphate buffered saline (PBS), and transferred to drug-free differentiation medium for the remainder of the experiment. Medium was changed on days 4 and 7. On day 10, the wells were supplemented with 1 mL of fresh differentiation medium. Megakaryocyte formation was characterized on day 14 via flow cytometry, cytocentrifuging (Cytospin) with Wright-Giemsa staining, transmission electron microscopy (TEM), or RNA isolation. Relative megakaryocyte yields were calculated by comparing seed-matched drug and vehicle conditions.
Flow cytometric analysis
Megakaryocytes formed from RUNX1-FPD patient-derived iPSCs were stained with CD41a and CD45 antibodies and assayed with a BD LSRFortessa cell analyzer.22 Primary MNC-derived megakaryocytes were characterized via flow cytometry for surface markers CD41a, CD42b, and CD61, with a BD FACSCanto II flow cytometer. On day 14 of in vitro megakaryocyte differentiation, the cells were washed with 0.1% FBS/PBS (fluorescence-activated cell sorting buffer) and incubated with antibodies for 30 minutes on ice in the dark. Stained cells were washed twice and resuspended in fresh buffer for analysis. Data were processed with FlowJo 10 software. Antibodies and their dilutions are listed in supplemental Table 5.
Electron microscopy
At day 14 of in vitro differentiation of cells from patient FPD-1, ∼106 cells were prepared for TEM (supplemental Figure 2). An equal volume of Karnovsky fixative was added to each well and incubated at 37°C for 10 minutes, followed by centrifugation. Medium and fixative were removed and replaced with pure fixative for 1 hour at room temperature. The cells were scraped from the well, transferred to a microcentrifuge tube, and stored at 4°C until TEM with a JEOL 120 kV JEM-1400 microscope.
Megakaryocyte granule quantification
TEM images were scored manually by counting the number of dense and α-granules per cell, based on standardized morphological classification.24 Dense granules were identified as small (∼0.25 µm), dark gray/black, circular puncta located within the vesicles. The α-granules were scored as larger (∼0.75 µm) gray vesicles, with defined borders and sandy-appearing internal composition. The cells were binned by quartile: no granules (−), 1 to 10 granules per cell (+), 1 to 20 granules per cell (++), and >20 granules per cell (+++). Unidentifiable granules or granule-like components were not scored.
Results
Transient expression of exogenous RUNX1 results in a sustained increase of endogenous RUNX1 transcript and protein
Given the reported ability of RUNX1 reported to autoactivate expression from its P1 promoter,15 we reasoned that transient expression of exogenous RUNX1 could result in prolonged elevation of endogenous RUNX1 messenger RNA (mRNA) and protein levels.16 To test this prediction, we used the human embryonic kidney cell line 293T, because it exhibits minimal levels of endogenous RUNX1 expression,25 potentially permitting changes in its expression to become more apparent. To distinguish exogenously transfected RUNX1 from endogenous RUNX1, a FLAG epitope tag was attached to the C-terminus of RUNX1 (RUNX1-FLAG). Levels of total RUNX1 protein increased, with a peak at days 3 to 4, after transient transfection of a vector expressing RUNX1-FLAG and were then maintained at fairly uniform levels throughout the 10-day time course (Figure 1A; top and bottom panels). In contrast, levels of transiently expressed RUNX1-FLAG decreased (as detected by antibodies to the FLAG epitope tag; Figure 1A; middle panel), whereas levels of internal control β-actin remained constant over the time course (Figure 1A; bottom panel). Semiquantitative RT-PCR with primers specific to sequences encoding the FLAG epitope confirmed diminution of the transiently transfected RUNX1-FLAG transcript (Figure 1B), and measurement of total RUNX1 transcript by qRT-PCR for RUNX1 mRNA (Figure 1C) mirrored the RUNX1 protein levels apparent on western blot analysis. RUNX1-FLAG expression vectors possessed a constitutive promoter lacking RUNX1 binding sites and therefore did not maintain an autoactivation circuit for transfected RUNX1-FLAG. We suspect that, as transiently expressed RUNX1-FLAG dissipates over time, newly increased quantities of endogenous RUNX1 protein will subsequently maintain elevated expression via autoactivation, but additional studies are necessary for confirmation.
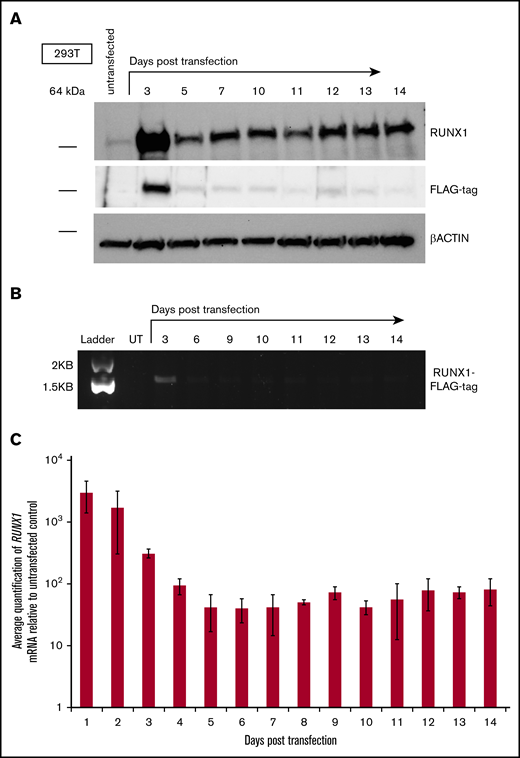
Effect of transient expression of exogenous RUNX1 on expression of endogenous RUNX1 in 293T cells. (A) Western blot analysis of total RUNX1 protein in 293T cells transiently transfected with FLAG epitope-tagged RUNX1 expression vector (RUNX1-FLAG), over a 10-day time course. Immunostaining performed with antibodies directed against RUNX1, FLAG tag, and β-actin control. (B) Agarose gel electrophoresis showing semiquantitative RT-PCR measurement of RUNX1-FLAG transcript, corresponding to the transfection performed in panel A. UT, untransfected. (C) Average fold change of RUNX1 transcript in 293T cells transiently transfected with RUNX1-FLAG plasmid, relative to untransfected control, as measured by qRT-PCR. Error bars show standard deviation of 3 repetitions.
Effect of transient expression of exogenous RUNX1 on expression of endogenous RUNX1 in 293T cells. (A) Western blot analysis of total RUNX1 protein in 293T cells transiently transfected with FLAG epitope-tagged RUNX1 expression vector (RUNX1-FLAG), over a 10-day time course. Immunostaining performed with antibodies directed against RUNX1, FLAG tag, and β-actin control. (B) Agarose gel electrophoresis showing semiquantitative RT-PCR measurement of RUNX1-FLAG transcript, corresponding to the transfection performed in panel A. UT, untransfected. (C) Average fold change of RUNX1 transcript in 293T cells transiently transfected with RUNX1-FLAG plasmid, relative to untransfected control, as measured by qRT-PCR. Error bars show standard deviation of 3 repetitions.
Inhibitors of RUNX1 proteolytic degradation influence levels of exogenously transfected RUNX1 protein and endogenous RUNX1 transcript
Because RUNX1-FPD typically results from haploinsufficiency of RUNX1, we wanted to determine whether retarding degradation of RUNX1 protein could stably elevate its steady-state levels. Phosphorylation of RUNX1 regulates its stability via ubiquitin-dependent proteasomal degradation. RUNX1 is phosphorylated by the cyclin-dependent kinases (CDKs) CDK1, CDK2, and/or CDK6,26-28 which subsequently renders it vulnerable to degradation mediated by the CDC20-containing anaphase promoting complex (APC),27,28 and, to a lesser extent, the Skp, Cullin, F-box (SCF)-SKP2 complex27 during the G2/M and S cell cycle phases, respectively. The RUNX1 degradation pathway is summarized in Figure 2A. To inhibit the initial phosphorylation step of the degradation pathway, we tested the following CDK inhibitors developed for clinical use: roscovitine, (targeting CDK1 and CDK2), R547 (CDK1 and CDK2), and terameprocol (CDK1).29 The APC and SCF E3 ligase complexes include the cullin-RING ubiquitin ligases Cul1 and ANAPC2, respectively, and are dependent on the neddylation system, an enzymatic cascade initiated by NEDD8 activating enzyme (NAE). We therefore tested the NAE inhibitor, pevonedistat.30 To inhibit the final step in the RUNX1-degradation pathway, we evaluated the 26S proteasome inhibitor bortezomib, as well as the second-generation inhibitors carfilzomib and orally administered ixazomib and oprozomib.31 We examined 293T cells transiently transfected with RUNX1-FLAG, followed by treatment with inhibitors. Western blot analysis revealed increases in transfected RUNX1-FLAG protein, indicating that RUNX1 is susceptible to drug-induced inhibition of protein degradation, especially with proteasome inhibitors and, to a lesser extent, the CDK inhibitor, terameprocol, but not pevonedistat (Figure 2B). Drug concentrations are listed in supplemental Table 1. qRT-PCR for expression of exogenously transfected RUNX1-FLAG revealed commensurately elevated RUNX1 transcript (Figure 2C). Given that the RUNX1-FLAG expression vector has no RUNX1 binding site, we conclude that retarding RUNX1 degradation increases RUNX1 protein abundance, which, in turn elevates RUNX1 transcript levels.
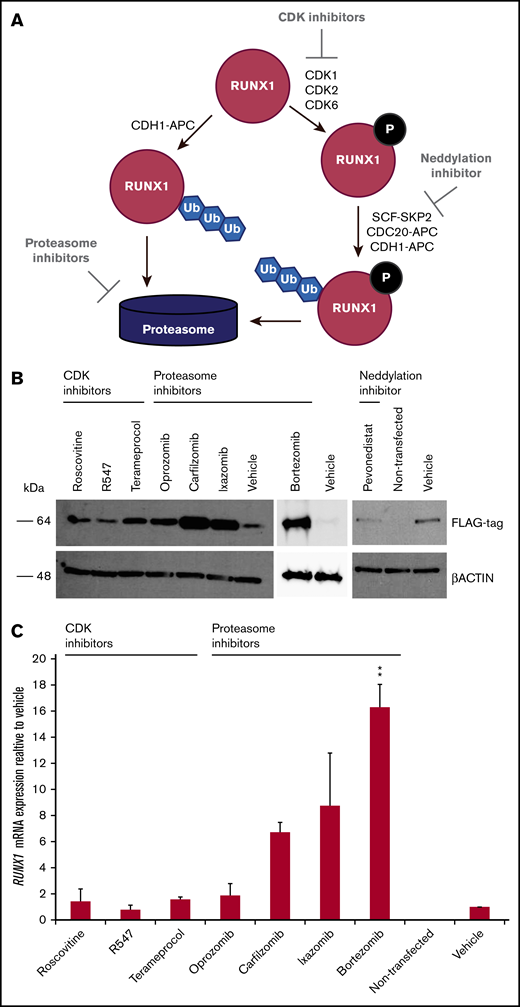
Effect of drugs inhibiting RUNX1 proteolytic degradation on RUNX1 protein and transcript levels in 293T cells transiently transfected with RUNX1-FLAG. (A) RUNX1 protein degradation pathway, showing the points at which inhibitors act. (B) Western blot analysis exhibiting elevated levels of exogenous RUNX1-FLAG protein expression in transiently transfected 293T cells after 24 hours of drug treatment vs vehicle (DMSO) treatment. (C) qRT-PCR revealing commensurately elevated levels of RUNX1 transcript in drug-treated 293T cells transiently transfected with RUNX1-FLAG. Error bars show standard deviation of 2 repetitions. Drug concentrations for all experiments are listed in supplemental Table 1. **P < .01, using a 2-tailed Student t test assuming unequal variance.
Effect of drugs inhibiting RUNX1 proteolytic degradation on RUNX1 protein and transcript levels in 293T cells transiently transfected with RUNX1-FLAG. (A) RUNX1 protein degradation pathway, showing the points at which inhibitors act. (B) Western blot analysis exhibiting elevated levels of exogenous RUNX1-FLAG protein expression in transiently transfected 293T cells after 24 hours of drug treatment vs vehicle (DMSO) treatment. (C) qRT-PCR revealing commensurately elevated levels of RUNX1 transcript in drug-treated 293T cells transiently transfected with RUNX1-FLAG. Error bars show standard deviation of 2 repetitions. Drug concentrations for all experiments are listed in supplemental Table 1. **P < .01, using a 2-tailed Student t test assuming unequal variance.
Inhibitors of RUNX1 proteolytic degradation influence endogenous RUNX1 protein and transcript levels, even after treatment
We next evaluated how inhibition of RUNX1 degradation influences endogenous levels of RUNX1 protein. CDK and, especially, proteasome inhibitors, but not the tested neddylation inhibitor pevonedistat, increased levels of RUNX1 protein, even in nontransfected 293T cells (Figure 3A), which, as noted, ordinarily express low relative levels of RUNX1. Terameprocol treatment reduced levels of detectable RUNX1 protein phosphorylated at position S276 (supplemental Figure 3), as expected. Consistent with RUNX1 autoactivation, changes in RUNX1 mRNA levels (Figure 3B) generally correlated with drug-induced changes in RUNX1 protein levels. We investigated whether elevated RUNX1 expression persists after transient inhibition of degradation pathways. We treated 293T cells for 24 hours, then washed and continued to grow the cells in drug-free medium, extracting aliquots 24, 48, and 72 hours later. Tests of all 3 drugs (carfilzomib, ixazomib, and bortezomib) showed sustained elevation of RUNX1 protein (Figure 3C) and transcript levels (Figure 3D), relative to vehicle-treated controls, particularly at 24 hours after their removal, but continuing up to 72 hours, although diminishing somewhat.
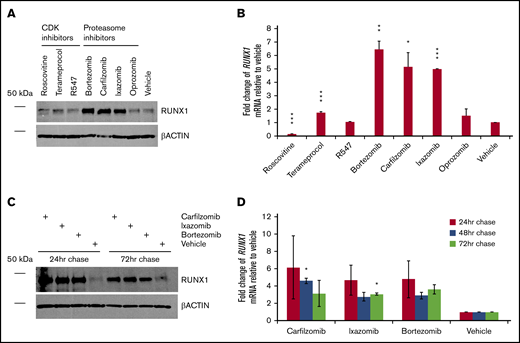
Persistence of RUNX1 expression after transient inhibition of its proteolytic degradation. (A) Nontransfected 293T cells were treated with the indicated drug or vehicle (DMSO) for 24 hours. Immediately afterward, protein lysates were subjected to western blot detection of endogenous RUNX1 compared with β-actin control. (B) Average fold change of endogenous RUNX1 mRNA expression relative to vehicle treatment, for drugs corresponding to panel A. (C) Nontransfected 293T cells were treated with the indicated drug or vehicle for 24 hours. At 24, 48, and 72 hours after the conclusion of drug treatment, protein lysates from aliquots were subjected to western blot detection of endogenous RUNX1 compared with β-actin control. (D) Average fold change of endogenous RUNX1 mRNA expression relative to vehicle treatment, for drugs corresponding to panel C. Error bars show standard deviation. *P < .05; **P < .01; ***P < .001, using a 2-tailed Student t test assuming unequal variance.
Persistence of RUNX1 expression after transient inhibition of its proteolytic degradation. (A) Nontransfected 293T cells were treated with the indicated drug or vehicle (DMSO) for 24 hours. Immediately afterward, protein lysates were subjected to western blot detection of endogenous RUNX1 compared with β-actin control. (B) Average fold change of endogenous RUNX1 mRNA expression relative to vehicle treatment, for drugs corresponding to panel A. (C) Nontransfected 293T cells were treated with the indicated drug or vehicle for 24 hours. At 24, 48, and 72 hours after the conclusion of drug treatment, protein lysates from aliquots were subjected to western blot detection of endogenous RUNX1 compared with β-actin control. (D) Average fold change of endogenous RUNX1 mRNA expression relative to vehicle treatment, for drugs corresponding to panel C. Error bars show standard deviation. *P < .05; **P < .01; ***P < .001, using a 2-tailed Student t test assuming unequal variance.
We also evaluated inhibitors in the hematopoietic cell lines K562 and MEG-01 (supplemental Figure 4). We observed variable effects, depending on class of inhibitor, cell line, and time course. RUNX1 mRNA levels generally correlated with RUNX1 protein levels. Exceptions may indicate drug-specific, posttranslational modifications, such as phosphorylation, that render total RUNX1 protein levels unchanged but increase activity. That these cell lines are leukemia-derived may make them less useful for evaluating the preleukemic phase of RUNX1-FPD. We therefore focused subsequent studies on RUNX1-FPD patient–derived iPSCs and primary cells.
Inhibitors of RUNX1 proteolytic degradation influence megakaryocyte output during differentiation of RUNX1-FPD iPSCs
iPSCs from patients with RUNX1-FPD display aberrant megakaryocytic differentiation in vitro, including reduced megakaryocytic colony formation and granule abnormalities.32 We therefore determined whether treatment of patient-derived iPSCs with drugs inhibiting RUNX1 degradation could normalize differentiation. iPSCs were generated from a patient with RUNX1-FPD who was heterozygous for a germline RUNX1 mutation in the splice acceptor site of exon 4 (NM_00100189:c.533-1G>T), which enforces the use of a cryptic splice acceptor in exon 4 with a resultant frameshift that causes premature termination.8 We prepared an isogenic control using CRISPR-Cas9–based, homology-directed repair to correct the germline mutation. The iPSCs were differentiated along the hematopoietic lineage and treated with drugs inhibiting RUNX1 proteolytic degradation (Figure 4A). Flow cytometric analysis of the pan megakaryocyte surface marker CD41a33 revealed only small, nonsignificant changes in iPSC-derived megakaryocytes when treated for 2 days with bortezomib or terameprocol (Figure 4B-C). Megakaryocyte-specific CFU assays were performed to determine the clonogenic capacity of CD41a+ hematopoietic stem and progenitor cells after treatment. Megakaryocytic colonies were scored 12 days after plating in drug-free semisolid medium and showed that bortezomib treatment produced significantly more total colonies compared with vehicle-treated FPD-iPSCs (P = .0076; n = 3; Figure 4D). Notably, bortezomib and terameprocol treatment reduced colony formation compared with vehicle in the isogenic control containing wild-type RUNX1.
![Transient inhibition of RUNX1 proteolytic degradation during megakaryocytic differentiation of RUNX1-FPD iPSCs. (A) In vitro iPSC megakaryocyte differentiation with drug treatment. (B) Representative flow diagrams of iPSC-derived megakaryocyte progenitors (CD41a+/CD45−). (C) Relative fold change of megakaryocyte progenitors generated from in vitro iPSC differentiation (FPD-iPSCs [n = 3]; isogenic and wild-type N-2.12 control cells [n = 2]). (D) Scored iPSC-derived megakaryocyte colonies after 14 days of a CFU assay. Error bars show standard deviation. *P < .05, using a 2-tailed Student t test assuming unequal variance.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/5/3/10.1182_bloodadvances.2020002709/2/m_advancesadv2020002709f4.png?Expires=1769103732&Signature=g-YSVa0k3jzqU4sAFHEyIp3fMrkl51vDez2NiCq6uaF5vMLtu1Y44i~oRXEDcrm7oNBENll-O8ZFST5WM5zmAbz~7T7ioTkepa0~96oPb6oUVVU-50QL-~28N-qP8Cc~RWPpmNdNTwaen4Pnyl8ltFT6no45PrOR3xM04SEPz0-sMibzXM4xQhemd7EssCZEpWowreqNpJe4BwXHe~LLHrQnj65AKKXzQqTcNiJ9q6Qf5ijTBK5fqouUvo8bUTcxaSKvWKzPSIjJKGXVX24QWUwu0ZcGZAwM14qEz~IkplisGBb09HB8bcYdW8uSl6qTjufuqJ5HMZzQRY3nWEwbxA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Transient inhibition of RUNX1 proteolytic degradation during megakaryocytic differentiation of RUNX1-FPD iPSCs. (A) In vitro iPSC megakaryocyte differentiation with drug treatment. (B) Representative flow diagrams of iPSC-derived megakaryocyte progenitors (CD41a+/CD45−). (C) Relative fold change of megakaryocyte progenitors generated from in vitro iPSC differentiation (FPD-iPSCs [n = 3]; isogenic and wild-type N-2.12 control cells [n = 2]). (D) Scored iPSC-derived megakaryocyte colonies after 14 days of a CFU assay. Error bars show standard deviation. *P < .05, using a 2-tailed Student t test assuming unequal variance.
Transient inhibition of RUNX1 proteolytic degradation during megakaryocytic differentiation of RUNX1-FPD iPSCs. (A) In vitro iPSC megakaryocyte differentiation with drug treatment. (B) Representative flow diagrams of iPSC-derived megakaryocyte progenitors (CD41a+/CD45−). (C) Relative fold change of megakaryocyte progenitors generated from in vitro iPSC differentiation (FPD-iPSCs [n = 3]; isogenic and wild-type N-2.12 control cells [n = 2]). (D) Scored iPSC-derived megakaryocyte colonies after 14 days of a CFU assay. Error bars show standard deviation. *P < .05, using a 2-tailed Student t test assuming unequal variance.
Inhibitors of RUNX1 proteolytic degradation influence megakaryocytic maturation in RUNX1-FPD primary cells
We determined whether inhibition of RUNX1 proteolytic degradation influences in vitro megakaryocytic differentiation of bone marrow cells of patients with RUNX1-FPD. Primary MNCs were isolated from bone marrow collected from 3 affected members of a family with RUNX1-FPD (none of whom had leukemia) sharing a heterozygous germline RUNX1 exon 9 nonsense mutation (NM_001754:c.1242C>A, p.Y414X).23 The cells were treated with either bortezomib or terameprocol for the first 48 hours of in vitro megakaryocytic differentiation (Figure 5A). The cells were harvested at the conclusion of the differentiation protocol after 12-days of culture in drug-free medium and assayed by flow cytometry for expression of the pan megakaryocyte surface markers33 CD41a and CD61, as well as CD42b, a marker indicative of advanced megakaryocyte maturation (Figure 5B; supplemental Figure 1). Cells from each of the patients responded differently to drug treatment, and no patient or treatment met the threshold of significance (Figure 5C; summarized in supplemental Table 2). When results were averaged across all patients, bortezomib exposure did not alter the overall frequency of megakaryocytes, defined as CD41a+/CD61+ double-positive cells, compared with vehicle-treated controls (1.08- ± 0.29-fold change relative to vehicle); however, we observed a modest increase in immature (CD41a+/CD42b−) megakaryocytes at the expense of mature megakaryocytes (1.16- ± 0.18-fold vs 0.77- ± 0.09-fold). In contrast, exposure to terameprocol caused an increase in total megakaryocytes compared with vehicle-treated controls (1.14- ± 0.16-fold), along with a specific increase in mature (1.16- ± 0.23-fold), but not immature (1.04- ± 0.12-fold), megakaryocyte populations. The appearance of cells after Wright-Giemsa staining corroborated these findings (Figure 5D). Terameprocol-treated cells exhibited features consistent with proplatelet formation, such as cell surface blebbing,34 which was not detected in cells treated with bortezomib or vehicle. qRT-PCR analysis showed that there was a transition from MYH10 to MYH9 expression, which is indicative of megakaryocyte maturation,35 after treatment with bortezomib or terameprocol, and that treatment with bortezomib normalized RUNX1 expression (Figure 5E).
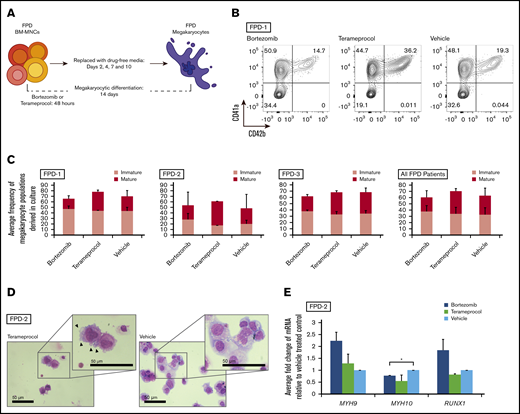
RUNX1-FPD primary bone marrow MNCs treated with inhibitors of RUNX1 proteolytic degradation early during in vitro megakaryocyte differentiation. (A) Differentiation protocol. (B) Representative flow cytometric analysis of megakaryocytes generated from patient FPD-1 at the conclusion of the differentiation protocol. (C) Graphic representation of flow cytometric analysis, as depicted in panel B, relative to vehicle-treated control, for each patient (FPD-1, -2, and -3, or all 3 analyzed collectively). Immature megakaryocyte populations correspond to CD41a+/CD42b− and mature populations correspond to CD41a+/CD42b+. Confidence interval represents standard deviation of 1 to 4 repetitions. (D) Cytocentrifuge preparation of cells (Wright-Giemsa stain) harvested at the conclusion of the differentiation protocol. Representative results obtained from FPD-2. Arrowheads indicate blebs consistent with proplatelet formation. (E) Myosin and RUNX1 expression after drug treatment in primary bone marrow samples. qRT-PCR for the indicated genes at day 14 of differentiation of primary bone marrow cells (FPD-2; n = 2). *P < .05, using a 2-tailed Student t test assuming unequal variance.
RUNX1-FPD primary bone marrow MNCs treated with inhibitors of RUNX1 proteolytic degradation early during in vitro megakaryocyte differentiation. (A) Differentiation protocol. (B) Representative flow cytometric analysis of megakaryocytes generated from patient FPD-1 at the conclusion of the differentiation protocol. (C) Graphic representation of flow cytometric analysis, as depicted in panel B, relative to vehicle-treated control, for each patient (FPD-1, -2, and -3, or all 3 analyzed collectively). Immature megakaryocyte populations correspond to CD41a+/CD42b− and mature populations correspond to CD41a+/CD42b+. Confidence interval represents standard deviation of 1 to 4 repetitions. (D) Cytocentrifuge preparation of cells (Wright-Giemsa stain) harvested at the conclusion of the differentiation protocol. Representative results obtained from FPD-2. Arrowheads indicate blebs consistent with proplatelet formation. (E) Myosin and RUNX1 expression after drug treatment in primary bone marrow samples. qRT-PCR for the indicated genes at day 14 of differentiation of primary bone marrow cells (FPD-2; n = 2). *P < .05, using a 2-tailed Student t test assuming unequal variance.
Inhibitors of RUNX1 proteolytic degradation influence dense granule formation in RUNX1-FPD primary cells
Deficient proplatelet formation, particularly involving dense granules,36 is a feature of RUNX1-FPD. We used TEM to more closely evaluate maturational changes in megakaryocyte morphology occurring with drug treatment. Using RUNX1-FPD primary cells with the protocol shown in Figure 5A, we found that vehicle-treated megakaryocytes often displayed some vacuoles, as well as α-granules with decreased content, compared with bortezomib- or terameprocol-treated samples (Figure 6A). We found that cells treated with either drug had a significantly greater abundance of dense granules (Figure 6B; bortezomib, P = .0025; terameprocol, P = .022). In contrast, α-granule abundance was not significantly altered after treatment with either drug (Figure 6C). These observations are in line with the dense granule defects that were previously observed in RUNX1-FPD-derived iPSCs and that were reversed after genome-editing correction of the germline RUNX1 mutation.32 These results suggest at least some restoration of cellular function after drug treatment.
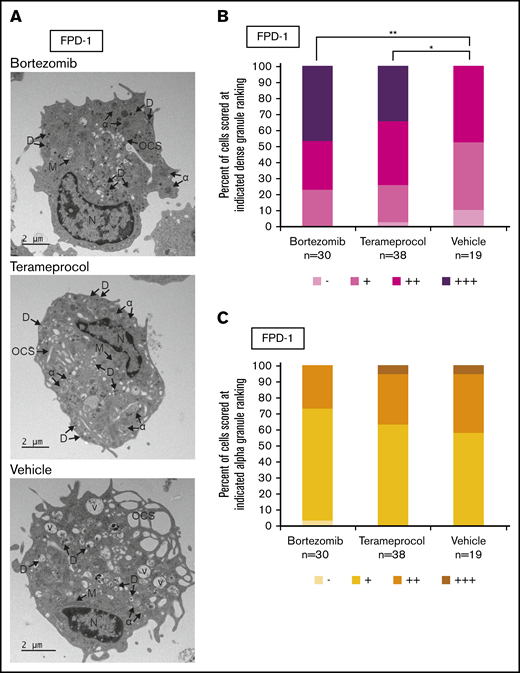
Effect of RUNX1 degradation inhibitor treatment on platelet granule formation during in vitro megakaryocyte differentiation of primary RUNX1-FPD bone marrow MNCs. Primary cells from a patient with RUNX1-FPD were treated and harvested as shown in Figure 5A. (A) Thin-section TEM images of cells treated with bortezomib, terameprocol, or vehicle. Arrows indicate ultrastructural components: dense granules (D), α-granules (α), open canalicular system (OCS), nucleus (N), mitochondria (M), and vacuoles (V). (B) Proportion of cells scored at given ranking for presence of dense granules. (C) Proportion of cells scored at given ranking for presence of α-granules. Rankings were designated as the following number of granules per cell: −, 0; +, 1 to 10; ++, 10 to 20; +++, >20. Unidentifiable granules were not included in the ranking. *P < .025; **P < .01, using a Bonferroni-adjusted χ2 test of independence.
Effect of RUNX1 degradation inhibitor treatment on platelet granule formation during in vitro megakaryocyte differentiation of primary RUNX1-FPD bone marrow MNCs. Primary cells from a patient with RUNX1-FPD were treated and harvested as shown in Figure 5A. (A) Thin-section TEM images of cells treated with bortezomib, terameprocol, or vehicle. Arrows indicate ultrastructural components: dense granules (D), α-granules (α), open canalicular system (OCS), nucleus (N), mitochondria (M), and vacuoles (V). (B) Proportion of cells scored at given ranking for presence of dense granules. (C) Proportion of cells scored at given ranking for presence of α-granules. Rankings were designated as the following number of granules per cell: −, 0; +, 1 to 10; ++, 10 to 20; +++, >20. Unidentifiable granules were not included in the ranking. *P < .025; **P < .01, using a Bonferroni-adjusted χ2 test of independence.
Primary cells from sporadic cases of AML with acquired RUNX1 mutations exhibit reduced RUNX1 expression
Last, we measured RUNX1 expression levels in MNCs from sporadic cases of AML. Eight had RUNX1 mutations and 12 did not. (We excluded germline origin in 7 of the 8 patients with AML with RUNX1 mutations by analyzing bone marrow mesenchymal cells, but 1 patient was unexpectedly found to have a RUNX1 mutation in non-blood cells, consistent with RUNX1-FPD.) Although a range of RUNX1 expression was evident across both groups, samples from patients with RUNX1 mutations exhibited a mean of 47% of total RUNX1 transcript levels compared with the mean of those lacking RUNX1 mutations (Figure 7A). We then treated cells from 2 patients with acquired RUNX1 mutations with bortezomib for 24 hours, followed by culture in drug-free medium for an additional 48 hours. In multiple replicates of each sample, RUNX1 expression was elevated (sporadic AML-1: 2.02- ± 0.54-fold, P = .082; sporadic AML-2: 1.56- ± 0.33-fold, P = .098), although we emphasize that statistical significance was not achieved (Figure 7B; supplemental Table 3).
![Basal RUNX1 expression and response to bortezomib in samples from sporadic cases of AML, with or without RUNX1 mutations. (A) Endogenous RUNX1 expression detected in MNCs from sporadic cases of AML, with or without RUNX1 mutations (mutant, n = 8; wild-type [WT], n = 12). (B) Relative fold change of RUNX1 transcript in samples from sporadic cases of AML with RUNX1 mutations after a 48-hour pulse-chase time course with bortezomib (n = 3). Case information is provided in supplemental Table 3. Error bars show standard deviation.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/5/3/10.1182_bloodadvances.2020002709/2/m_advancesadv2020002709f7.png?Expires=1769103732&Signature=4TazW46YbxNocSNYl1MHoRaxK222QMQ4F7cR4EyvB5yVjHE~sju4F5jb18wgJ4gLi4WCxVrGP5HyJqeiPfMG~m1C8V2GPnUkZhnr~3fVqCVo4LZ0CYBbnBekvJOIhBCDHduqOt1YpNNyTl6KKiVLGN9ZjgWhe3L28xShQia0Wd6ghUJ6mZdDWydTnitQWb-REuXPratzy2zkmmVKTJ7BkI6~i3zNRw-~lm2TALOU2Nxeuw9h-7ZMAwBsNd3nR~EfDtWUrJrstW8PKW9eq6mtYWHolJuIxDyw2nEEppZSLMX6ZRL2-VZy~k3fdh5OUt-W8cIWoarhQO9uZlBHUJYApw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Basal RUNX1 expression and response to bortezomib in samples from sporadic cases of AML, with or without RUNX1 mutations. (A) Endogenous RUNX1 expression detected in MNCs from sporadic cases of AML, with or without RUNX1 mutations (mutant, n = 8; wild-type [WT], n = 12). (B) Relative fold change of RUNX1 transcript in samples from sporadic cases of AML with RUNX1 mutations after a 48-hour pulse-chase time course with bortezomib (n = 3). Case information is provided in supplemental Table 3. Error bars show standard deviation.
Basal RUNX1 expression and response to bortezomib in samples from sporadic cases of AML, with or without RUNX1 mutations. (A) Endogenous RUNX1 expression detected in MNCs from sporadic cases of AML, with or without RUNX1 mutations (mutant, n = 8; wild-type [WT], n = 12). (B) Relative fold change of RUNX1 transcript in samples from sporadic cases of AML with RUNX1 mutations after a 48-hour pulse-chase time course with bortezomib (n = 3). Case information is provided in supplemental Table 3. Error bars show standard deviation.
Discussion
We showed that briefly retarding RUNX1 protein degradation with drugs targeting phosphorylation-dependent proteasomal degradation influences megakaryocyte formation by using in vitro and primary cell models of RUNX1-FPD. We tested CDK inhibitors, a neddylation inhibitor, and direct inhibitors of the proteasome. It is unclear why CDK but not neddylation inhibition had an effect, given that they both target APC and SCF-SKP2 complexes; however, the different results may be related to specific properties of the compound (pevonedistat) that we evaluated.
Not all the cell sources evaluated, including iPSCs and primary bone marrow cells, responded similarly to attempts to restore RUNX1 expression. A possible explanation may relate to the influence of additional mutations that arise during leukemogenesis or adaptation to cell culture. In primary cell samples, the proportion of malignant cells also varies from one person to the next. Another possibility (and a potential limitation of this strategy) is that RUNX1 expression from wild-type alleles, regardless of RUNX1 mutational status, may already be functioning near maximum transcriptional capacity, leaving little room for improvement. A distinct limitation of our approach relates to the underlying genetic heterogeneity of the disorder, involving numerous missense, deletion, and chain-terminating RUNX1 mutations. The strategy we propose would be applicable only to haploinsufficient or other reduced-activity mutations and would not be applicable to dominant-negative germline RUNX1 mutations, which comprise a minority of RUNX1-AML cases, because elevated expression of the mutant allele would concomitantly hinder wild-type protein function. Additional therapeutic considerations relate to clinical severity and secondary acquired somatic mutations, as exemplified by recently reported kindreds.37 Yet another explanation for inconsistent drug effects may relate to nonspecific targeting of proteolytic mechanisms involved in multiple pathways. Clinical translation of this strategy would benefit from attempts to selectively restore RUNX1 expression while perturbing abundance of as few other polypeptides as possible. Nevertheless, RUNX1 may be among only a handful of proteins regulating their own expression and may therefore respond to drugs preventing its degradation, both more sensitively and for a longer duration.
Accordingly, we found that elevated levels of RUNX1 transcript and protein, expected in kinetic and experimental modeling of an autoactivation circuit,16 persist beyond transient expression of exogenous RUNX1 or drug treatment, including through subsequent rounds of cell division. How this occurs is not entirely clear, but it may be instructive to examine MyoD transcriptional autoactivation. In several otherwise differentiated cell types, transient expression of exogenous MyoD is sufficient to promote skeletal muscle reprogramming with maintenance of MyoD expression from its endogenous locus.38 MyoD is a master regulator of cell lineage through cooperative interactions with factors required for epigenetic control of differentiation, including RUNX1.39 It seems reasonable to suppose that RUNX1 similarly helps maintain the transcriptional architecture necessary for lineage specification, including its ongoing expression. In fact, other RUNX factors, if not RUNX1 itself, maintain promoter occupancy during mitosis and ensuing cell division.40 Correspondingly, in mice, Runx1 expression is essential for embryonic hematopoiesis, but, once established, is dispensable.41
Recently, an opposite strategy for targeting RUNX1 in AML, involving repression of its expression, has been shown to eradicate RUNX1-mutant leukemic cells.42 It is worth distinguishing between germline RUNX1 mutations and those arising somatically in leukemic cells. In sporadic cases of leukemia, normal clones with 2 functioning copies of RUNX1 understandably may be resistant to reduced RUNX1 expression compared with heterozygously mutant cells with only a single normally functioning copy of RUNX1. In individuals with RUNX1-FPD, however, there is no reservoir of normal cells, so further repression of RUNX1 expression in preleukemic individuals may suppress hematopoiesis to such an extent that it counterproductively promotes selection for secondary mutations required for leukemogenesis.
Loss of the wild-type RUNX1 allele may not be uncommon.10,43 Our strategy will not work in the setting of biallelic RUNX1 loss. Our approach may prove most beneficial as a preventative measure, before acquisition of secondary mutations. The possibility that our strategy could impose selective pressure favoring loss of the wild-type allele should be considered. However, compared with AML associated with sporadic RUNX1 mutations, patients with RUNX1-FPD who have leukemia exhibit enrichment for somatic mutations involving the second RUNX1 allele,11 suggesting that reduced RUNX1 activity may favor mutations affecting the second allele and that restoring activity may prevent occurrence of mutation of the wild-type allele.
Nevertheless, it is worth noting that we observed reduced RUNX1 expression even among sporadic cases of AML lacking RUNX1 mutations. In fact, among the 5 lowest RUNX1-expressing sporadic cases of AML, 3 had no identifiable RUNX1 mutation, and, in a fourth case, the variant allele fraction for the RUNX1 mutation was only 7% (Figure 7A). It is possible that restoring RUNX1 expression in cases lacking germline or acquired RUNX1 mutations could prove beneficial.
An unexpected result was that in the iPSC model, bortezomib treatment increased megakaryocyte colony formation in cells with a heterozygous RUNX1 mutation but reduced megakaryocyte colony formation in isogenic controls in which the germline mutation was corrected via editing. Reduced colony formation may be due to cell death, redirection of progenitors to an alternate cell fate, or proliferation at the expense of differentiation. If the latter, then, for sporadic cases of AML, bortezomib treatment could simultaneously promote differentiation of RUNX1-mutant cells and confer a proliferative advantage to normal cells lacking a RUNX1 mutation. Further experimentation is needed to distinguish among these possibilities.
Finally, loss-of-function germline mutations in other autoactivating transcription factors degraded through the ubiquitin-proteasome pathway, such as GATA2, similarly predispose to leukemia,44 potentially extending this strategy to related disorders.
Data and renewable materials will be shared upon e-mail request to the corresponding author (horwitz@uw.edu).
Acknowledgments
This work was supported by Alex’s Lemonade Stand Foundation and RUNX1 Research Program grants (M.S.H. and E.P.P.).
Part of this manuscript appears in the dissertation of M.C.K., in partial fulfillment of requirements for a Doctor of Philosophy degree from the University of Washington.
Authorship
Contribution: M.C.K., M.R.H., D.L.F., P.G., E.P.P., S.B.K., P.S.B., and M.S.H. planned the experiments; M.C.K., M.R.H., D.J.A., J.J., A.G.K., J.D., S.C., M.D., S.B., and J.A.M. executed most of the experiments; M.C.K., M.R.H., and M.S.H. wrote the initial draft of the manuscript; and all authors edited the manuscript and approved the final version.
Conflict-of-interest disclosure: P.S.B. has benefitted from institutional research funding from AbbVie, Bristol-Myers Squibb, Pfizer, SecuraBio, Glycomimetics, Invivoscribe, JW Pharmaceutical, Novartis, and Trovagene and is an advisory board member for CVS Caremark. The remaining authors declare no competing financial interests.
The current affiliation for M.R.H. is Clinical Research Division, Fred Hutchinson Cancer Research Center, Seattle, WA.
The current affiliation for J.J. is Department of Cell Biology, Johns Hopkins School of Medicine, Baltimore, MD.
The current affiliation for P.S.B. is Division of Hematology and Oncology, Department of Medicine, and Chao Family Comprehensive Cancer Center, School of Medicine, University of California, Irvine, CA.
Correspondence: Marshall S. Horwitz, Institute for Stem Cell and Regenerative Medicine, Department of Pathology, University of Washington School of Medicine, 850 Republican St, Room N435, Seattle, WA 98109-4725; e-mail: horwitz@uw.edu.

