

Abstract
Poly(ADP‐ribose) polymerase 1 (PARP1) is a key mediator of various forms of DNA damage repair and plays an important role in the progression of several cancer types. The enzyme is activated by binding to DNA single-strand and double-strand breaks. Its contribution to chromatin remodeling makes PARP1 crucial for gene expression regulation. Inhibition of its activity with small molecules leads to the synthetic lethal effect by impeding DNA repair in the treatment of cancer cells. At first, PARP1 inhibitors (PARPis) were developed to target breast cancer mutated cancer cells. Currently, PARPis are being studied to be used in a broader variety of patients either as single agents or in combination with chemotherapy, antiangiogenic agents, ionizing radiation, and immune checkpoint inhibitors. Ongoing clinical trials on olaparib, rucaparib, niraparib, veliparib, and the recent talazoparib show the advantage of these agents in overcoming PARPi resistance and underline their efficacy in targeted treatment of several hematologic malignancies. In this review, focusing on the crucial role of PARP1 in physiological and pathological effects in myelodysplastic syndrome and acute myeloid leukemia, we give an outline of the enzyme’s mechanisms of action and its role in the pathophysiology and prognosis of myelodysplastic syndrome/acute myeloid leukemia and we analyze the available data on the use of PARPis, highlighting their promising advances in clinical application.
Introduction
Poly(ADP-ribose) polymerases (PARPs) are a family of enzymes that use the oxidized form of nicotinamide adenine dinucleotide (NAD+) to transfer ADP-ribose to other proteins (poly ADP-ribosylation). The family consists of at least 18 enzymes that are encoded by different genes and all share a conserved catalytic domain.1 Therefore, some isoforms, such as poly(ADP-ribose) polymerase 1 (PARP1), are most known for their implication in different cellular processes. PARP1 is an ∼113 kDa nuclear protein and the first member of the PARP family identified. It is connected to a plethora of cellular procedures such as DNA repair, transcriptional and posttranscriptional regulation of gene expression, control of protein degradation, and cell death.2 PARP1 is overexpressed in many cancers such as testicular and other germ cell tumors, neuroblastoma, malignant lymphoma, Ewing sarcoma, breast cancer, and colon cancer.3-5 It also contributes to progression of endometrial cancer, breast cancer (BRCA)-mutated ovarian cancer, and BRCA-mutated serous ovarian cancer.6 Although, most studies focus on the DNA damage detection and repair effects of the molecule, PARP1 has more recently been studied in the context of the regulation of chromatin structure and transcription and linked to DNA methylation, imprinting, and chromosome organization.7-9
Evaluating the DNA damage response (DDR) machinery as a therapeutic target is a promising strategy for designing novel pharmaceutical agents. DDR in hematological malignancies has been extensively studied but not fully understood. It has been reported that the BRCA1 expression level was found reduced in acute myelogenous leukemia (AML) samples. When AML was treated with DNA damaging agents, the loss of BRCA1 function led to the accumulation of genomic alterations, and even to synthetic lethality.10 Consequently, it is important to understand the roles of PARP1 in DNA repair to proceed to developing successful therapeutic regimens for treating different cancers.11 PARP inhibitors (PARPis) are already available and have shown significant benefits in a variety of malignancies. A study trying to illustrate the importance of oncogenic transcription factors for AML progression12 demonstrated for the first time a potential utility of PARPi-induced lethality for leukemia, driven by AML1-ETO and PML-RARa. AML cells with low expression of key members of the DDR pathway, such as Rad51, ATM, BRCA1, and BRCA2, displayed obvious sensitivity to PARPis.12 Furthermore, they showed that combining a PARPi with a GSK3 inhibitor proved an effective therapeutic strategy for PARPi-resistant AML. In this review, we give an outline of PARP1 mechanism of action7 (Figure 1), focusing on its crucial role in physiological and pathological effects in hematologic malignancies. Moreover, we analyze the available data on the use of PARPis in AML and/or myelodysplastic syndromes (MDS).
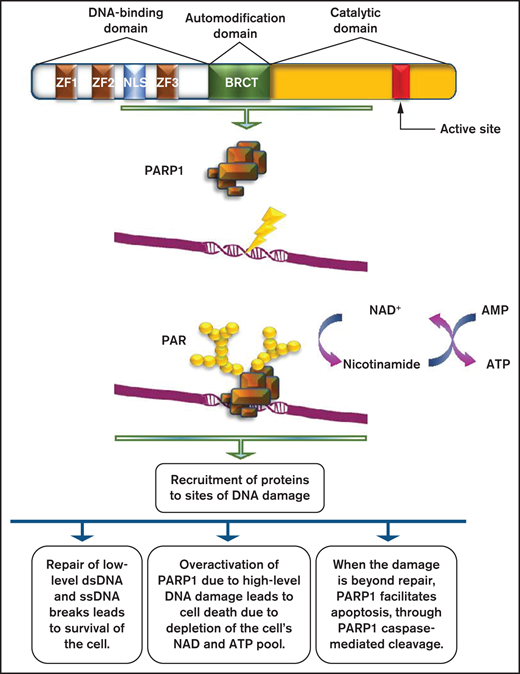
Role of PARP1 in DNA damage repair. PARP1 consists of a DNA-binding domain with 3 zinc finger motifs, an automodification domain that contains the BRCA1 C terminus (BRCT) domain, and a carboxy-terminal catalytic domain, which contains the active site of the enzyme. PARP1 is usually activated by DNA damage occurring as a result of the DNA damage response. The net result of its activation is the production of PAR chains, with the use of nicotinamide adenine dinucleotide (NAD+) as substrate. PARylation results in the recruitment of several proteins with multiple roles on DNA damage repair.
Role of PARP1 in DNA damage repair. PARP1 consists of a DNA-binding domain with 3 zinc finger motifs, an automodification domain that contains the BRCA1 C terminus (BRCT) domain, and a carboxy-terminal catalytic domain, which contains the active site of the enzyme. PARP1 is usually activated by DNA damage occurring as a result of the DNA damage response. The net result of its activation is the production of PAR chains, with the use of nicotinamide adenine dinucleotide (NAD+) as substrate. PARylation results in the recruitment of several proteins with multiple roles on DNA damage repair.
PARP1 biochemical activities
In human cells, normal metabolic activities as well as environmental factors can lead to DNA damage, resulting in a range from 20,000 to 1 million individual molecular damages per cell per day.13,14 Consequently, the DNA repair machine responds to lesions by activating apoptosis.15 When normal repair processes fail to respond, nonrepairable DNA damage may arise, including double-strand breaks (DSBs) of DNA. Activation of PARP1 is a usual event in the DDR. Posttranscriptionally, PARPs attach to a polymer termed poly(ADP-ribose) (PAR), to PARP1 itself, and to other histone and nonhistone proteins within a dynamic process called poly(ADP ribosyl)ation (PARylation).16,17 DNA damage may produce extensive auto PARylation, resulting in the inhibition of PARP1 binding to DNA and to a decrease of its catalytic activities.6 Several kinds of DNA damage may cause rapid recruitment of PARP1 to the sites of damage through DNA binding. Single base alternations or nucleotide damage are the most common type of DNA damage. They can modify base-pairing nature and result in spontaneous mutations that are strongly associated with cancer predisposition. Single-strand breaks (SSBs) are rapidly detected and bound by PARP1.9,10 The rate of SSB repair (SSBR) is likely to be increased with the use of the molecule. SSBR activation could be mediated by the induction of accumulation of SSBR components such as DNA ligase 3 (LIG3), DNA polymerase β and bifunctional polynucleotide kinase 3′-phosphatase and/or their stabilization at SSBs,18 thus inducing the repair process. DNA-damaging agents, such as ionizing and ultraviolet radiation or impairments from by-products of the cell own metabolism are among the most common causes of DSB production.19 DDR proteins such as the ataxia telangiectasia mutated (ATM) kinase target DSBs. ATM consists of PAR-binding domains, and the stimulation of PAR activity depends on its interaction with them.20 However, there are other critical pathways for the activation of the DDR because PARP1-lack of function only delays the recruitment of certain proteins but does not completely suspend it.
Parthanatos
PARP1 function ranges from supporting survival to inducing death. Parthanatos (from the Greek Θανατoς, “death”), a form of programmed cell death, shares cytological and morphological features with apoptosis and necrosis, but in contrast to apoptosis, it is the result of a caspase-independent molecular mechanism.21,22 It is mediated by the pathologically high levels of PARP1 activity. The main event is the translocation of the apoptosis-inducing factor from mitochondria to the nucleus,23 leading to dissociation of histones and DNA degradation. PAR toxicity depends on the length and complexity of the PAR polymer. PAR polymers with a size greater than 60 ADP-ribose units are more toxic than less complex polymers. Delivery of >60 ADP-ribose polymer kills cells in a dose-dependent manner. The toxicity of PAR polymers is emphasized in studies of poly-(ADP) ribose glycohydrolase (PARG). PARG is responsible for encoding four isoforms, the nuclear 110-kDa isoform, the cytoplasmic 102-kDa and 99-kDa isoforms (detected only in humans), and the 59-kDa isoform (60 kDa in mice).24 Different sizes of PAR polymer can bind to proteins through either covalent or noncovalent binding. That several PARPs but only 2 PARGs have been identified so far makes it clear that the polymers produced by the PARP family members are structurally distinct, a fact that plays a role in the different functions of the several PARPs. Noncovalent binding of PAR to proteins is stable, whereas in vitro PAR binding is resistant to strong acids, chaotropes, detergents, and high salt concentrations. Limitation of PARP1 overactivation affects apoptosis-inducing factor release from the mitochondria, indicating that there is a nuclear-mitochondrial interaction that occurs in PARP1-mediated cell death. Because parthanatos subtends PARP1-mediated cell death through the actions of the PAR polymer, the next steps involve the identification of specific PAR targets and the designation of how PAR binding leads to cytotoxicity.
Methylation
Epigenetics is defined as heritable changes in gene expression that do not go along with changes in DNA sequence.25 Hypermethylation of CpG islands in gene promoters, histone deacetylation, and genome hypomethylation are common events in tumorigenesis. DNA methylation and histone deacetylation may be affected by hypomethylating agents (HMAs) and histone deacetylase inhibitors.26 PARP1 is related to DNA methylation and is correlated to gene expression silencing. Based on the enzyme’s involvement and regulating gene expression, a study reported its association with chromatin structure modification. The activity of enzyme was inhibited by a histone variant, thus leading to reactivation of a gene on the inactive X chromosome, establishing the participation of PARP1 in silencing conservation.27 Inhibitors of PARP1 have shown to act synergistically with HMAs. They inhibit DNA methylation by inactivating and trapping DNA methyltransferase (DNMT) to DNA, facilitating the repair of damaged DNA by the base excision repair machinery. PARP1 may affect methylation by regulating the expression of the DNA methyltransferase-1 (DNMT1) gene. Binding to the gene promoter and protecting it from methylation, it leads to induction of the DNMT1 activation.28 Inhibition of DNMT1 expression causes hypomethylation of the whole genome. This correlation of PARP1 with methylation is a key point in DNA repair, since hypermethylation is a major event in the pathogenesis of MDS and AML, and hypomethylating agents are an effective treatment option for these patients.
Targeting genetic abnormalities in MDS and AML
Gene mutations that are likely associated with AML can be classified into 9 distinct functional categories: the signaling genes (FLT3, KIT), the DNA-methylation-associated genes (DNMT3A, TET2, IDH1/2), the chromatin-modifying genes (ASXL1, EZH2, MLL), the nucleophosmin encoding gene (NPM1), the myeloid transcription factor genes (CEBPA, RUNX1), the transcription factor fusion genes (PML-RARA, RUNX1-RUNX1T1, CBFB-MYH11), the tumor suppressor genes (TP53, WT1), the spliceosome–complex genes (SF3B1, SRSF2, U2AF1), and the cohesin-complex genes (SMC1, SMC3, STAG2, RAD21). NPM1 and biallelic CEBPA mutations are associated with a better prognosis. In contrast, patients with mutations of FLT3-ITD, RUNX1, ASXL1, and TP53 are considered to present with a worse prognosis.29 That genomic alterations are present in AML and other hematological malignancies allows the presumption that these DNA changes might have as an origin the aberrant DSB repair.30-34 As an example, predisposition to develop MDS and AML is evident in patients with Fanconi anemia; a chromosomal disorder known for the mutation of homologous recombination (HR) components (BRCA2 [FANCD1], FANCO [RAD51C], FANCJ [BRIP1], and FANCN [PALB2]).35 Gene mutations in Fanconi anemia have been identified in therapy-related AML and MDS, predisposing to several malignancies.36,37
Patients with high-risk MDS have few treatment options and are often not eligible for intensive chemotherapy (ICT) because of comorbidities. They are treated with less aggressive therapies, including low-dose cytarabine and HMAs. ICT has been reported to induce a complete remission in ∼50% of higher-risk MDS patients, which is significantly lower than that of AML patients. The duration of response is usually short, and patients with adverse karyotypes or TP53 mutations have lower response rates.38 ICT can be used as a bridge to transplantation for patients failing 5-azacytidine (AZA).39
Venetoclax, is an orally selective inhibitor of the B-cell lymphoma 2 protein that, when acting synergistically with AZA, increases both response and prolonged survival as it was observed in a phase III trial.40,41 Indeed, prior trials trying to evaluate the efficacy of their combination comparing to venetoclax alone in AML patients, showed promising results.42,43 This combination therapy, which led to an astonishing complete remission rate in the venetoclax plus HMA cohort, resulted in its approval by the US Food and Drug Administration (FDA) in 2018.44 In a retrospective study with high-risk MDS patients, it was shown that an HMA plus venetoclax leads to high response rates.45 However, the addition of venetoclax to HMAs is associated with potent myelosuppression because of the bone marrow dysplasia and cytopenias, thus needing further investigation of this combination in clinical trials.41 At the time being, a phase I and 2 phase II trials evaluating venetoclax in combination with AZA in treatment-naïve higher risk MDS (NCT02942290), AML elderly patients with no previous treatment (NCT03466294) and MRD+ patients after AML/MDS allogeneic hematopoietic stem cell transplantation (NCT04809181) are under investigation.46-48
Glasdegib is a potent oral inhibitor of the Hedgehog signaling pathway, producing rapid and complete tumor regression as a single agent or in combination with chemotherapy, reduced expression of key leukemia stem cell regulators, and decreased leukemia stem cell populations in patient-derived AML cells.49-51
Lenalidomide is widely used in MDS. The combination of lenalidomide and AZA has significant clinical activity and can induce cytogenetic responses in patients with del(5q). However, controlled studies have failed to show a clinical benefit of adding lenalidomide to azacytidine in mixed cohorts of all cytogenetic subtypes. Guadecitabine is a novel azanucleoside, with clinical activity in refractory patients but the usefulness of this drug needs to be investigated in larger trials.52,53
Immune checkpoint inhibitors as monotherapy seem to have modest clinical activity in MDS, although there are limited published data.54,55 Studies have evaluated the clinical usefulness of combining AZA with immune checkpoint inhibitors.56
Midostaurin, enasidenib, and ivosidenib, 3 kinase inhibitors that are orally bioavailable, were approved in 2017 and 2018 for the treatment of AML. Midostaurin has been approved for targeting the FLT3-mutated AML combined with the “7 plus 3” regimen.57 Gilteritinib, a small molecule type I FLT3 inhibitor is being studied both in newly diagnosed AML and in refractory or relapsed AML. It was found to have efficacy alone or in combination with chemotherapy or azacytidine, compared with azacytidine as monotherapy.58 Enasidenib (isocitrate dehydrogenase 2 [IDH2] inhibitor) and ivosidenib (IDH1 inhibitor) have been approved for the treatment of adults with IDH1- or IDH2-mutated relapsed/refractory AML.59,60 Moreover, BCR-ABL1 kinase inhibitors have been approved for targeting the rare subtype of BCR-ABL1+ AML. Published preclinical data regarding the development of PARPis are listed and analyzed in Table 1.
Preclinical data on the development of PARPi
| Reference | Genetics/studied parameters | Disease | Sensitivity to PARPi/results |
|---|---|---|---|
| Esposito et al, 2015 | Synthetic lethality of oncogenic transcription for leukemia treatment | AML | Sensitivity to PARPis of AML cells with low expression of members of the DDR pathway. AML cells driven by repressive transcription factors, including AML1-ETO and PML-RARα fusion oncoproteins, are sensitive to PARPis. Sensitivity to PARPis of AML cells with low expression of Rad51, ATM, BRCA1, and BRCA2. Genetic or pharmacological inhibition of HOXA9 impairs DDR and sensitizes MML leukemia to PARPis. |
| Zampieri et al, 2009 | DNA methylation | Ovarian cancer, breast cancer | PARPis with HMAs lead to synergistic inhibition. Inactivation and trapping of DNMT to DNA facilitates the role of BER machinery. |
| Lord et al, 2017 | Synthetic lethality, DNA repair | Ovarian cancer, breast cancer | PARPis trap PARP1 on DNA, preventing autoPARylation and PARP1 release from the site of damage of BRCA-mutant cells. |
| Boussios et al, 2012 | Synthetic lethality, DNA repair | Ovarian cancer, breast cancer | Tumors carrying mutations in BRCA1/2 implicated in homologous repair deficiency are particularly sensitive to PARPis. |
| Meng et al, 2014 | Apoptosis, knockdown of PARP1 and/or PARP2 | AML | Synergistic action of PARPi with death ligands results in enhanced expression of DR5 and Fas and sensitivity to treatment with multiple death ligands (agonistic anti-Fas antibody, recombinant human TRAIL, and agonistic anti-DR5 antibody). |
| Faraoni et al, 2018 | Apoptosis resistance, modulation of FAS and TRAIL receptors | AML | AML ΒΜ samples express FAS and DR5 transcripts at lower levels than normal BM. Apoptosis triggered by olaparib is associated with a dose-dependent up-regulation |
| Maifrede et al, 2017 | PARP1 knockdown | AML bearing MLL translocations | inhibitors of PARP1 enhance the therapeutic effect of cytotoxic drugs against MLL leukemias. |
| Molenaar et al, 2018 | Correlation of IDH1/IDH2 mutations to DNA damage and responses to PARPis | AML | IDH1/2MUT cells are sensitive to PARPi as monotherapy or/and in combination with DNA-damaging agents. Concomitant administration of IDH1/2MUT inhibitors during cytotoxic therapy decrease the efficacy of both agents in IDH1/2MUT AML. |
| Faraoni et al, 2014 | Αpoptosis, in vitro sensitivity to olaparib | AML | Olaparib induced cell death in the majority of AML samples (88%) and tested cell lines. Olaparib preferentially killed leukemic blasts and did not affect the viability of normal BM and CD34− peripheral blood cells. |
| Nieborowska-Skorska et al, 2017 | DNA repair | MPN | PARPi combination with ruxolitinib-mediated inhibition of DSB repair and/or hydroxyurea causes accumulation of lethal DSBs, resulting in elimination of MPN cells. |
| Patel et al, 2019 | DNA repair, genomic instability | MPN | In veliparib and busulfan treated SET2 and HEL cells, veliparib decreased busulfan’s IC50. Combination treatment of SET2 cells caused G2M arrest in 53% of cells, compared with 30% with veliparib alone and 35% with busulfan alone. |
| Muvarak et al, 2016 | DNA damage-related binding between DNMTs and PARP1 | AML, breast cancer | Combining DNMTi and PARPi (talazoparib) increases tight binding of PARP1 in chromatin, frequency of DSBs, and synergistic cytotoxicity while it decreases clonogenicity |
| Zhao et al, 2017 | Synthetic lethality | AML driven by MLL fusion proteins | Combining olaparib with DNMT inhibitor induce cell-cycle block and apoptosis. Olaparib can sensitize MLL leukemic cells to both DNMT inhibitors and chemotherapy agents. |
| Reference | Genetics/studied parameters | Disease | Sensitivity to PARPi/results |
|---|---|---|---|
| Esposito et al, 2015 | Synthetic lethality of oncogenic transcription for leukemia treatment | AML | Sensitivity to PARPis of AML cells with low expression of members of the DDR pathway. AML cells driven by repressive transcription factors, including AML1-ETO and PML-RARα fusion oncoproteins, are sensitive to PARPis. Sensitivity to PARPis of AML cells with low expression of Rad51, ATM, BRCA1, and BRCA2. Genetic or pharmacological inhibition of HOXA9 impairs DDR and sensitizes MML leukemia to PARPis. |
| Zampieri et al, 2009 | DNA methylation | Ovarian cancer, breast cancer | PARPis with HMAs lead to synergistic inhibition. Inactivation and trapping of DNMT to DNA facilitates the role of BER machinery. |
| Lord et al, 2017 | Synthetic lethality, DNA repair | Ovarian cancer, breast cancer | PARPis trap PARP1 on DNA, preventing autoPARylation and PARP1 release from the site of damage of BRCA-mutant cells. |
| Boussios et al, 2012 | Synthetic lethality, DNA repair | Ovarian cancer, breast cancer | Tumors carrying mutations in BRCA1/2 implicated in homologous repair deficiency are particularly sensitive to PARPis. |
| Meng et al, 2014 | Apoptosis, knockdown of PARP1 and/or PARP2 | AML | Synergistic action of PARPi with death ligands results in enhanced expression of DR5 and Fas and sensitivity to treatment with multiple death ligands (agonistic anti-Fas antibody, recombinant human TRAIL, and agonistic anti-DR5 antibody). |
| Faraoni et al, 2018 | Apoptosis resistance, modulation of FAS and TRAIL receptors | AML | AML ΒΜ samples express FAS and DR5 transcripts at lower levels than normal BM. Apoptosis triggered by olaparib is associated with a dose-dependent up-regulation |
| Maifrede et al, 2017 | PARP1 knockdown | AML bearing MLL translocations | inhibitors of PARP1 enhance the therapeutic effect of cytotoxic drugs against MLL leukemias. |
| Molenaar et al, 2018 | Correlation of IDH1/IDH2 mutations to DNA damage and responses to PARPis | AML | IDH1/2MUT cells are sensitive to PARPi as monotherapy or/and in combination with DNA-damaging agents. Concomitant administration of IDH1/2MUT inhibitors during cytotoxic therapy decrease the efficacy of both agents in IDH1/2MUT AML. |
| Faraoni et al, 2014 | Αpoptosis, in vitro sensitivity to olaparib | AML | Olaparib induced cell death in the majority of AML samples (88%) and tested cell lines. Olaparib preferentially killed leukemic blasts and did not affect the viability of normal BM and CD34− peripheral blood cells. |
| Nieborowska-Skorska et al, 2017 | DNA repair | MPN | PARPi combination with ruxolitinib-mediated inhibition of DSB repair and/or hydroxyurea causes accumulation of lethal DSBs, resulting in elimination of MPN cells. |
| Patel et al, 2019 | DNA repair, genomic instability | MPN | In veliparib and busulfan treated SET2 and HEL cells, veliparib decreased busulfan’s IC50. Combination treatment of SET2 cells caused G2M arrest in 53% of cells, compared with 30% with veliparib alone and 35% with busulfan alone. |
| Muvarak et al, 2016 | DNA damage-related binding between DNMTs and PARP1 | AML, breast cancer | Combining DNMTi and PARPi (talazoparib) increases tight binding of PARP1 in chromatin, frequency of DSBs, and synergistic cytotoxicity while it decreases clonogenicity |
| Zhao et al, 2017 | Synthetic lethality | AML driven by MLL fusion proteins | Combining olaparib with DNMT inhibitor induce cell-cycle block and apoptosis. Olaparib can sensitize MLL leukemic cells to both DNMT inhibitors and chemotherapy agents. |
AML-ETO, acute myeloid leukemia-eight twenty-one oncoprotein; ATM, ataxia-telangiectasia mutated; BER, base excision repair; BM, bone marrow; DR5, death receptor 5; FAS, FS-7-associated surface antigen; HEL, hevein-like preprotein; IC50, half-maximal inhibitory concentration; MML, myelomastocytic leukemia; PML-RARα, promyelocytic leukemia/retinoic acid receptor α; TRAIL, tumor necrosis factor-related apoptosis inducing ligand; SET2, nucleosomal histone H3-selective methyltransferase.
PARPis
Synthetic lethal targeted therapy
Almost 15 years ago, 2 groups described the “synthetic lethal (SL) interaction” between PARP inhibition and BRCA1 or BRCA2 mutations, suggesting a new treatment of patients with BRCA-mutant tumors.61,62 SL as a concept was initially introduced a century ago by geneticists to describe the situation in which a defect in either 1 of 2 genes has little impact on the cell/organism, but the combination of defects in both genes results in death.63 It is quite important to achieve a better understanding of what causes/triggers an SL interaction, which are the factors that determine the robustness of SL interactions, and how these interactions can be predicted. For example, it has been suggested that protein–protein interaction networks are used to identify robust SL interaction effects associated with different pairs of genes.64 Moreover, proteins with similar functions seem to share SL interactions, leading to computational approaches that identify SL relationships.65
Clinical development of PARPi
PARPis are small molecules that compete with the oxidized form of nicotinamide adenine dinucleotide (NAD+) at the catalytic pocket of PARPs66 (mainly PARP1 and PARP2) (Figure 2), resulting in the inhibition of DNA repair enzymatic activities regulated by PARylation.67 Some PARPis can trap PARP1 and PARP2 on the so-called endogenous DNA breaks.68,69 Currently, PARPis such as olaparib, rucaparib, and niraparib are approved by the FDA and the European Medicine Agency for the treatment of ovarian cancer, whereas veliparib is in the latest stage of clinical development.70 Talazoparib was approved by the FDA for the treatment of metastatic germline BRCA1/2-mutated breast cancer in October 2018. They have been studied both as single agents and in combination with chemotherapy, antiangiogenic agents, and ionizing radiation.71 The PARP trapping depends on a toxic allosteric effect that varies among PARPis (talazoparib > niraparib > olaparib = rucaparib > veliparib).72 The mechanisms involved in PARPi cytotoxicity vary depending on the type of the defective DNA repair pathway and on the status of tumor cell proliferation.56
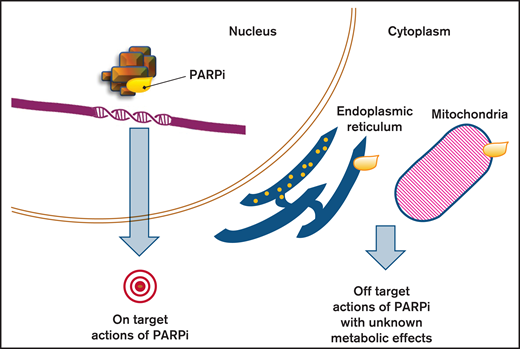
Effects of PARP inhibition. PARPis are able to create their own interaction network with proteins outside the nucleus, beside the sole blockage of PARP (on target action of PARPis). By inducing signaling pathways and impacting secondary proteins, they can affect cell functions and cause metabolic responses that constitute the off-target actions of PARPi.
Effects of PARP inhibition. PARPis are able to create their own interaction network with proteins outside the nucleus, beside the sole blockage of PARP (on target action of PARPis). By inducing signaling pathways and impacting secondary proteins, they can affect cell functions and cause metabolic responses that constitute the off-target actions of PARPi.
In addition to DNA repair, PARPs are able to control cellular functions that are important for tumor survival by regulating the chromatin structure and modifying the signaling molecules that interfere with gene transcription.73-77 Studies performed in primary AML blasts78 and tumor cell lines79,80 revealed that the PARPi cytotoxic effect could be due to upregulation of death receptor 5 (DR5) and FS-7-associated surface antigen (FAS) death receptors that respectively requires the activation of SP1 and NF-KB. As a result, leukemia cells are sensitized to produce endogenously Fas ligand (FASL) and TNF-related apoptosis inducing ligand (TRAIL). In conclusion, PARPis can cause tumor cytotoxicity via several pathways and mechanisms that may be different from those associated with DNA damage sensing and repair.81
The clinical studies initially focused on hereditary tumors such as breast, prostate, ovarian, and pancreatic cancers. These tumors are associated with pathogenic mutations in the BRCA genes.82-84 Olaparib was approved in 2014 as maintenance therapy for recurrent, BRCA-mutated ovarian cancer, or for the treatment of BRCA-mutated advanced ovarian cancer (after 3 or more lines of chemotherapy).85 In 2018, olaparib was finally approved by the FDA in first-line maintenance setting of BRCA-mutated advanced ovarian cancer. Niraparib and rucaparib have shown promising results when used as maintenance treatment of recurrent platinum-sensitive ovarian cancer after completion of platinum-based chemotherapy.86,87
Olaparib and talazoparib were recently approved (2018) for HER2-negative metastatic breast cancer in patients with deleterious/suspected deleterious germline mutations of BRCA genes (after at least 2 lines of chemotherapy).88,89 Some other PARPis that are being investigated in clinical trials are veliparib, pamiparib, fluzoparib, and 2X-121.90-92
PARPi activity as monotherapy in AML and MDS
Almost a decade ago, it was suggested that PARPi therapy could be beneficial for MDS and AML. More specifically, microsatellite instability was associated with downregulation of HR repair genes in MDS/AML. In fact, mutations in CtIP (high-risk MDS) and in CtIP and meiotic recombination protein 11 (primary AML samples) had as a result the leukemia cells to be sensitive to PARPis.93 Additionally, it was reported that samples from myeloproliferative neoplasms (MPN) patients, with a tendency to evolve to AML, were more sensitive to olaparib and veliparib compared with normal bone marrow cells. Additionally, myeloblasts with nonmutated JAK2 were more sensitive to PARPi than JAK2-V617F mutated samples.94 This effect was the result of JAK2-V617F ability to activate transcription through signal transducer and activator of transcription and to inhibit apoptosis.95
The t(8;21) (RUNX1-RUNX1T1) is a repeated chromosomal translocation in AML. A relevant study showed that transformed RUNX1-RUNX1T1 primary hematopoietic cells (mice) were very sensitive to olaparib and veliparib compared with bone marrow cells. PARPi had little effect in the presence of the t(9;11) (p21; q23)(mixed lineage leukemia [MLL]-MLLT3≈MLL-AF9) and t(1;19)(TCF3-PBX1≈E2A-PBX) translocation.12 Another study demonstrated that olaparib inhibited the proliferation and quiescence of MLL-AF9 AML stem cells proposing that it is possible for PARPi to be used for MLL-rearranged leukemia.96
IDH1/2 mutations in primary AML cells demonstrated induction of HR defects and decrease in ATM expression that renders AML cells sensitive to PARPi. The IDH1/2 inhibitors can protect cells against PARPi because they restore ATM expression and decrease DNA damage. Thus, combining PARPi with IDH1/2 inhibitors is better to be avoided in IDH1/2 mutated AML.97
The levels of PARP1 protein were considered to affect tumor response to PARPis. PARP1 and PARP298-100 were studied in primary AML and MDS.101,102 However, the expression of PARP1/2 was not directly associated with sensitivity to PARPis. High levels of the MPL proto-oncogene thrombopoietin receptor (upregulated in RUNX1/RUNXT1 AML) correlate with PARP expression.87,103 MPL stimulation by thrombopoietin results in the activation of AKT1 and ERK1/2 pathways leading to proliferation and apoptosis resistance of leukemia cells.83 PARP1 is an important mediator of translocation common in AML (via the Alt-EJ pathway).104 However, because data for MDS and AML treated with PARPi are still scarce, some concerns have arisen regarding delayed adverse events from PARPi. An observational clinical study (NCT04326023) with 178 enrolled patients, using the VigiBase database, tried to investigate potent adverse events related to PARPis. It was observed that PARPis increased the risk of MDS and AML vs placebo treatment, suggesting that these adverse events need further evaluation.105 Currently, a phase II clinical trial (NCT03953898) compares the efficacy of olaparib in IDH mutated MDS and refractory/relapsed (R/R) AML patients to that of standard chemotherapy.106 Alongside, an ongoing phase I pilot study (NCT03974217) examines if mutations in the cohesin complex can represent a therapeutic target for talazoparib, a new experimental drug, in this subset of patients.107 Results of clinical trials on the use of PARPis in several malignancies are presented and analyzed in Table 2.
Results from clinical trials of PARPi in several malignancies
| Clinical trial (EudraCT number)/reference | Disease | Therapy | Potential mechanism | Results |
|---|---|---|---|---|
| NCT00753545 L edermann et al, 2014 | Ovarian cancer | Olaparib 400 mg BID vs placebo | Synthetic lethality DNA repair | BRCA+: median PFS significantly longer in the olaparib group OS not significantly different between groups Serious adverse events reported in 25 (18%) patients under olaparib and 11 (9%) under placebo. |
| NCT01891344 Swisher et al, 2017 | Ovarian cancer | Rucaparib 600 mg PO, BID | Homologous recombination deficiency DNA methylation Genomic LOH | BRCA+: PFS longer in high LOH Grade ≥3 treatment-related adverse events: anemia (45 [22%] patients) and elevations in ALT/AST (25 [12%]) |
| NCT01618136 Plummer et al, 2020 | Ovarian cancer, B-cell malignancies, malignant solid tumors, triple-negative breast cancer, advanced melanoma | E7449 50-800 mg QD, PO E7449 plus TMZ QD, PO E7449 plus carboplatin and paclitaxel QD, PO | PARP-DNA trapping Synthetic lethality | Antitumor activity of E7449 in 13 patients, durable in 8. The 2X-121 DRP identified patients achieving PR and durable SD. E7449: good tolerability, promising antitumor activity and significant concentration-dependent PARP inhibition. |
| NCT043260230 Morice et al, 2020 | MDS, AML (review of randomized controlled trials) | Olaparib, rucaparib, niraparib, talazoparib, veliparib | PARPis significantly increased the risk of MDS and AML compared with placebo treatment with no between-study heterogeneity. | |
| NCT03953898 Reference number106 | R/R AML, MDS | Olaparib | Inhibition of cancer cell growth by blocking enzymes needed for cell growth. | No results Still recruiting |
| NCT03974217 Reference number107 | Leukemia | Talazoparib | Synthetic lethality (leukemia cells with a mutation in cohesin may be dependent on PARP activity to survive; when inhibiting PARP with talazoparib the leukemia cells die) | No results Still recruiting |
| NCT00588991 Reference number122 | R/R AML, high-risk MDS, aggressive myeloproliferative disorder | Veliparib Carboplatin Topotecan Hydrochloride | DNA repair cytotoxicity of multiple classes of chemotherapy drugs, including topoisomerase I inhibitors and platinating agents. | No results/not recruiting Veliparib/topotecan/carboplatin combination warrants further investigation |
| NCT04207190 Reference number123 | R/R AML | Gemtuzumab- ozogamicin, talazoparib, talazoparib tosylate | PARP1 trapping Potential ability of talazoparib to enhance levels of DNA damage induced by GO therapy. | Still recruiting |
| NCT02878785 Reference number124 | Untreated AML, R/R AML | Decitabine, talazoparib | SSB repair | Active, not recruiting Suggestion that talazoparib will increase the effects of decitabine in leukemia cells |
| Clinical trial (EudraCT number)/reference | Disease | Therapy | Potential mechanism | Results |
|---|---|---|---|---|
| NCT00753545 L edermann et al, 2014 | Ovarian cancer | Olaparib 400 mg BID vs placebo | Synthetic lethality DNA repair | BRCA+: median PFS significantly longer in the olaparib group OS not significantly different between groups Serious adverse events reported in 25 (18%) patients under olaparib and 11 (9%) under placebo. |
| NCT01891344 Swisher et al, 2017 | Ovarian cancer | Rucaparib 600 mg PO, BID | Homologous recombination deficiency DNA methylation Genomic LOH | BRCA+: PFS longer in high LOH Grade ≥3 treatment-related adverse events: anemia (45 [22%] patients) and elevations in ALT/AST (25 [12%]) |
| NCT01618136 Plummer et al, 2020 | Ovarian cancer, B-cell malignancies, malignant solid tumors, triple-negative breast cancer, advanced melanoma | E7449 50-800 mg QD, PO E7449 plus TMZ QD, PO E7449 plus carboplatin and paclitaxel QD, PO | PARP-DNA trapping Synthetic lethality | Antitumor activity of E7449 in 13 patients, durable in 8. The 2X-121 DRP identified patients achieving PR and durable SD. E7449: good tolerability, promising antitumor activity and significant concentration-dependent PARP inhibition. |
| NCT043260230 Morice et al, 2020 | MDS, AML (review of randomized controlled trials) | Olaparib, rucaparib, niraparib, talazoparib, veliparib | PARPis significantly increased the risk of MDS and AML compared with placebo treatment with no between-study heterogeneity. | |
| NCT03953898 Reference number106 | R/R AML, MDS | Olaparib | Inhibition of cancer cell growth by blocking enzymes needed for cell growth. | No results Still recruiting |
| NCT03974217 Reference number107 | Leukemia | Talazoparib | Synthetic lethality (leukemia cells with a mutation in cohesin may be dependent on PARP activity to survive; when inhibiting PARP with talazoparib the leukemia cells die) | No results Still recruiting |
| NCT00588991 Reference number122 | R/R AML, high-risk MDS, aggressive myeloproliferative disorder | Veliparib Carboplatin Topotecan Hydrochloride | DNA repair cytotoxicity of multiple classes of chemotherapy drugs, including topoisomerase I inhibitors and platinating agents. | No results/not recruiting Veliparib/topotecan/carboplatin combination warrants further investigation |
| NCT04207190 Reference number123 | R/R AML | Gemtuzumab- ozogamicin, talazoparib, talazoparib tosylate | PARP1 trapping Potential ability of talazoparib to enhance levels of DNA damage induced by GO therapy. | Still recruiting |
| NCT02878785 Reference number124 | Untreated AML, R/R AML | Decitabine, talazoparib | SSB repair | Active, not recruiting Suggestion that talazoparib will increase the effects of decitabine in leukemia cells |
BID, twice per day; DRP, drug response predictor; EudraCT, European Union Drug Regulating Authorities Clinical Trials Database; GO, gemtuzumab ozogamicin; LOH, loss of heterozygosity; OS, overall survival; PFS, progression-free survival; PO (per os), oral administration; PR, partial response; QD, once a day; SD, stable disease.
Candidate biomarkers of sensitivity to PARPi
Finally, special attention has been devoted to the discovery of predictive biomarkers focusing on the identification of patients that are likely to benefit from PARPi. Knowledge of the homologous recombination deficiency (HRD) status seems to be crucial for treatment decision. PARPis selectively target HRD, especially in tumors from patients bearing the deleterious mutations of BRCA1 and 2. Up to 55% of sporadic epithelial ovarian cancer are homologous recombination repair-deficient for several reasons, including BRCA1 promoter methylation, meiotic recombination protein 11 mutation and BRCA2-interacting transcriptional suppressor overexpression.108,109 Reliable candidate biomarkers for the identification of patients with defects in the HR pathway would expedite the clinical development of PARPi-based therapies. BRCA1/2 loss-of-function mutation impairs homologous recombination repair and induces PARP hyperactivation reflected by an increased abundance of PARs.110,111 HRD may occur without BRCA mutation in the context of “BRCAness.”112 BRCA2-interacting transcriptional suppressor genes and ETS fusion genes that are reported in different tumors inhibit BRCA2.111,113 In ovarian cancer, the importance of platinum sensitivity, which seems to be one of the most credible biomarkers is distinct, but because this information is not to our knowledge at the start of chemotherapy, we need to rely more on the HRD and BRCA status to identify patients that are likely to respond to PARPis.114 In AML, despite the important efforts in understanding the molecular basis of the disease, there are still more to be elucidated. A study performed in AML cell lines observed a direct correlation of RAD51 and an inverse correlation of γH2AX with olaparib half-maximal inhibitory concentration.100 Additionally, the PARPi caused a dose-dependent increase in the number of γH2AX+ cells, which suggested a possible sensitivity to olaparib.100 These data support the role of the repair proteins RAD51 and γH2AX as potential markers of sensitivity of AML cells to olaparib. In conclusion, multiple molecular signatures in combination with a personalized approach should be considered for the prediction of patients who may benefit from PARPi therapy.
PARPi-based combination therapies
Combination therapies evaluate the effects of PARPi interaction with agents that target genetic alterations present in AML/MDS and MPN. Agents with lower toxicity in comparison with chemotherapy are better to be combined with PARPis. The JAK2-V617F mutation is known for unrestrained HR activity resulting in genetic instability. It is the most frequent genetic alteration in MPN and ruxolitinib (JAK1/2 inhibitor) has been approved for the treatment myelofibrosis and polycythemia vera in patients resistant to hydroxyurea.115 Ruxolitinib treatment sensitizes MPN cells to SL caused by talazoparib.116 The JAK2-V617F MPN model was also studied for the combination of a PARPi with busulfan (component of regimens of allo-HSCT).117 It was assumed that combining busulfan with a PARPi might lead to increased efficacy. More precisely, when busulfan and veliparib were combined, the result was synergistic cytotoxicity in mutated MPN cells.118
FLT3-ITD-mutated AML is characterized by increased genomic instability (through Alt-EJ repair factors, LIG3, and PARP1), associated with induction of MYC expression.119-121 Additionally, when FLT3-ITD cells are mutated, they present an increase in ROS production and evidence of interchromosomal HR which may result in loss of heterozygosity, which is common in myeloid malignancies.122,123 When quizartinib, a tyrosine kinase inhibitor, inhibits FLT3-ITD, there is a decrease in HR and NHEJ protein expression, an increase in DSB formation and sensitization of FLT3-ITD+ AML cells to olaparib and talazoparib. More precisely, although quizartinib mainly exhibits cytostatic effects in proliferating cells,124 PARPis exhibit cytotoxic effects in NHEJ-deficient AML cells. This is an important conclusion, because therapeutic options for AML usually fail to eliminate quiescent drug-refractory leukemia stem cells. As a result, many patients relapse after allo-HSCT.125
Another case of combination therapy is PARPi synergizing with the AZA and decitabine used in MDS and AML. In this case, there is reactivation of hypermethylated tumor suppressor genes involved in the differentiation, transformation, and induction of DNA damage. Some groups have shown that low doses of 5-azacitidine or decitabine together with talazoparib or olaparib have as a result synergistic cytotoxicity in AML cells (because of increased DNA damage and delayed DNA repair).126,127
Finally, with the PARPis being under investigation in vitro, in vivo, and in clinical trials as monotherapy or in combination with chemotherapy, progress in their development has led to a better understanding of the contribution of different mechanisms of action and to improvement of their therapeutic potential.128 However, because of the concerning potential adverse events associated with MDS/AML, especially with the BRCA-mutated patients, further investigations should be carried out for prolonged toxicity. An active phase I clinical trial (NCT00588991) is trying to study the side effects and best dose of veliparib combined with chemotherapy in patients with high-risk MDS, R/R AML, or aggressive MPN to address the optimal inhibition of cancer cell growth.129 To date, talazoparib is being studied in a phase I/Ib clinical trial in combination with gemtuzumab ozogamicin in CD33+ patients with R/R AML, to assess if the addition of talazoparib in the existing therapy increases its efficacy.130 In a phase II interventional trial (NCT02878785), talazoparib is given in combination with decitabine, to find out if these 2 agents work better together than decitabine alone for the treatment of AML.131
Conclusion
To date, thanks to novel technologies, accumulating facts from preclinical studies point out PARPi as promising therapies entering many clinical trials. Because of the different profiles of each agent, patient-specific factors should be taken into consideration when deciding which inhibitor to use as the preferable option.132 Research and development of novel targets related to other PARP family members may also contribute to the revealing of new drugs and the further comprehension of the biological and clinical role of PARPi.93 A recent study suggested that the positive correlation of PARP1, PARP2, PARP3, and TRPM2 genes in physiological cells, is disturbed in patients with AML.133 Consequently, the need for further research of the mutual expression and regulation of different PARP family members is mandatory to identify possible biomarkers, provide more perspectives for optimal PARPi-based combination outline, and outspread their therapeutic outlook.
Authorship
Contribution: All authors participated in designing the review article; C.-N.K. and D.T. participated in drafting the manuscript; P.T.D. and N.-A.V. contributed in the critical review of the article; and all authors have read and approved the manuscript.
Conflict-of-interest disclosure: P.T.D. reports personal fees for presentations for Roche, Celgene, and Novartis. N.-A.V. reports investigational grants and personal fees for presentations and advisory roles from Celgene/Genesis Pharma. The remaining authors declare no competing financial interests.
Correspondence: Christina-Nefeli Kontandreopoulou, Hematology Unit, First Department of Internal Medicine, National and Kapodistrian University of Athens, “Laikon” General Hospital, Athens 11527, Greece; e-mail: elina_knt@hotmail.com.

