

Key Points
Gene expression and mutational analysis confirm the differences among the 3 peripheral TCL subclasses: AITL, PTCL-NOS, and PTCL-TFH.
The expression of a gene set, including B-cell genes, is an IPI-independent prognostic factor for AITL cases.

Abstract
Peripheral T-cell lymphoma (PTCL) is a clinically aggressive disease, with a poor response to therapy and a low overall survival rate of approximately 30% after 5 years. We have analyzed a series of 105 cases with a diagnosis of PTCL using a customized NanoString platform (NanoString Technologies, Seattle, WA) that includes 208 genes associated with T-cell differentiation, oncogenes and tumor suppressor genes, deregulated pathways, and stromal cell subpopulations. A comparative analysis of the various histological types of PTCL (angioimmunoblastic T-cell lymphoma [AITL]; PTCL with T follicular helper [TFH] phenotype; PTCL not otherwise specified [NOS]) showed that specific sets of genes were associated with each of the diagnoses. These included TFH markers, cytotoxic markers, and genes whose expression was a surrogate for specific cellular subpopulations, including follicular dendritic cells, mast cells, and genes belonging to precise survival (NF-κB) and other pathways. Furthermore, the mutational profile was analyzed using a custom panel that targeted 62 genes in 76 cases distributed in AITL, PTCL-TFH, and PTCL-NOS. The main differences among the 3 nodal PTCL classes involved the RHOAG17V mutations (P < .0001), which were approximately twice as frequent in AITL (34.09%) as in PTCL-TFH (16.66%) cases but were not detected in PTCL-NOS. A multivariate analysis identified gene sets that allowed the series of cases to be stratified into different risk groups. This study supports and validates the current division of PTCL into these 3 categories, identifies sets of markers that can be used for a more precise diagnosis, and recognizes the expression of B-cell genes as an IPI-independent prognostic factor for AITL.
Introduction
Peripheral T-cell lymphoma (PTCL) is a clinically aggressive disease, with a poor response to therapy and an approximate 5-year overall survival (OS) of only 30%.1 Nodal PTCL is not considered a single disease but rather a group of distinct disorders, including angioimmunoblastic T-cell lymphoma (AITL), PTCL not otherwise specified (NOS), and PTCL with T follicular helper (TFH) phenotype. Gene expression profiling studies have identified proliferation as a prognostic marker2 and delineated biological and prognostic subgroups within PTCL-NOS (PTCL-GATA3 and PTCL-TBX21).3
Histological study of PTCL has revealed significant stromal participation, with the presence of follicular dendritic cells (FDCs), eosinophils, plasma cells, and high endothelial venules, in which B cells and Epstein-Barr virus play a role in PTCL pathogenesis. Molecular studies have confirmed that monoclonal mutated T cells comprise a minor part of the tumor mass in most cases and, definitively, in AITL cases.4,5
Differential diagnosis of these 3 main subtypes of PTCL involves a combination of clinical, histological, and immunohistochemical markers but frequently requires more specific markers and has relatively low reproducibility.6 Several projects have since evaluated whether gene expression analysis could improve diagnostic accuracy by delineating specific T-cell lymphoma classes or by identifying more precise prognostic models.2,3,7-10 Nevertheless, the need for reliable diagnostic and prognostic markers in PTCL/AITL remains partially unmet.
We have analyzed a series of 105 cases with a diagnosis of PTCL using a customized NanoString platform that includes 208 genes related to T-cell differentiation, oncogenes and tumor suppressor genes, deregulated pathways, and stromal cell subpopulations. Specifically, the platform includes genes expressed by the cell types present in PTCL and AITL specimens, together with normal T-cell populations, for the purposes of facilitating the deconvolution of the T-cell lymphoma microenvironment, and developing an integrated view of the cell composition of PTCL tumoral specimens. We have also analyzed the mutational profile using a custom panel targeting 62 genes in 76 cases distributed in AITL, PTCL-TFH, and PTCL-NOS.
The data generated in this manner were used to identify the signature characteristics of the different types of PTCL (AITL, PTCL-NOS, and PTCL-TFH) and identify prognostic markers.
Results were validated using a separate group of 54 patients with PTCL.
Methods
Study design and patients
One hundred five patients with PTCL were included in the study, which was developed in collaboration with several Spanish hospitals whose work complied with the clinical protocols of the Grupo Español de Linfomas y Trasplante Autólogo de Médula Ósea (GELTAMO) Group, under the supervision of the Fundación Jiménez Díaz (FJD) local ethics committee, and in accordance with the Declaration of Helsinki.
Formalin-fixed, paraffin-embedded (FFPE) tissue sections from diagnostic biopsies were collected at the time of diagnosis before CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone), CHOP-like, or CHOEP (cyclophosphamide, doxorubicin, etoposide, vincristine, and prednisone, for treating patients younger than 60 years) regimens. We analyzed 66 patients with AITL, 21 patients with PTCL-NOS, and 18 patients with PTCL-TFH. The main clinical characteristics of the patients are shown in supplemental Table 1. Diagnoses were made using whole sections. All cases were reviewed, and a consensus diagnosis was made by 2 expert hematopathologists (S.M.R.-P., M.A.P.) following the 2017 World Health Organization Guidelines. All cases were nodal PTCL.
The validation series of 54 patients with PTCL comprised 27 AITL, 18 PTCL-NOS, and 9 PTCL-TFH cases. Clinical and molecular features, follow-up time, and current status are summarized in supplemental Table 1. Samples were obtained following the same requirements as those described for the discovery series.
Moreover, the mutational profile was analyzed using a custom panel that targeted 62 genes in 76 cases distributed in 44 patients with AITL, 18 patients with PTCL-TFH, and 14 patients with PTCL-NOS (supplemental Appendix).
nCounter gene-expression assay
Gene-expression profiling was performed with nCounter Technology (NanoString Technologies). Total RNA from 105 FFPE sections from diagnostic samples was isolated using a truXTRAC FFPE total NA kit (Covaris Inc., Woburn, MA) following the manufacturer's instructions (supplemental Appendix).
Targeted next-generation sequencing
Genomic DNA was extracted from FFPE using a truXTRAC FFPE DNA Kit (Covaris Inc.) following the manufacturer's instructions.
The SureSelect target enrichment custom panel (3110861) was designed using the SureDesign (Agilent Technologies, Santa Clara, CA) web-based tool. The 62 genes included have previously been described as mutated in PTCL (supplemental Table 3). The designs covered coding exons of the selected genes. The targeted regions (according to Human Assembly GRCh37/hg19) were captured using a SureSelect Target Enrichment Low Input System (Agilent Technologies), as described in the manufacturer’s instructions. Captured libraries were diluted to 1.3 pM for Illumina clustering, and paired-end sequencing was performed on a NextSeq500 sequencer (Illumina Inc., San Diego, CA).
Somatic single nucleotide variants (SNVs) were identified in each sample following the best practices GATK workflow for calling somatic mutations, which involves aligning reads with the Burrows-Wheeler Aligner (version 0.7.17) and SNV detection and filtering using Mutect211 (supplemental Appendix).
In general, we excluded variants with a variant alelle frequency (VAF) of <5% because we expect these variants to be artifacts due to the sensitivity of the NGS technique. In addition, we have excluded variants with a VAF of >40% because earlier published findings led us to expect them to be germline mutations. Previously identified specific mutations, such as RHOAG17V and IDH2 (R172), were accepted with a threshold of 1% VAF.
Tissue microarrays and immunostaining
Immunohistochemistry with specific antibodies was performed for clinical diagnosis (antibodies listed in supplemental Table 4). Specifically, we used PD1, CXCL13, and ICOS as TFH markers. All PTCL-TFH cases expressed at least 2 TFH cell markers.
Tissue microarrays were designed as previously described, using two 0.6-mm tissue cores per case taken from archival FFPE tumor blocks (supplemental Appendix).
Survival analysis
First, we divided the data of the 159 patients into a discovery (training) set (n = 105) and a validation set (n = 54) at random in a 2:1 ratio. Survival data were available for 105 patients with PTCL from the discovery series and for 54 patients with PTCL from the validation series (supplemental Appendix). Survival data were available for 75 of the 76 patients sequenced.
Statistical analysis
All statistical analyses were performed using R 3.5.2 (R Foundation for Statistical Computing, Vienna, Austria), IBM SPSS Statistics for Windows version 21 (IBM Corp., Armonk, NY), and GraphPad Prism 8.4.0 (San Diego, CA) (supplemental Appendix).
Results
General features of the PTCL series
The clinical characteristics of the discovery series are summarized in supplemental Table 1. The series includes 105 patients diagnosed with PTCL, subdivided into 66 patients with AITL, 21 patients with PTCL-NOS, and 18 patients with PTCL with the TFH phenotype, all of whom were treated with curative intention following the guidelines of the GELTAMO group.12
Most cases were diagnosed as AITL (n = 66), the others being roughly equally split between PTCL-NOS (n = 21) and PTCL-TFH (n = 18). The median age for all patients was 67 years (range: 26-88 years) (supplemental Table 1). Mostly, the initial therapeutic approaches were CHOP, CHOP-like, and CHOEP regimens for the most common PTCL subtypes.
Survival probability for the whole series was 47.92% at 32 months, with a median follow-up of 32 months (range: 1-177 months).
Immunohistochemical analyses were performed in whole sections of all 105 samples. The tissue microarrays and immunohistochemistry were performed as a part of the histopathological review. We did not find any significant differences between histological diagnosis and either OS or time to progression (TTP) (data not shown).
Mutational analysis
Of the 76 sequenced cases, 44 were classified as AITL, 18 as PTCL-TFH, and 14 as PTCL-NOS. A total of 46 368,287 mapped reads were generated, and 81.8% of all the bases were aligned to the complete human genome (UCSC hg19, GRCh37, February 2009). The mean coverage was 993X (range: 31-1379) for tumor samples. The mean coverage depth for each tumor sample was calculated by HsMetrics (Picard Tools, Broad Institute, Cambridge, MA). This tool was used to analyze the results of the target-capture sequencing experiments. Twenty-seven of the 76 sequenced tumor cases from the entire series (35.53%) had values greater than the mean. The mean coverage in the coding region was the same as the mean coverage, and the same number of samples was greater than the mean.
Figure 1 and supplemental Table 5 summarize the details of the SNVs. After filtering, we observed a complex genomic landscape, defined by the presence of an average of 4 SNVs per sample (range: 1-20). We identified SNVs in 52 of the 62 genes (83.87%) analyzed. As described previously,3,13-15 some cases have more than 1 SNV per gene, this phenomenon being most frequently observed in TET2 gene, although we also observed multiple mutations in VAV1, RHOA, NOTCH1, MTOR, JAK1, ZEB1, ATM, DNMT3A, and ARID1B. Only 2 AITL samples had no SNVs remaining after filtering.
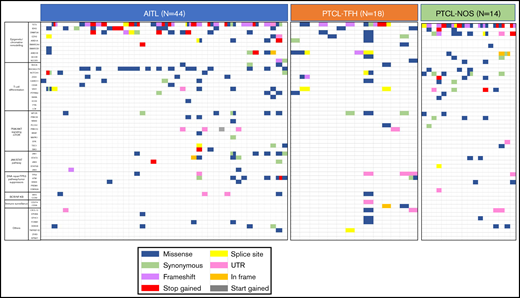
Distribution of mutations in the 62 analyzed genes in the whole series of 76 tumor samples. Analysis of genomic alterations by target sequencing panel for primary lymphomas in patients with AITL, PTCL-TFH, and PTCL-NOS. A total of 46 368,287 mapped reads were generated, and 81.8% of the total bases were aligned to the complete human genome (UCSC hg19, GRCh37, February 2009). The mean coverage was 993X (range: 31-1379) for tumor samples. Rows correspond to sequenced genes; columns represent individual PTCL patients. Color coding: blue, missense; green, synonymous; purple, frameshift; red, stop gained; yellow, splice sit; pink, UTR; orange, in frame; gray, start gained.
Distribution of mutations in the 62 analyzed genes in the whole series of 76 tumor samples. Analysis of genomic alterations by target sequencing panel for primary lymphomas in patients with AITL, PTCL-TFH, and PTCL-NOS. A total of 46 368,287 mapped reads were generated, and 81.8% of the total bases were aligned to the complete human genome (UCSC hg19, GRCh37, February 2009). The mean coverage was 993X (range: 31-1379) for tumor samples. Rows correspond to sequenced genes; columns represent individual PTCL patients. Color coding: blue, missense; green, synonymous; purple, frameshift; red, stop gained; yellow, splice sit; pink, UTR; orange, in frame; gray, start gained.
The most recurrently mutated genes in the overall series were TET2 (61.84%), DNMT3A (30.26%), RHOAG17V (23.68%), IDH2 (21.05%), ARID1B (17.1%), VAV1 (15.78%), MTOR (14.47%), NOTCH1 (13.15%), and ZEB1 (10.52%), although the order of their frequencies differed between tumor groups. Mutations in the DNA methylation regulator TET2 gene were the most common among the 3 nodal T-cell lymphoma types.
The main difference among the 3 nodal PTCL classes was the higher frequency of RHOAG17V mutations (P < .0001). They were present approximately twice as frequently in cases of AITL (34.09%) as in PTCL-TFH (16.66%) but could not be detected in PTCL-NOS. Unlike previous results,15 the DNMT3AR882 variant was identified in the 3 subgroups although at low frequencies in AITL (3/44), PTCL-TFH (1/18), and PTCL-NOS (1/14).
We ascribed the mutated genes to 10 pathways (Figure 1). The epigenetic/chromatin remodeling pathway was mutated in 77.22% of all cases, and the T-cell differentiation and PI3K/AKT signaling mTOR pathways were mutated in 60.52% and 31.57% of cases, respectively. With respect to the lymphoma-type subdivision, epigenetic/chromatin remodeling was the most frequently affected pathway in PTCL-NOS (85.71%), AITL (77.27%), and PTCL-TFH (72.22%). However, the T-cell differentiation pathway was more frequently altered in AITL and PTCL-TFH (68.18% and 66.66%, respectively) than in PTCL-NOS subtype (28.57%). In the PTCL-NOS subtype, genetic alterations were also present in the immune surveillance pathway (21.42%) but were absent from the other subtypes. Additional data are shown in Figure 2 and supplemental Table 5.
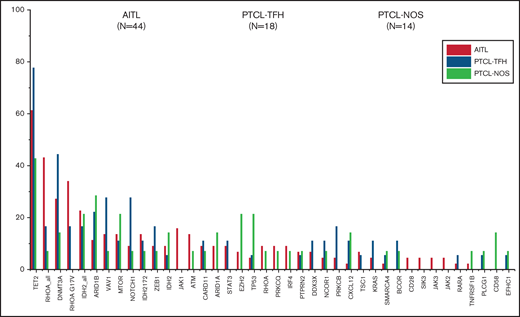
Most frequently mutated genes (%) in the whole cohort. They are subdivided as AITL (orange), PTCL-TFH (green), and PTCL-NOS (purple).
Most frequently mutated genes (%) in the whole cohort. They are subdivided as AITL (orange), PTCL-TFH (green), and PTCL-NOS (purple).
To study the prognostic impact of gene mutations on the PTCL series, we analyzed the data by Cox proportional hazards regression. We found no significant differences within the overall series or when specific tumor types were analyzed separately.
Unsupervised gene expression analysis
We studied the gene expression profile using the NanoString Technologies platform. The unsupervised analysis revealed clusters of coregulated genes, which corresponded to the tumor stroma or the neoplastic cells, as shown in Figure 3.
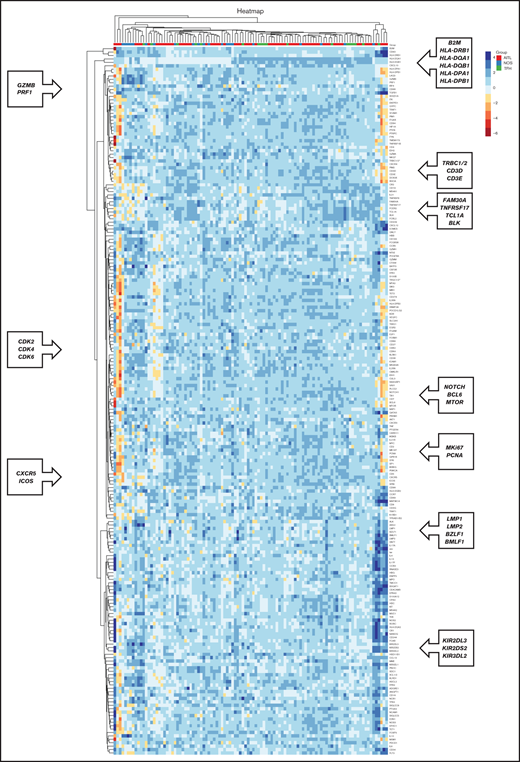
Co-regulated genes, as classified by unsupervised hierarchical clustering analysis, based on the genes included in the NanoString platform. Heatmap depicting gene clusters of coregulated genes that allow the recognition of signatures expressed by tumoral and stromal cells. Each column represents sample categories (AITL in red, PTCL-NOS in blue, and PTCL-TFH in green). Each row represents the expression level of a gene, depicted according to the color scale shown.
Co-regulated genes, as classified by unsupervised hierarchical clustering analysis, based on the genes included in the NanoString platform. Heatmap depicting gene clusters of coregulated genes that allow the recognition of signatures expressed by tumoral and stromal cells. Each column represents sample categories (AITL in red, PTCL-NOS in blue, and PTCL-TFH in green). Each row represents the expression level of a gene, depicted according to the color scale shown.
Some of the clusters of coregulated genes corresponded to the following:
Major histocompatibility complex–associated genes (B2M, HLA-DRB1, HLA-DPA1, HLA-DPB1, HLA-DQA1, and HLA-DQB1)
Cytotoxic T cells (PRF1 and GZMB)
T cells (CD3D, CD3E, and TRBC1/2)
B cells (FAM30A, TNFRSF17, TCL1A, and BLK)
Proliferation genes (CDK2, CDK4, CDK6, MKi67, and PCNA)
TFH cells (CXCR5 and ICOS)
Epstein-Barr virus (BMLF1, BZLF1, LMP1, and LMP2)
Natural killer cells (KIR2DS2, KIR2DL2, and KIR2DL3)
Other coregulated genes of interest were those defining FDCs, macrophages, and the NF-κB pathway (classic and alternative), among others (Figure 3).
Unsupervised analysis did not reveal any well-defined clusters related to the tumor subtypes of patients with PTCL. However, most of the AITL cases were members of 2 main clusters, with cases of PTCL-NOS and PTCL-TFH interspersed among the other groups (Figure 3).
Supervised gene expression analysis and genes differentially expressed in PTCL entities
We compared the gene signatures of the different types of PTCL (AITL, PTCL-NOS, and PTCL-TFH).
Comparison between AITL and PTCL-NOS identified 21 differentially expressed genes (FDR < 0.05 and log2 FC > 2). Differential expression analyses showed 8 significantly upregulated and 4 significantly downregulated genes in AITL (FDR < 0.01 and log2 FC > 2; Figure 4A). Supplemental Table 6 shows the top 20 genes. Genes defining AITL in this study are those that recognize the TFH phenotype (CXCL13, CD10 [MME]), FDCs (CR2), mast cells (HDC), alternative NF-κB (MAP3K14 [NIK]), and others, whereas PTCL-NOS shows a higher level of expression of cytotoxic genes (NKG7, GZMB, PRF1) and important immunoregulators, such as IL-10.

Volcano plots, Venn diagram and trend plot. The upregulated/unchanged/downregulated genes for (A) AITL vs PTCL-NOS, (B) AITL vs PTCL-TFH, and (C) PTCL-TFH vs PTCL-NOS represented by volcano plots. (D) Venn diagram of the 3 entities. (E) Trend plot of cell type scores.
Volcano plots, Venn diagram and trend plot. The upregulated/unchanged/downregulated genes for (A) AITL vs PTCL-NOS, (B) AITL vs PTCL-TFH, and (C) PTCL-TFH vs PTCL-NOS represented by volcano plots. (D) Venn diagram of the 3 entities. (E) Trend plot of cell type scores.
Eleven genes were differentially expressed (FDR < 0.05 and log2 FC > 2) between AITL and PTCL-TFH subtypes when we performed a differential gene expression analysis using solver Advanced Data Analysis (NanoString Technologies). We found 8 significantly upregulated genes in the AITL subtype (FDR < 0.01 and log2 FC > 2; Figure 4B). Supplemental Table 7 indicates the top 20 genes. The analysis showed that AITL cases were characterized by a higher level of expression of genes defining specific T-cell subpopulations such as CD8A, EOMES, CD63, CD34, TGFB1, and CCR7.
When comparing PTCL-TFH and PTCL-NOS, we found 4 genes with a higher level of expression in PTCL-NOS (GZMB, NKG7, BATF3, and IL-10 with FDR < 0.05 and log2 FC > 2), whereas the genes upregulated in the PTCL-TFH group identified the TFH phenotype (PD1, CXCL13, and ICOS) and GATA3 (Figure 4C). Supplemental Table 8 shows the top 20 genes.
Differential expression analysis identified significant differences among the 3 main tumor subtypes. The Venn diagram shows the number of genes that overlapped and differed among these 3 entities (Figure 4D). All 3 conditions seem to have a relatively large group of genes (AITL: 115 genes; PTCL-NOS: 148 genes; PTCL-TFH: 130 genes) that was specific to each entity.
Genes found to be differentially expressed among the PTCL lymphoma types identified signatures for cell types that characterize each of these tumors. For example, AITL cases had enhanced expression of FDCs, DCs, mast cells, and major histocompatibility complex–associated gene sets, whereas elevated expression of a neutrophil-associated gene set was found in the PTCL-NOS group (Figure 4E).
Box plot comparisons of cell types in the AITL, PTCL-NOS, and PTCL-TFH sample sets showed significantly higher B-cell, T-cell, and vascular scores in the AITL cases (Figure 5A-C). AITL cases and the other PTCL subtypes also showed notably variable expression of this B-cell gene set (FAM30A, TNFRSF17, TCL1A, and BLK). The level of expression of a proliferation-associated gene set was higher in the PTCL-TFH subtype (Figure 5D). Finally, a TFH gene set was enriched in AITL and PTCL-TFH (Figure 5E).
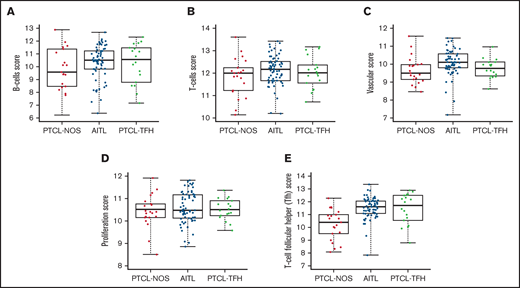
Box plots of cell type scores of AITL, PTCL-NOS, and PTCL-TFH. Scores for (A) B cells, (B) T cells, and (C) vascular, (D) proliferation, and (E) T-cell follicular helper (TFH) cells.
Box plots of cell type scores of AITL, PTCL-NOS, and PTCL-TFH. Scores for (A) B cells, (B) T cells, and (C) vascular, (D) proliferation, and (E) T-cell follicular helper (TFH) cells.
Prognostic significance of the gene expression signatures
To study the prognostic impact of the genes and clinical variables on the PTCL series, we analyzed the data by Cox proportional hazards regression. More informative models emerged when specific tumor types were analyzed separately (Model 2, Model 3, and Model 4 for each tumor subtype) than when analyzed together (Model 1; P = .055) (supplemental Table 9).
In AITL cases, analyzing the univariate Cox proportional hazards ratios identified International Prognostic Index (IPI) and age and 5 genes whose expression was significantly associated with differences in OS and TTP probability (supplemental Tables 10 and 11, respectively). These correspond to genes expressed by B cells (BLK, CD19, MS4A1, and SDC1) and IDH1 (P < .05). The Cox multivariate analysis yielded an integrated model including IPI, BLK, CD19, and MS4A1.
In the PTCL-NOS subtype, analyzing the univariate Cox proportional hazards ratios identified IPI and 6 genes whose expression was significantly associated with differences in OS and TTP probability (supplemental Tables 12 and 13, respectively). These were genes involved in proliferation (MIB1, EZF1, and SP1), expressed by monocytes (ARG1), related to the NF-κB pathway (IKBKB), and of importance in T-cell lymphoma pathogenesis (CARD11) (P < .05). The Cox multivariate analysis yielded an integrated model that included MIB1 and EZF1.
In the PTCL-TFH subgroup, the univariate Cox proportional hazards ratio analysis identified IPI and 6 genes whose expression was significantly associated with differences in OS and TTP probability (supplemental Tables 14 and 15, respectively). These were genes expressed by cytotoxic T cells (GZMA, PRF1, KLRB1, and NKG7), monocytes (CD163), and TFH (CXCR5) cells (P < .05). The Cox multivariate analysis yielded an integrated model that included IPI, GZMA, and KLRB1.
In a subsequent analysis, taking the median risk score as the cut-point, 66 patients with AITL in the discovery set were divided into high-risk (greater than the median risk score) and low-risk (less than or equal to the median) groups. The 21 patients with PTCL-NOS and 18 PTCL-TFH cases from the discovery series were analyzed in the same way. Kaplan-Meier (K-M) survival curve analyses were conducted for each patient group, which showed that both the OS and the TTP of the 3 groups of lymphoma types in the high-risk group were significantly lower than for the low-risk group (P < .05; Figure 6A-F). OS probabilities for AITL cases, stratified using the integrated model, were 75% (HR, 0.35; 95% CI, 0.14-0.85) for the low-risk group and 30% (HR, 2.73; 95% CI, 1.37-6.86) for the high-risk group. TTP survival probability was 70% (HR, 0.32; 95% CI, 0.16-0.92) for the low-risk group and 20% (HR, 2.08; 95% CI, 1.42-6.68) for the high-risk group. K-M survival curves showed 75% and 30% OS for the low-risk and high-risk groups, respectively. The TTP survival probabilities were 70% for the low-risk group and 20% for the high-risk group. These percentages were selected using the differences between the log-rank test typically reported for the Cox regression model, which is estimated by maximizing likelihood or partial likelihood.
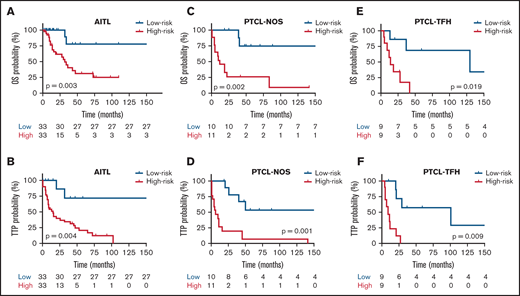
Kaplan-Meier survival analysis in relation to each prognostic model for OS and TTP. Red and blues lines indicate the high- and low-risk groups, respectively. The vertical bar represents the OS and the TTP probability (%), whereas the horizontal bar represents the follow-up time in months. Patients at risk at the corresponding time point are shown. Statistical significance (P) was that associated with the log-rank test. (A-B) AITL, (C-D) PTCL-NOS, and (E-F) PTCL-TFH.
Kaplan-Meier survival analysis in relation to each prognostic model for OS and TTP. Red and blues lines indicate the high- and low-risk groups, respectively. The vertical bar represents the OS and the TTP probability (%), whereas the horizontal bar represents the follow-up time in months. Patients at risk at the corresponding time point are shown. Statistical significance (P) was that associated with the log-rank test. (A-B) AITL, (C-D) PTCL-NOS, and (E-F) PTCL-TFH.
Separate K-M survival curve analyses were performed for prognostic models, including B-cell genes and IPI in the AITL group. Taking the median risk score as the cut-point, 21 patients with PTCL-NOS in the discovery series were divided into high-risk (11 samples, greater than the median risk score) and low-risk (10 samples, less than or equal to the median) groups. Expression of these genes and IPI identified significantly shorter OS and TTP in the high-risk group than in the low-risk group (P < .01) (Figure 6A-B). Analyses were also performed for models of proliferation genes in the PTCL-NOS subtype. Taking the median risk score as the cut-point, 18 patients with PTCL-TFH in the discovery series were divided into high-risk (9 cases) and low-risk (9 samples) groups. Expression of these genes identified significantly shorter OS and TTP in the high-risk than in the low-risk group (P < .01) (Figure 6C-D. Finally, IPI and cytotoxic T-cell genes in PTCL-TFH cases were analyzed. Expression of these genes identified significantly shorter OS and TTP in the high-risk than in the low-risk group (P < .01) (Figure 6E-F).
We also specifically analyzed the potential prognostic value of GATA3 and TBX21 expression in the cases in the PTCL-NOS category. No significant association was found with differences in survival probability (log-rank test, P = .082 for OS and P = .087 for TTP). However, the number of patients with this diagnosis was relatively small (21).
Mutational data were not found to be associated with differences in either OS or TTP.
Validation of the prognostic models in an independent PTCL series
To validate the clinical outcome prediction models, a validation series including FFPE samples from 54 patients with PTCL (27 AITL, 18 PTCL-NOS, and 9 PTC-TFH cases) was analyzed.
The characteristics of the validation set are summarized in supplemental Table 1. All patients were treated with CHOP, CHOP-like, or CHOEP regimens. The median age of the patients was 64.5 years (range: 31-88 years). The proportional survival of the whole series was 44.46% at 83 months, with a median follow-up of 83 months (range: 1-237 months).
The prognostic models were then applied to the validation series. To assess whether each model can be validated, we first scored 54 patients and divided them into high- and low-risk groups according to the scores, with the median risk score as the cut-point. In the AITL group, analysis of OS identified the following HRs: low risk, 0.29 (95% CI, 0.12-0.71) and high risk, 3.15 (95% CI, 1.39-5.05). The TTP group had the following HR values: low risk, 0.27 (95% CI, 0.11-0.51) and high risk, 3.17 (95% CI, 1.25-6.19). In the AITL entity, the difference in survival between the high- and low-risk groups was analyzed with a 2-sided log-rank test, which gave roughly similar significant results for OS and TTP (P < .0001) (Figure 7A-B, respectively) from a K-M survival analysis. However, we could not validate the specific prognostic models in the PTCL-NOS and PTCL-TFH validation series (data not shown). Taken together, we found robust evidence that validated the prognostic model in an independent group of patients with AITL.
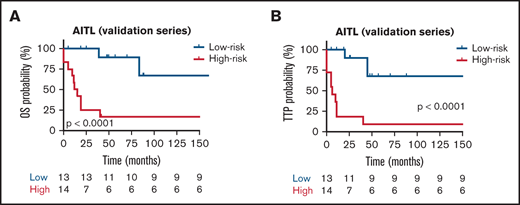
Kaplan-Meier survival analysis for OS and TTP in the validation series. Red and blues lines indicate the high- and low-risk groups, respectively. The vertical bar represents the TTP probability (%), whereas the horizontal bar represents the follow-up time in months. Patients at risk at the corresponding time point are shown. Statistical significance (P) was that associated with the log-rank test. (A-B) AITL.
Kaplan-Meier survival analysis for OS and TTP in the validation series. Red and blues lines indicate the high- and low-risk groups, respectively. The vertical bar represents the TTP probability (%), whereas the horizontal bar represents the follow-up time in months. Patients at risk at the corresponding time point are shown. Statistical significance (P) was that associated with the log-rank test. (A-B) AITL.
Discussion
PTCL subclassification and prognostication still lack biologically sound, reproducible markers and instead rely mainly on clinical and analytical characteristics.16,17 The exceptions are Ki6718 and the subclassification into GATA3/TBX21 PTCL-NOS.9,19 This study identifies a set of genes that may be useful for stratifying patients with PTCL and AITL according to their specific risks, thereby aiding the clinical management of these patients. The study was performed in paraffin-embedded samples and thus could be routinely applied.
Unsupervised clustering (Figure 3) shows the existence of coregulated gene sets that define specific components of the stroma and phenotypic features of the neoplastic cells, enabling the deconvolution of the complex tumor samples in terms that allow individual discrete cell subpopulations or subtle changes in the neoplastic phenotype to be identified.
Our results provide a degree of validation of the existence of consistent differences among the 3 major lymphoma subtypes, thus confirming the subclassification proposed by the World Health Organization,20 corroborated through molecular analysis.3 They also identify specific gene sets that may be used for differential diagnosis and reflect differences in the neoplastic cells and/or the tumor microenvironment. Mutational data also confirm differences among the 3 subclasses. There was a higher frequency of RHOAG17V mutations (P < .0001) in AITL cases, as was also reported recently by Yoon et al.21
It was also possible to identify a distinct subgroup of AITL cases, enriched in B cells, with a more favorable prognosis (75% at 60 months). A subset of AITL cases with a more indolent clinical course had been demonstrated in previous clinical and molecular studies.9,22,23 A favorable role for the presence of B cells in T-cell lymphoma prognosis was initially proposed by Iqbal and coworkers9 after their use of Affymetrix HG-U133 chips in a series of fresh AITL specimens. Our study confirmed these observations using a tool that can be applied in routine FFPE specimens based on the recognition of a B-cell signature that includes TCL1A, PNOC, CD19, CD20, and SPIB genes. An attempt to reproduce this finding, which was present in 49 of 105 (46.7%) cases, using immunohistochemistry for CD20 in paraffin-embedded sections was not successful.
Of the other PTCL subtypes, PTCL-NOS analysis showed that the expression of a set of proliferation-related genes was associated with shorter OS and TTP, as revealed by univariate and multivariate analyses. This was not confirmed in the validation group because the difference was too small to be statistically significant for the low number of cases, but the result was nevertheless consistent with those of other researchers.18,24,25
PTCL-TFH group survival analysis showed that in contrast to the findings in AITL, the presence of a B-cell gene signature was not associated with changes in survival probability. In contrast, the expression of cytotoxic/natural killer markers (GZMA, PRF1, and KLRB1) was associated with shorter OS and TTP although these findings were not confirmed in the validation series.
These series of cases show that the expression of GATA3/TBX21 is not associated with changes in survival probability, in contrast with the findings of Iqbal and coworkers.9,19,21 Our findings are consistent with those of Drieux et al.,7 although both studies featured relatively small numbers of cases with a diagnosis of PTCL-NOS.
The mutational analysis of the gene panel containing 62 selected genes allowed us to confirm already known variants in PTCL. The mutational profile of AITL, PTCL-NOS, and PTCL-TFH identified variants in TET2, DNMT3A, and RHOA, in line with results of previous studies.15,21 However, unlike some previous publications,15,21,26-28 we observed IDH2 variants in all PTCL groups, albeit with different frequencies, and the DNMT3AR882 variant was identified in the 3 subgroups but at low frequencies in patients with AITL (3/44), PTCL-TFH (1/18), and PTCL-NOS (1/14). The main difference among the 3 nodal PTCL classes was the higher frequency of RHOAG17V mutations. They were present approximately twice as frequently in AITL cases (34.09%) as in PTCL-TFH (16.66%) but could not be detected in cases of PTCL-NOS.21,29,30
In summary, this study identifies gene clusters expressed by the different microenvironmental and neoplastic components of PTCL-NOS, PTCL-TFH, and AITL, including specific gene sets, which can be used with routinely processed paraffin-embedded samples to provide a more specific diagnosis and prognosis.
Acknowledgments
The authors thank Kevin Troulé for his bioinformatic support at the Bioinformatics Unit of the Spanish National Cancer Research Center (CNIO) in Madrid and Ignacio Mahillo for his statistical support at the Biostatistics and Epidemiology Unit of Instituto de Investigación Sanitaria-Fundación Jiménez Díaz University Hospital in Madrid. The authors especially thank all the Biobanks and Pathology Departments for their exceptional work collecting and organizing samples. Additionally, the authors are indebted to the patients who contributed to this study.
This work was supported by grants from Instituto de Salud Carlos III, from Ministerio de Economía, Industria y Competitividad, Asociación Española Contra el Cáncer (AECC), Comunidad Autónoma de Madrid and Centre for Biomedical Network Research on Cancer (CIBERONC): SAF2013-47416-R, CIBERONC-ISCIII (CB16/12/00291), ISCIII-MINECO-AES-FEDER (Plan Estatal I + D + I 2013-2016), AECC PROYE18054PIRI, CAM B2017/BMD-3778, PIC97/2017_FJD, PIE15/0081, PIE16/01294, and PI19/00715. R.A.-A. is the recipient of PFIS predoctoral fellowship. L.T.-R. is funded by Marie Skłodowska-Curie Individual Fellowship (No 882597). M.R. is supported by CIBERONC (CB16/12/00291) P.M. has a Miguel Servet contract funded by the ISCIII (CP16/00116). L.d.l.F. was supported by the ISCIII contract CA18/00017.
Authorship
Contribution: M.R., R.A.-A., R.M., and M.A.P. conceived and designed the study; M.R., R.A.-A., S.M.R.-P., L.C., T.V., R.C., M.S.-B., I.B., C.B., J.F.G., M.M., M.G.-C., P.M.-A., F.C., D.C., and R.M. acquired the data; R.A.-A., J.B., and M.R. conducted experiments; M.R., L.T.-R., R.A.-A., L.d.l.F., and P.M. performed data analysis; M.R., L.T.-R., R.A.-A., S.M.R.-P., R.M.-A., I.F.-M., L.T.-R., M.S.-B., L.K., C.S., A.G., R.M., P.M., and M.A.P. interpreted data; R.A.-A., J.B., L.C., I.B., L.d.l.F., and P.M. provided technical assistance; M.R., L.T.-R., and M.A.P. wrote the manuscript. All authors critically read and approved the final manuscript.
Conflict-of-interest disclosure: M.A.P. declares having received lecture fees and advisory board fees from Millenium/Takeda, Jansen, NanoString, Kyowa Kirin, Gilead, and Celgene. The remaining authors declare no competing financial interests.
Correspondence: Miguel A. Piris, Department of Pathology, Hospital Universitario Fundación Jiménez Díaz, Instituto de Investigación Sanitaria Fundación Jiménez Díaz, Avenida Reyes Católicos, 2, E-28040, Madrid, Spain; e-mail: miguel.piris@quironsalud.es.

