

Key Points
ROS generation is essential for thrombus formation in HIT.
NOX2 inhibition prevents thrombus formation in a murine model of HIT.

Abstract
Heparin-induced thrombocytopenia (HIT) is associated with severe and potentially lethal thrombotic complications. NETosis was recently shown to be an important driver of thrombosis in HIT. We investigated the role of reactive oxygen species (ROS) and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 2 (NOX2) and their contributions to thrombus development in HIT. We showed that neutrophil activation by HIT immune complexes induced ROS-dependent NETosis. Analysis of thrombi formed in a microfluidics system showed ROS production in both platelets and neutrophils, and abundant neutrophil extracellular traps (NETs) and ROS distributed throughout the clot. Neutrophil-targeted ROS inhibition was sufficient to block HIT-induced NETosis and thrombosis using human blood. Inhibition of NOX2 with diphenyleneiodonium chloride or GSK2795039 abrogated HIT-induced thrombi in vivo using FcγRIIa+/hPF4+-transgenic mice. Thrombocytopenia in mice remained unaffected by ROS inhibition. Increased ROS production in activated neutrophils was also confirmed using fresh blood from patients with active HIT. Our findings show that ROS and NOX2 play a crucial role in NETosis and thrombosis in HIT. This enhances our understanding of the processes driving thrombosis in HIT and identifies NOX2 as a potential new therapeutic target for antithrombotic treatment of HIT.
Introduction
Heparin-induced thrombocytopenia (HIT) is a serious and potentially life-threatening immune reaction to heparin, a widely used anticoagulant. The clinical features of HIT include thrombocytopenia and venous and/or arterial thrombosis.1 Thrombotic complications such as myocardial infarction, thrombotic stroke, pulmonary embolism and arterial occlusion leading to limb gangrene may result in disastrous consequences and even death.2 Severe thrombosis and high mortality rates continue to be a medical challenge despite current therapies with nonheparin anticoagulants such as argatroban and danaparoid.3-5 Advancing our understanding of the pathogenesis of HIT will enable identification of key targets and development of new and more effective therapies for thromboses in HIT and their devastating consequences.
HIT is mediated by a pathogenic antibody that recognizes and forms a complex with platelet factor 4 (PF4) and heparin or polyanions.6 The HIT immune complex interacts with FcγRIIa receptors, leading to cell activation. HIT was traditionally considered a platelet disorder, in which engagement of the HIT immune complex with FcγRIIa triggers platelet activation and aggregation. Platelet activation results in release of procoagulant factors and microparticles that promote coagulation and perpetuate the cycle of platelet and coagulation activation.7 The pathogenesis of HIT has evolved over recent years and it has become increasingly clear that thrombosis in HIT extends beyond platelet activation, involving monocytes,8 endothelial cells,3 and neutrophils.9,10
Substantial evidence supports the concept that neutrophil activation is a major driver of thrombosis in HIT.9 Direct engagement of the HIT immune complex with FcγRIIa on neutrophils results in release of neutrophil extracellular traps (NETs), and NETosis is required for thrombus formation in HIT.9,10 In a protein arginine deiminase 4 (PAD4) knockout HIT mouse model,9 platelet/fibrin-rich thrombi in lungs were reduced significantly. Although PAD4, a key enzyme required for NET formation in mice, was shown to be necessary for thrombosis, upstream molecular pathways leading to NETosis are yet to be fully characterized. It has been shown that the signaling pathways and NET requirements are agonist-specific.11,12 For example, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) activity is required for phorbol 12-myristate 13-acetate (PMA) and some bacteria-induced NETs but is dispensable for NET formation in ionophore-stimulated neutrophils.11 The contribution of reactive oxygen species (ROS) to HIT pathogenesis has also not been previously assessed.
Our study investigated the requirement of ROS and the NOX pathways in HIT-induced thrombosis. We clearly show that HIT immune complexes can directly activate neutrophils to produce ROS and cause mitochondrial dysfunction. We provide strong evidence that in HIT, neutrophils undergo NETosis in a NOX-dependent manner, and inhibition of NOX activity reduces NETs and thrombosis in the murine HIT model. Enhanced levels of ROS production and NETosis were confirmed in circulating neutrophils in patients with acute HIT. Dissecting the mechanisms and pathways involved in thrombosis in HIT has identified NOX as a potential therapeutic target to reduce thrombosis and to improve clinical outcome of patients with HIT.
Materials and methods
Reagents
The following reagents were obtained: Hoechst 33342, 2,4-dinitrophenol (DNP), and L-012 (Sigma-Aldrich, St. Louis, MO); diphenyleneiodonium chloride (DPI; Abcam, Cambridge, United Kingdom); GSK2795039 (MedChemExpress, Monmouth Junction, NJ); anti-CD15 AF647, anti-myeloperoxidase (MPO) phycoerythrin (PE), and anti-rabbit secondary antibody BV421 (BD Biosciences, San Jose, CA); and CellROX, MitoSOX, dihydrorhodamine 123 (DIH123), 3,3′-dihexyloxacarbocyanine iodide (DiOC6), and Sytox Green (nucleic acid stain; Invitrogen, Carlsbad, CA).
Human samples
Blood samples were collected with informed consent from healthy donors and from patients with diagnosis of HIT following the criteria previously described.1 All HIT patient samples were positive by HIT antibody enzyme-linked immunosorbent assay and 14C-serotonin release assays. The study was approved by the University of New South Wales (NSW) Human Research Ethics Committee (HC12093) and the South Eastern Sydney Local Health District Human Research Ethics Committee (Sydney, NSW, Australia; 17/349 LNR/18/POWH/45) in accordance with the Declaration of Helsinki.
Antibodies
KKO hybridoma cells were kindly provided by Gowthami Arepally (Duke University, Durham, NC). Cells were cultured in Dulbecco modified Eagle medium containing 10% fetal bovine serum in a humidified incubator at an aeration of 5% CO2 at 37°C. Cells were cultured for 24 hours in serum-free Dulbecco modified Eagle medium before collection of antibody-containing supernatant. KKO and total immunoglobulin G (IgG) from healthy donor and patient plasma were purified by protein G agarose affinity chromatography.
Detection of ROS and NETs in vitro
Neutrophils from healthy donors were isolated from EDTA-anticoagulated blood using the EasySep direct human neutrophil isolation kit from StemCell Technologies (Vancouver, BC, Canada). Purity was assessed by flow cytometry. Cells were treated with HIT or control IgG (3 mg/mL) or KKO or isotype control (0.1 mg/mL), purified PF4 (10 μg/mL), and heparin (0.5 U/mL). Lumi-aggregometry (Chronolog 700 aggregometer; Chrono-Log, Havertown PA) was performed as described previously.13 For whole-blood assays, fresh citrate-anticoagulated blood from healthy donors or HIT patients was used. Low-density granulocytes (LDGs) were isolated using density gradient medium Lymphoprep.14 For ROS-inhibition studies, samples were treated in the absence or presence of ROS inhibitors DNP (750 µM) or DPI (25 µM) prior to HIT IgG activation. Neutrophils and NETs were stained using anti-CD15, anti-MPO, and anti-Cit H3/anti-rabbit IgG.9,15 CellROX, MitoSOX, dihydrorhodamine 123, and DiOC6 were used for ROS detection.
NET assessment by confocal microscopy
Isolated neutrophils were stained with Hoechst 33342 and anti-CD15 antibody and seeded into 8-well Nunc Laboratory-Tek II chambers. Neutrophils were incubated with DNP or DPI for 30 minutes prior to the addition of HIT or normal IgG, heparin, and purified PF4. Sytox Green was added prior to imaging using a confocal laser-scanning microscope (Leica TCS SP8) running Leica LAS X software.
Microfluidics studies
Microfluidics experiments with Vena8 Fluoro+ biochip microchannels were conducted as previously described.9 Briefly, microchannels were coated overnight with fibrinogen or von Willebrand factor (VWF) at 4°C, washed with phosphate-buffered saline (PBS), and blocked with PBS/1% bovine serum albumin. Fresh citrate-anticoagulated whole blood from healthy donors was diluted 1/2 in PBS, incubated at 37°C for 30 minutes with DPI (50 µM), then treated with purified IgG (HIT or normal IgG 3 mg/mL, KKO 0.25 mg/mL) and heparin (0.5 U/mL). For neutrophil-targeted ROS-inhibition assays, neutrophils were depleted from whole blood using CD15 microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany). Separately, autologous purified neutrophils were treated with or without DPI, washed, and reconstituted into neutrophil-depleted blood. To measure ROS, NET release, and neutrophil and platelet accumulation, samples were labeled with Hoechst 33342, Sytox Green, anti-CD41 or AP2, anti-CD15, and CellROX. Blood was perfused at a shear stress of 67 dyne/cm2 (1500 s−1 ) for up to 30 minutes and the biochip was imaged using a Q-Imaging EXi Blue camera (Q-Imaging, Surry, BC, Canada) connected to a fluorescent microscope (Zeiss Axio Observer.A1) driven by Venaflux software (Cellix Ltd). Quantitative analysis of DNA, platelet, and neutrophil accumulation was calculated with Image-Pro Premier 9.1 software (Media Cybernetics, Rockville, MD). Imaging by confocal microscopy was conducted on selected microchannels.
In vivo studies
Double-transgenic FcγRIIa+/hPF4+ mice expressing the R131 isoform of human FcγRIIa and human PF416 aged between 8 and 12 weeks were used for animal studies. The HIT condition was recreated by IV injection of the HIT-like monoclonal antibody KKO (400 µg per mouse) and intraperitoneal injection of heparin (1 U/g). ROS inhibitors were administered by intraperitoneal injection as follows: DPI (3 mg/kg) 30 minutes before and 30 minutes after, or GSK2795039 (100 mg/kg) 60 minutes before KKO and heparin injection. Anti-CD42c Dylight-649 antibody (Emfret, Germany) was used to label mouse platelets in vivo. For select studies, L-012 (25 mg/kg) was administered intraperitoneally 5 minutes prior to euthanasia. Extracted lungs were imaged immediately using the IVIS Spectrum running Living Image 4.5.5 software (Perkin Elmer, MA).9,17-19 Platelet counts were analyzed by flow cytometry and calculated relative to time 0, prior to IgG and heparin injection. All animal experiments were approved by the Animal Care and Ethics Committee of the University of New South Wales, Australia.
Statistics
Data sets were tested for normality using the Shapiro-Wilk normality test. Differences between 2 groups were compared using the Student t test. Multiple comparisons were analyzed by 1-way analysis of variance (ANOVA) with posttest correction for multiple comparisons as described in the figure legends. Each mouse was used as a biological replicate in animal studies and each individual healthy donor for in vitro experiments was considered a biological replicate. Statistical analyses were performed using GraphPad Prism 8 (La Jolla, CA). P values <.05 were considered statistically significant.
Results
HIT IgG stimulates ROS production in neutrophils
Because neutrophil activation is essential for thrombosis in HIT,9,10 we sought to determine whether ROS is involved in HIT IgG-induced neutrophil activation. To recreate the HIT condition in vitro, purified neutrophils were stimulated with IgG from HIT patients or KKO, PF4, and heparin. ROS production was examined using a luminol-based assay. PMA, a known ROS inducer,20-22 was used as a positive control. Treatment with KKO showed a significant accumulation of luminescence over time compared with treatment with isotype control (Figure 1), indicating enhanced ROS generation by KKO-treated neutrophils. Similar results were obtained following incubation of neutrophils with 3 different HIT patient IgGs when compared with normal IgG (Figure 1). Maximum ROS neutrophil production under different conditions was shown in Figure 1. These data show that HIT immune complexes are strong inducers of ROS.
![HIT IgG stimulates ROS production in neutrophils. Luminescence accumulation by neutrophils isolated from (A-C) 3 healthy donors incubated with PMA (25 nM, 50 nM, and 100 nM), or heparin (Hep) and purified PF4 and normal IgG (mouse isotype control [mIgG] or normal human IgG [nIgG]) or HIT IgG (KKO, or IgG from 3 HIT patients, HIT1 to HIT3). (D) Quantification of peak luminescence induced by neutrophils stimulated by PMA, normal IgG, or HIT IgG. Data expressed as mean plus or minus the standard error of the mean (SEM) (n = 3). *P < .05, **P < .01, ***P < .001, and ****P < .0001 by Student t test for comparisons between KKO and mIgG; 1-way ANOVA with Bonferroni correction was used for multiple comparisons (HIT1-3 and nIgG).](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/5/23/10.1182_bloodadvances.2020003093/5/m_advancesadv2020003093f1.png?Expires=1767705057&Signature=iLf-4raffNCujAdMnd~jk8YoYCtTTNdbjLXkKGjL6SWirlzulBEBeoBt3zXJrG5SDge6G2EGrH9bAXdNCU0Q0D~ITeCdwGMFzd8zIY1AKHm2Tfpvo24WzHEFQvTMEJ8jbQjXZEnS6GB~0ahNfmQqOr1H3~sWvxcXprfjPPAQ1UJfGY4oAGVOWq0vf8QVsWFhUa06aLSTQ3QGQGBfcfnBj2dR4Wvk-BVikradvz3RPcyoTC3s0wpIcJuAXR3GvjdwSOBfI8KcsK0wLW4s6W9cvnEInrGxHXX3aDlbqtWMIllh3yZT8M0OBldld7CqCElaZqjvnFSmm5tthxhA3gEq7g__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
HIT IgG stimulates ROS production in neutrophils. Luminescence accumulation by neutrophils isolated from (A-C) 3 healthy donors incubated with PMA (25 nM, 50 nM, and 100 nM), or heparin (Hep) and purified PF4 and normal IgG (mouse isotype control [mIgG] or normal human IgG [nIgG]) or HIT IgG (KKO, or IgG from 3 HIT patients, HIT1 to HIT3). (D) Quantification of peak luminescence induced by neutrophils stimulated by PMA, normal IgG, or HIT IgG. Data expressed as mean plus or minus the standard error of the mean (SEM) (n = 3). *P < .05, **P < .01, ***P < .001, and ****P < .0001 by Student t test for comparisons between KKO and mIgG; 1-way ANOVA with Bonferroni correction was used for multiple comparisons (HIT1-3 and nIgG).
HIT IgG stimulates ROS production in neutrophils. Luminescence accumulation by neutrophils isolated from (A-C) 3 healthy donors incubated with PMA (25 nM, 50 nM, and 100 nM), or heparin (Hep) and purified PF4 and normal IgG (mouse isotype control [mIgG] or normal human IgG [nIgG]) or HIT IgG (KKO, or IgG from 3 HIT patients, HIT1 to HIT3). (D) Quantification of peak luminescence induced by neutrophils stimulated by PMA, normal IgG, or HIT IgG. Data expressed as mean plus or minus the standard error of the mean (SEM) (n = 3). *P < .05, **P < .01, ***P < .001, and ****P < .0001 by Student t test for comparisons between KKO and mIgG; 1-way ANOVA with Bonferroni correction was used for multiple comparisons (HIT1-3 and nIgG).
Mitochondrial dysfunction in HIT
Mitochondria are the major source of intracellular ROS23-25 and mitochondrial dysfunction leads to an increase in ROS production.26-29 Mitochondrial dysfunction was examined using the voltage-sensitive dye DiOC6. Using fresh whole blood from healthy donors, our data showed a marked reduction of DiOC6 fluorescence in neutrophils in KKO- or HIT IgG-treated blood compared with the normal controls (Figure 2A), indicating a loss in mitochondrial membrane potential. Substantial decreases in DiOC6 fluorescence were detected in blood treated with heparin and IgG from 3 HIT patients (Figure 2B), whereas no changes were observed for PBS, heparin alone, or heparin plus normal IgG (Figure 2B). Treatment with DPI, an inhibitor of NOX, restored mitochondrial membrane potential back to control levels (Figure 2A-B). The generation of ROS was further examined using DIH123, a ROS indicator that fluoresces when oxidized by ROS. A strong heparin-dependent increase in DIH123 fluorescence was observed in blood treated with KKO and 3 patient HIT IgGs but not with control IgG (Figure 2C). DPI reversed this effect (Figure 2D). These data demonstrate that HIT IgGs and KKO induce increased ROS generation in neutrophils through mitochondrial dysfunction.
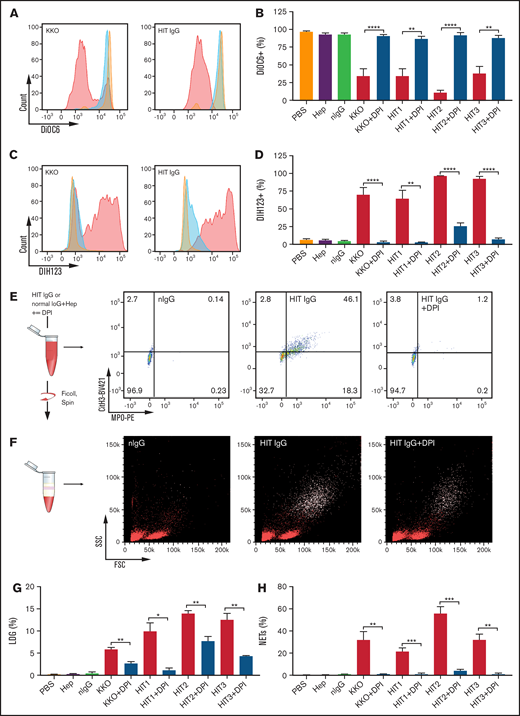
ROS is required for HIT IgG-induced NETosis. (A) Representative histograms of DiOC6 fluorescence in the neutrophil (CD15+) population when stimulated with normal IgG (orange), KKO (left), or HIT IgG (right) plus heparin (0.5 U/mL) in the absence (red) or presence (blue) of DPI, using flow cytometry. (B) Quantification of mitochondrial membrane potential in control or HIT immune complex–activated neutrophils from healthy individuals, with and without NOX inhibitor DPI (n = 4). (C) Representative histograms of DIH123 staining in the neutrophil (CD15+) population when stimulated with normal IgG (orange), KKO (left), or HIT IgG (right) in the absence (red) or presence (blue) of DPI. (D) Quantification of ROS production in control or HIT immune complex–activated neutrophils from healthy individuals, with and without NOX inhibitor DPI (n = 4 except HIT3, n = 3). Representative dot plots of (E) NETosis (MPO+Cit H3+) and (F) the LDG population (CD15+ events, white dots). Quantification of (G) percentage of LDG (n = 3) and (H) NETosis in blood treated with control or HIT IgG complexes, in the absence and presence of DPI (n = 4). Data expressed as mean plus or minus SEM. *P < .05, **P < .01, ***P < .001, and **** P < .0001 by Student t test . FSC, forward scatter; MPO-PE, myeloperoxidase-phycoerythrin; Ms IgG, mouse IgG (used as control for KKO); Norm, normal; SSC, side scatter.
ROS is required for HIT IgG-induced NETosis. (A) Representative histograms of DiOC6 fluorescence in the neutrophil (CD15+) population when stimulated with normal IgG (orange), KKO (left), or HIT IgG (right) plus heparin (0.5 U/mL) in the absence (red) or presence (blue) of DPI, using flow cytometry. (B) Quantification of mitochondrial membrane potential in control or HIT immune complex–activated neutrophils from healthy individuals, with and without NOX inhibitor DPI (n = 4). (C) Representative histograms of DIH123 staining in the neutrophil (CD15+) population when stimulated with normal IgG (orange), KKO (left), or HIT IgG (right) in the absence (red) or presence (blue) of DPI. (D) Quantification of ROS production in control or HIT immune complex–activated neutrophils from healthy individuals, with and without NOX inhibitor DPI (n = 4 except HIT3, n = 3). Representative dot plots of (E) NETosis (MPO+Cit H3+) and (F) the LDG population (CD15+ events, white dots). Quantification of (G) percentage of LDG (n = 3) and (H) NETosis in blood treated with control or HIT IgG complexes, in the absence and presence of DPI (n = 4). Data expressed as mean plus or minus SEM. *P < .05, **P < .01, ***P < .001, and **** P < .0001 by Student t test . FSC, forward scatter; MPO-PE, myeloperoxidase-phycoerythrin; Ms IgG, mouse IgG (used as control for KKO); Norm, normal; SSC, side scatter.
HIT IgG-mediated neutrophil activation and NETosis are dependent on NOX-derived ROS
As we described previously,9 LDGs, often described as activated neutrophils30 or NETting neutrophils,31 were found in fresh blood of acute HIT patients. In the present study, we treated healthy donor blood with KKO or HIT IgG in the presence of heparin. We observed a significant increase in neutrophils undergoing NETosis in whole blood (Figure 2E) and in the LDG population (supplemental Figure 1A) when treated with KKO or HIT IgG and heparin compared with controls. Importantly, the LDG population in fresh acute HIT patient blood (supplemental Figure 2A) generated large amounts of ROS (supplemental Figure 2B) and NETs (supplemental Figure 2C). The increase in LDG (Figure 2F-G) and NETting neutrophils (Figure 2E,H) induced by HIT immune complexes was abrogated by DPI, indicating that this is dependent on NOX activity.
The contribution of mitochondrial respiration and NOX to HIT IgG-induced ROS generation and their respective requirement for HIT IgG-induced NETosis was further analyzed in the presence of DPI and of the mitochondrial uncoupler (ROS inhibitor), DNP. DNP and DPI have previously been shown to be effective at inhibiting A23187- and PMA-induced NETosis, respectively.11,32 Isolated neutrophils were treated with DNP or DPI prior to stimulation with HIT IgG and heparin. As expected, direct activation of neutrophils with HIT IgG and heparin resulted in a significant release of extracellular DNA when monitored by live cell microscopy (supplemental Figure 3A). Treatment with DNP showed no reduction in extracellular DNA release in response to HIT IgG treatment, indicating no inhibition of NETosis. In contrast, blockage of NOX with DPI significantly impaired NET release as extracellular DNA levels were similar to normal IgG-treated controls (supplemental Figure 3B).
The efficiency of DNP (supplemental Figure 4A) and DPI (supplemental Figure 4B) in blocking ROS production was confirmed in HIT IgG-induced LDGs. By using MitoSOX, a superoxide indicator that concentrates within the mitochondria, LDGs formed following treatment of neutrophils with KKO or HIT IgG were shown to generate variable levels of superoxide depending on the antibody (supplemental Figure 4A).
Together, these results indicate that HIT-stimulated neutrophils generate abundant ROS and undergo NETosis in a NOX-dependent manner.
Thrombus formation in HIT requires neutrophil-derived ROS
To examine the role of ROS in thrombosis in HIT, thrombi were generated using fresh blood, KKO, and heparin in the microfluidics system in the presence or absence of DPI. Thrombi imaged by confocal microscopy showed an abundance of platelets, neutrophils, ROS, and released DNA within the clot (Figure 3A). ROS generation in neutrophils and platelets was confirmed by the overlap of ROS staining in neutrophils and platelets (Figure 3B). Enhanced levels of ROS production were also observed in circulating platelets and neutrophils in patients with acute HIT (supplemental Figure 2D-E). Clot component and distribution analysis of the thrombus formed in the microfluidics assay is shown in Figure 3C.
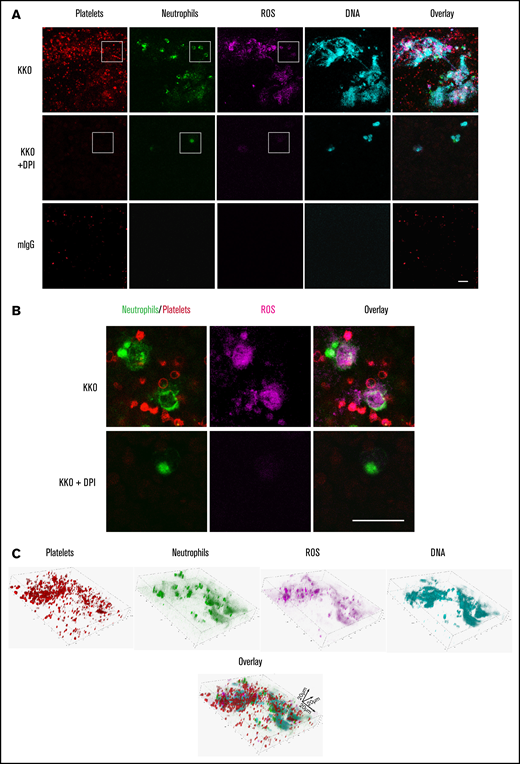
HIT IgG induces NETosis and ROS-containing thrombi. (A) Representative confocal microscopy images from n = 3 experiments of KKO-induced thrombi in whole blood showing platelets (AP2 AF594, red), neutrophils (anti-CD15 FITC, green), ROS (CellROX, magenta), and DNA release (Hoechst 3342, blue). Scale bar, 20 µm. (B) Magnified image of dotted area in panel A showing KKO-induced ROS in neutrophils and platelets. Scale bar, 20 µm. (C) Confocal microscopy analysis of KKO-induced thrombus in microfluidics channels. 3-Dimensional reconstruction and distribution of DNA (Hoechst 3342, blue), neutrophils (anti-CD15 FITC, green), platelets (AP2, AF594, red), and ROS (CellROX deep red, magenta) in thrombus.
HIT IgG induces NETosis and ROS-containing thrombi. (A) Representative confocal microscopy images from n = 3 experiments of KKO-induced thrombi in whole blood showing platelets (AP2 AF594, red), neutrophils (anti-CD15 FITC, green), ROS (CellROX, magenta), and DNA release (Hoechst 3342, blue). Scale bar, 20 µm. (B) Magnified image of dotted area in panel A showing KKO-induced ROS in neutrophils and platelets. Scale bar, 20 µm. (C) Confocal microscopy analysis of KKO-induced thrombus in microfluidics channels. 3-Dimensional reconstruction and distribution of DNA (Hoechst 3342, blue), neutrophils (anti-CD15 FITC, green), platelets (AP2, AF594, red), and ROS (CellROX deep red, magenta) in thrombus.
To determine the effect of ROS inhibition on the degree of DNA and cell accumulation in HIT-induced thrombi, blood flowing on VWF-coated microchannels (using a microfluidics system) was stained, imaged, and quantitatively analyzed. Thrombus component deposition was quantified as the percentage area coverage vs time. As anticipated, thrombi induced by KKO (Figure 4A) and HIT IgG (Figure 4B) showed marked deposition of extracellular DNA, platelets, and neutrophils. Pretreatment of blood with DPI resulted in a drastic reduction in all 3 components and consequently a significant decrease in clot size. NOX inhibition causing NETosis suppression and thrombus size reduction confirmed the central role of ROS in HIT-associated thrombosis.
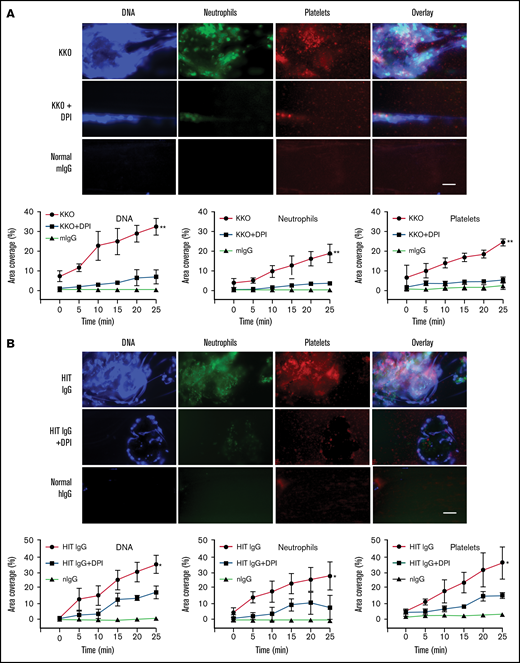
Inhibiting ROS abrogates clot formation and release of NETs under flow conditions. (A) Representative fluorescent images of whole blood treated with isotype control and heparin, or KKO and heparin plus vehicle control or DPI. Extracellular DNA (Sytox, blue), neutrophils (anti-CD15 AF594, green), and platelets (anti-CD41 V450, red). Quantification of area coverage of extracellular DNA, neutrophils, and platelets from KKO-induced thrombi with and without DPI. Scale bar, 50 µm. (B) Whole blood treated with normal human IgG (hIgG) and heparin or a representative HIT IgG and heparin plus vehicle control or DPI. Staining and quantification as described in panel A. Scale bar, 50 µm. Data expressed as mean plus or minus standard deviation (SD) (n = 3). *P < .05, **P < .01, and ****P < .0001 by 1-way ANOVA with Tukey correction for multiple comparisons.
Inhibiting ROS abrogates clot formation and release of NETs under flow conditions. (A) Representative fluorescent images of whole blood treated with isotype control and heparin, or KKO and heparin plus vehicle control or DPI. Extracellular DNA (Sytox, blue), neutrophils (anti-CD15 AF594, green), and platelets (anti-CD41 V450, red). Quantification of area coverage of extracellular DNA, neutrophils, and platelets from KKO-induced thrombi with and without DPI. Scale bar, 50 µm. (B) Whole blood treated with normal human IgG (hIgG) and heparin or a representative HIT IgG and heparin plus vehicle control or DPI. Staining and quantification as described in panel A. Scale bar, 50 µm. Data expressed as mean plus or minus standard deviation (SD) (n = 3). *P < .05, **P < .01, and ****P < .0001 by 1-way ANOVA with Tukey correction for multiple comparisons.
Next, we examined whether neutrophil-targeted ROS disruption was sufficient to inhibit thrombosis. Neutrophils were separated from whole blood, treated with DPI or vehicle control, washed, and then reconstituted into autologous neutrophil-depleted blood. The reconstituted blood was then treated with normal or HIT IgG and heparin; it flowed through the microfluidics channels and was imaged using fluorescent microscopy. As expected, large clots abundant in extracellular DNA, neutrophils, and platelets were observed in HIT IgG-treated blood reconstituted with vehicle-treated neutrophils (Figure 5) but not in similarly reconstituted blood treated with normal IgG. Interestingly, blood reconstituted with neutrophils pretreated with DPI showed a significant reduction in DNA, neutrophil, and platelet deposition following HIT IgG treatment. These data demonstrate a direct link between neutrophil-derived ROS and its requirement for thrombus formation in HIT.
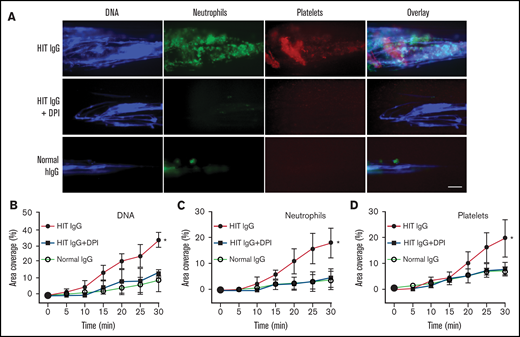
Neutrophil-targeted ROS inhibition blocks NETs and thrombosis in whole blood. Neutrophil-depleted blood was reconstituted with autologous neutrophils pretreated with vehicle control or DPI, then treated with normal IgG or HIT IgG plus heparin. (A) Representative fluorescent images showing extracellular DNA (Sytox, blue), neutrophils (anti-CD15 AF594, green), and platelets (anti-CD41 V450, red). Quantification of area coverage of (B) DNA, (C) neutrophils, and (D) platelets under different treatment conditions. Scale bar, 50 µm. Data expressed as mean plus or minus SD (n = 3). *P < .05; ns, not significant, by 1-way ANOVA with Tukey correction for multiple comparisons.
Neutrophil-targeted ROS inhibition blocks NETs and thrombosis in whole blood. Neutrophil-depleted blood was reconstituted with autologous neutrophils pretreated with vehicle control or DPI, then treated with normal IgG or HIT IgG plus heparin. (A) Representative fluorescent images showing extracellular DNA (Sytox, blue), neutrophils (anti-CD15 AF594, green), and platelets (anti-CD41 V450, red). Quantification of area coverage of (B) DNA, (C) neutrophils, and (D) platelets under different treatment conditions. Scale bar, 50 µm. Data expressed as mean plus or minus SD (n = 3). *P < .05; ns, not significant, by 1-way ANOVA with Tukey correction for multiple comparisons.
ROS inhibition suppresses thrombus formation in HIT but not thrombocytopenia or hypothermia
Having shown that thrombus formation in HIT involves ROS activity in vitro, we explored whether inhibiting ROS in vivo was sufficient to prevent thrombosis in a murine model of HIT. HIT can be recapitulated using KKO and heparin9,10,16,33 in double transgenic mice expressing human PF4 and human FcγRIIa.16 L-012 was administered to HIT mice, and extracted lungs were imaged using the IVIS Spectrum CT scanner to detect luminescence. KKO-treated mice without DPI treatment demonstrated strong accumulation of luminescence in the lungs; however, lungs extracted from mice treated with DPI showed a significant reduction in luminescence, indicating ROS inhibition (supplemental Figure 5A).
To verify that neutrophil activation was blocked in FcγRIIa+/hPF4+ mice following ROS inhibition, NETosis markers such as the presence of activated neutrophils (LDGs) and cell-free DNA were examined. A proportion of LDGs in KKO-treated mice (supplemental Figure 5B) was found to also be ROS+ (supplemental Figure 5C). There was a significant reduction in circulating LDG and ROS+ cells in the DPI-treated HIT mice to levels seen in control mice. Similarly, the abundance of cell-free DNA in circulation in KKO-treated mice was reduced with DPI treatment compared with mice without (supplemental Figure 5D). These in vivo findings are consistent with our microfluidics observations in which ROS inhibition blocked neutrophil activation.
Pulmonary thromboembolism was detected using an IVIS Spectrum CT scanner by measuring fluorescently labeled mouse platelets in lungs extracted from KKO- or normal IgG-treated mice in the presence or absence of DPI. There was strong fluorescence within the lungs from mice treated with KKO and heparin, indicating dense platelet accumulation and thrombosis (Figure 6A). In support of the microfluidics findings, inhibition of ROS using DPI showed a significant decrease in fluorescence in the lungs, suggesting that DPI was effective in suppressing HIT-induced thrombosis in vivo.
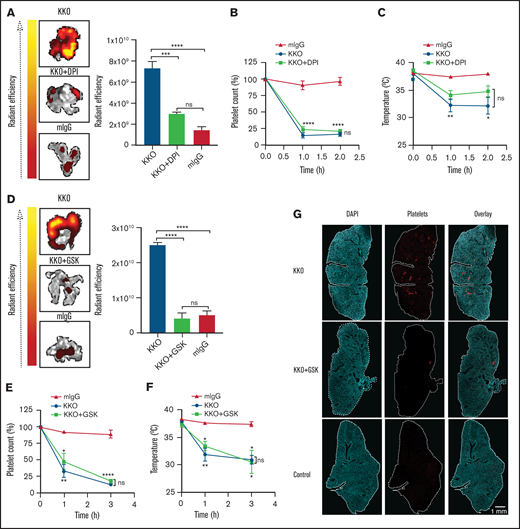
Inhibiting ROS protects mice from HIT-induced thrombosis but not thrombocytopenia. FcγRIIa+/hPF4+ double transgenic mice were injected IV with normal IgG or KKO. Heparin was injected intraperitoneally. Representative images and graphical representation of fluorescence intensity from platelet accumulation in mouse lungs from mice treated with normal IgG, KKO, and vehicle control, (A) KKO and DPI (n = 4), or (D) KKO and GSK2795039 (n = 3). (B,E) Platelet counts are represented as percentage of platelets relative to basal levels (n = 4). Rectal temperature of FcγRIIa+/hPF4+ mice treated with KKO or normal IgG, with and without (C) DPI (n = 4) or (F) GSK2795039 (n = 3). (G) Confocal images of lung lobe from mice treated with heparin and KKO, KKO and GSK2795039, or control IgG. 4′,6-Diamidino-2-phenylindole (DAPI)-stained nuclei (blue), platelet-rich thrombi (red). Scale bar, 1 mm.
Inhibiting ROS protects mice from HIT-induced thrombosis but not thrombocytopenia. FcγRIIa+/hPF4+ double transgenic mice were injected IV with normal IgG or KKO. Heparin was injected intraperitoneally. Representative images and graphical representation of fluorescence intensity from platelet accumulation in mouse lungs from mice treated with normal IgG, KKO, and vehicle control, (A) KKO and DPI (n = 4), or (D) KKO and GSK2795039 (n = 3). (B,E) Platelet counts are represented as percentage of platelets relative to basal levels (n = 4). Rectal temperature of FcγRIIa+/hPF4+ mice treated with KKO or normal IgG, with and without (C) DPI (n = 4) or (F) GSK2795039 (n = 3). (G) Confocal images of lung lobe from mice treated with heparin and KKO, KKO and GSK2795039, or control IgG. 4′,6-Diamidino-2-phenylindole (DAPI)-stained nuclei (blue), platelet-rich thrombi (red). Scale bar, 1 mm.
Other clinical features of HIT include thrombocytopenia and systemic reactions such as hypothermia. Despite protection of KKO-treated FcγRIIa+/hPF4+ mice from thrombosis with administration of DPI, pretreatment with DPI did not prevent the development of thrombocytopenia (Figure 6B) or hypothermia (Figure 6C). This indicates that thrombocytopenia/hypothermia and thrombosis are mediated by distinct mechanisms and inhibiting ROS production does not affect platelet destruction or development of hypothermia. Similar results were observed with the NOX2-specific inhibitor GSK2795039: treatment with GSK2795039 protected mice from thrombosis (Figure 6D) but not thrombocytopenia (Figure 6E) or hypothermia (Figure 6F).
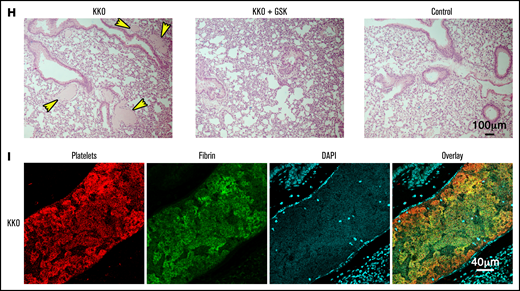
Examination of lung sections from mice treated with KKO and heparin confirmed the presence of platelet- and fibrin-rich clots (Figure 6G-I). Conversely, lungs of GSK2795039-treated mice showed a significant reduction in clots to levels similar to control mice (Figure 6G-H), confirming that NOX2 plays a critical role in HIT-induced thrombosis. Together, these data indicate that ROS inhibitors are potent suppressors of thrombosis in HIT and potential therapeutic agents for HIT.
Collectively, our data demonstrate that HIT IgG activates neutrophils to undergo NETosis via NOX- and ROS-dependent pathways and that NOX2 and ROS are essential for NETosis and thrombus formation in HIT.
Discussion
Severe thrombosis is a significant contributor to morbidity and mortality in patients with HIT and its control continues to be a clinical challenge even with nonheparin anticoagulant therapies. Recent research has provided convincing evidence that thrombosis in HIT is driven by neutrophil activation and NETosis.9,10 This study further validates that the pathogenesis of thrombosis induced by blood immune complexes is distinct from the mechanism of thrombocytopenia.34-36 In this study, we sought to explore the cellular processes/pathways involved in HIT-induced thrombosis in order to identify new potential therapeutic targets.
Several studies have investigated the signaling intermediates of ROS generated by well-characterized NETosis stimuli.22,37-40 NETosis induced by some stimuli such as PMA, calcium ionophore, and immobilized immune complexes41 is ROS-dependent, whereas others, including soluble immune complexes involved in autoimmunity and inflammation are ROS-independent.42-45 The discrepancies in the requirement of ROS in previous studies involving immune complexes may have been due to the use of artificial immune complexes.41,45 In this study, the significance of ROS in HIT IgG-induced NET formation was determined using clinically relevant immune complexes (IgG purified from HIT patients’ plasma plus heparin and human PF4).
The main sources of ROS include the NADPH oxidases46 and the mitochondrial electron transport chain.47 These processes can also result in feed forward processes, called ROS-induced ROS production, further augmenting ROS production.48 Mitochondrial ROS is essential in NETosis stimulated by calcium ionophores32 and ribonucleoprotein-containing immune complexes.49 Conversely, PMA, fungi, bacteria, and systemic lupus erythematosus antibodies are NOX2-dependent NETosis stimuli.20-22 Other studies have shown that PMA-induced NETosis is also independent of PAD411 and mitochondrial ROS.32 These findings emphasize the diverse requirements and pathways engaged in NETosis depending on the stimuli and disease condition. HIT-induced NETosis is a recent finding and the key mediators and pathways that govern this process had not yet been fully investigated. Our current data demonstrate that HIT IgG can directly activate neutrophils to generate ROS, explaining the elevated levels of ROS observed ex vivo in circulating neutrophils of HIT patients. We found that HIT IgG induced both cytoplasmic and mitochondrial ROS production in neutrophils, as determined by CellROX and MitoSOX staining, respectively, and it was associated with a decrease in mitochondrial membrane potential. Despite the aberrant mitochondrial function in activated neutrophils, the disruption of mitochondrial respiration was dispensable as its inhibition with DNP did not prevent NETosis. However, blockage of NOX2 was sufficient to inhibit NET release by neutrophils induced by HIT IgG and heparin. NOX2 therefore appears to be required in HIT-induced NETosis.
The role of ROS was investigated in terms of its requirement for the development of HIT-associated thrombosis, thrombocytopenia, and systemic reaction (such as hypothermia). We recently showed that inhibiting neutrophil activation and NETosis by targeting PAD4 successfully abolished thrombosis in a HIT mouse model9 but had no effect on the development of thrombocytopenia and hypothermia. Only blocking of HIT immune complexes to FcγRIIa receptors by aglycosylated IV.39 prevented thrombosis, thrombocytopenia, and hypothermia. The current findings are consistent with these observations. ROS and NOX inhibition were effective in suppression of thrombosis but insufficient in protecting mice from thrombocytopenia and hypothermia, thus confirming that thrombocytopenia and thrombosis are distinct manifestations of HIT. Importantly, the data presented here provide strong evidence that ROS inhibitors, DPI and GSK2795039, a novel small molecule that selectively inhibits NOX2,50 prevent the formation of LDGs, impede NETosis, and consequently suppress thrombosis in vitro and in vivo. DPI was able to significantly inhibit HIT-induced platelet activation (supplemental Figure 1B), suggesting that ROS also plays a role in platelet activation; however, DPI was insufficient in protecting platelets from destruction in vivo. DPI continues to be the most potent and reliable NOX inhibitor available to date,51 despite its reported potential off-target effects. To strengthen our findings on NOX inhibition, we additionally used another potent and specific NOX2 inhibitor, GSK2795039, to confirm that NOX2 was critical for HIT-induced thrombosis, particularly in vivo. Platelet activation and the resulting ROS production likely contribute to thrombosis, although microfluidics data confirmed that neutrophil-targeted ROS inhibition was sufficient in blocking thrombosis, emphasizing the central role neutrophil activation plays in thrombosis. Neutrophil-specific knockout of NOX will be of future interest.
In summary, we demonstrate a novel mechanistic connection between ROS signaling and FcγRIIa activation in HIT by immune complexes. Our data show that NOX2-derived ROS in neutrophils is required for NETosis and contributes significantly to the prothrombotic state in HIT. HIT-induced neutrophil activation, mitochondrial dysfunction, NETosis, and thrombosis under flow conditions were dependent on NOX2-derived ROS. This was supported by our study using a murine HIT model, showing attenuation of thrombi when NOX2 was inhibited. Dissecting the mechanism of HIT-induced NETosis and thrombosis has identified NOX2 as a signaling intermediate, which could be a potential novel therapeutic target in HIT. Our work provides a newfound understanding of the requirements of NOX and ROS in the pathogenesis of HIT-induced thrombosis. This has important implication in the development of potential new antithrombotic treatments for HIT.
Acknowledgments
The authors thank Gowthami Arepally (Duke University) for providing KKO hybridoma cells. The authors acknowledge Kathryn Evans for technical assistance with animal experiments, and Rose Wong and Shiying Zheng for assistance with patient samples. The authors also acknowledge the National Imaging Facility, a National Collaborative Research Infrastructure Strategy (NCRIS) capability, at the Mark Wainwright Imaging Analytical Centre, University of New South Wales.
This work was supported by grants from the Australian Health and Medical Research Council (project grant 1109745 and program grant 1052616 [B.H.C.]).
Authorship
Contribution: H.H.L.L. conceived the study, designed and performed experiments, analyzed and interpreted data, and wrote the manuscript; J.P. provided research guidance, designed and performed confocal microscopy experiments, analyzed and interpreted data, and contributed to the manuscript; Z.A. designed and performed microfluidics experiments and analyzed and interpreted data; S.E.M. provided FcγRIIa+/hPF4+-transgenic mice and contributed to the manuscript; H.H.L.L., J.P., and F.Y. designed and performed animal experiments; and B.H.C. conceived and supervised the study, provided input to data interpretation, and critically reviewed and edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Beng H. Chong, Haematology Research Unit, St George & Sutherland Clinical School, Research & Education Centre, Level 3, 4-10 South St, Kogarah, NSW 2217, Australia; e-mail: beng.chong@unsw.edu.au.

