

Key Points
HAVCR2Y82C mutation was found in 51% of SPTCL cases and was associated with younger age, systemic illness, and shorter RFS.
HAVCR2Y82C SPTCLs were enriched in inflammatory signaling, and HAVCR2WT SPTCLs showed higher CCR4 expression in the microenvironment.

Abstract
Recent studies identified germline mutations in HAVCR2 (encoding T-cell immunoglobulin mucin 3) as a genetic factor that predisposes to subcutaneous panniculitis-like T-cell lymphoma (SPTCL). However, the differences between HAVCR2-mutated (HAVCR2MUT) and HAVCR2 wild-type (HAVCR2WT) SPTCLs remain unclear. A nationwide cohort of 53 patients with SPTCL diagnosed at 8 Korean institutions was established. Whole-exome sequencing and RNA-sequencing were performed on 8 patients in the discovery set. In the validation set, targeted gene sequencing or direct sequencing of HAVCR2 was performed. Of 49 patients with available HAVCR2 status, 25 (51.0%) were HAVCR2Y82C. HAVCR2Y82C was associated with younger age (P = .001), development of hemophagocytic lymphohistiocytosis or hemophagocytic lymphohistiocytosis–like systemic illness (P < .001), and short relapse-free survival (RFS) (P = .023). Most mutated genes in SPTCLs were involved in immune responses, epigenetic modifications, and cell signaling. Mutations in UNC13D, PIAS3, and KMT2D were more frequent in HAVCR2WT SPTCLs. At the gene expression level, HAVCR2Y82C SPTCLs were enriched in genes involved in IL6-JAK-STAT3 signaling and in tumor necrosis factor-α signaling via NF-κB. CCR4 was significantly upregulated in HAVCR2WT SPTCLs both at the messenger RNA level and at the protein level. We established a risk stratification system for SPTCL by integrating clinical and histopathological features, including age and HAVCR2 mutation status. This risk stratification system was strongly associated with RFS (P = .031). In conclusion, the HAVCR2Y82C mutation was common in Korean patients with SPTCL and was associated with unique clinicopathological and genetic features. Combining clinicopathological parameters could aid in predicting prognosis for patients with SPTCL.
Introduction
Subcutaneous panniculitis-like T-cell lymphoma (SPTCL) is a rare T-cell non-Hodgkin lymphoma (NHL) with a cytotoxic phenotype. SPTCLs account for <1% of all NHLs worldwide1 and 0.3% of all malignant lymphomas in the Republic of Korea.2 SPTCL typically affects young individuals, with a median patient age of 36 years and a female sex bias.3 Histopathologically, SPTCL is characterized by CD8-positive T cells infiltrating into subcutaneous adipose tissue, with rimmed individual fat cells in a lace-like pattern. Differential diagnoses include lupus panniculitis (LP) and other T-cell NHLs with cutaneous involvement, such as primary cutaneous γδ T-cell lymphoma (PCGDTCL) and mycosis fungoides (MF).4 Patients with SPTCL have an excellent prognosis, with a 5-year overall survival (OS) rate of >80%; however, ∼20% of patients with SPTCL develop hemophagocytic lymphohistiocytosis (HLH), which can significantly affect patient survival (5-year OS of 46%).3
Recent genetic studies revealed that recurrent germline mutations in HAVCR2 were present in 25% to 85% of patients with SPTCLs.5-7HAVCR2 encodes T-cell immunoglobulin mucin 3 (TIM-3) protein, a critical checkpoint molecule that regulates inflammatory responses.8 Y82C, I97M, and T101I are common recurrent germline HAVCR2 mutations in SPTCLs, impairing TIM-3 protein folding, cellular expression, and function.5 Defective TIM-3 leads to persistent inflammatory responses and potentially causes HLH. Compared with wild-type HAVCR2 (HAVCR2WT) SPTCLs, HAVCR2-mutated (HAVCR2MUT) SPTCLs are more frequent in younger individuals and are often associated with HLH; nevertheless, these findings could not be confirmed in a cohort of 13 Asian subjects with SPTCLs.6
Although identification of germline mutations that cause SPTCLs provided a deeper insight into the mechanisms underlying SPTCLs, their pathogenesis has not been fully elucidated. Except for HAVCR2 mutations, no recurrent genetic alterations have been associated with SPTCLs, and the biological mechanisms underlying HAVCR2WT SPTCLs remain largely unknown. In addition, it is unclear whether HAVCR2 alterations are specific to SPTCLs or if they are also found in other diseases with panniculitic presentation.
To address these unresolved questions, we established a nationwide multicenter cohort composed of Korean patients with SPTCLs and performed the first genetic study on this population. After a comprehensive review of the clinical and histopathological features, we investigated the mutational spectrum of SPTCLs using whole-exome sequencing (WES), targeted DNA sequencing, and direct sequencing. We compared the mutational patterns of SPTCLs according to HAVCR2 status and performed RNA-sequencing–based gene expression analysis to identify dysregulated pathways and biological differences between HAVCR2MUT and HAVCR2WT SPTCLs, followed by validation on the clinical tissue samples.
Methods
Patients and samples
A nationwide multicenter cohort consisting of 53 patients with SPTCL was established (supplemental Table 1); the patients diagnosed at 8 Korean institutions between 1995 and 2020 were screened, and those with available tissue samples for the study were included. Seven patients with LP, 4 patients with PCGDTCL, and 8 patients with MF diagnosed at Seoul National University Hospital (SNUH) were also included; among them, all patients with LP and 3 patients with PCGDTCL exhibited panniculitis-like presentation. All cases were reviewed and diagnosed by experienced hematopathologists in each institute using the revised fourth World Health Organization classification guidelines.1 Available hematoxylin and eosin slides, immunohistochemistry (IHC) slides (CD3, CD20, CD4, CD8, CD56, granzyme B, TIA-1, Ki-67, T-cell receptor βF1 [TCRβF1], and TCRγ), and Epstein-Barr virus in situ hybridization samples were reviewed by an experienced hematopathologist at SNUH. Clinical information, including bone marrow involvement, HLH or HLH-like systemic illness, relapse-free survival (RFS), OS, and treatment regimens, were collected from the medical records of each institution. HLH was defined according to the HLH-2004 criteria.9 However, not all institutes were able to run every laboratory test listed in HLH-2004, and thus we designated “HLH-like systemic illness” for those with incomplete criteria for HLH-2004 but clinically regarded as HLH warranting intensive treatment (supplemental Table 2).
This study was approved by the Institutional Review Board of SNUH (approval no. 1809-143-977).
WES and targeted sequencing
WES was performed by using 9 formalin-fixed, paraffin-embedded samples from 8 patients with SPTCLs, including 1 patient with disease recurrence (patient SP03); all these patients were diagnosed at SNUH (discovery set). Matched non-neoplastic tissue samples were available from 2 patients (SP01 and SP04). Sequencing metrics are summarized in supplemental Table 3.
To evaluate the mutational landscape of SPTCLs, MFs, PCGDTCLs, and LPs, we created a customized panel comprising 208 genes (supplemental Table 4) based on the following criteria: genes with mutations found in >2 patients in the discovery set of this study or previously reported studies on SPTCL5,6,10,11 (eg, HAVCR2, PIAS3, PLCG2); genes with mutations found in at least 1 patient in the discovery set and known to have functional implications in inflammatory responses or T-cell biology (eg, IFNL2, F5, GDF1); genes with mutations previously reported in CTCLs12 ; and other genes that affect the pathogenesis of lymphoid neoplasms (eg, RHOA, TET2, MYD88). Targeted gene sequencing (TGS) was performed for a total of 32 patients: 20 patients with SPTCL, 8 with MF, 3 with PCGDTCL, and 1 with LP. Details are provided in the supplemental Methods and supplemental Table 5.
RNA-sequencing and gene expression analysis
RNA-sequencing was performed on 8 samples in the discovery set (supplemental Table 6); 4 were HAVCR2Y82C, and 4 were HAVCR2WT. Details are provided in the supplemental Methods.
Genes with median transcripts per million values of <5 were excluded from further analysis. Gene set enrichment analysis (GSEA)13 between the HAVCR2Y82C and HAVCR2WT groups was performed by gene set permutation due to the small sample size.14 Gene sets from MSigDB (http://software.broadinstitute.org/gsea/msigdb)15 and the SignatureDB collection (https://lymphochip.nih.gov/signaturedb/)16 were used. A cutoff false discovery rate (FDR) q-value ≤0.25 was used to define significant enrichment.
Next, we defined differentially expressed genes (DEGs) between the HAVCR2Y82C and HAVCR2WT groups. DESeq2 analysis17 was performed on the raw read count matrix after discarding genes with median read counts <5. Genes with an adjusted q-value <0.05 and a log2FC > 2.0 or < -2.0 were regarded as statistically significant.
Direct sequencing
For those patients who were not suitable for high-throughput sequencing, direct sequencing of HAVCR2 exon 2 was performed, covering all of the previously reported variants in patients with SPTCL (Y82C, I97M, T101I).5-7 Details are provided in the supplemental Methods and supplemental Table 7.
IHC and T-cell clonality test
IHC results were retrieved from the pathology report of each participating institution. During a central review process conducted by SNUH, immunostainings for TCRβF1, TCRγ, and T-cell clonality test were performed if necessary. To validate the findings from the gene expression analysis, IHC was performed for CCR4, Foxp3, and pSTAT3 on the 4-μm-thick whole sections of formalin-fixed, paraffin-embedded tissue samples. CCR4, Foxp3, and pSTAT3 immunostains were digitally scanned and quantified. The positivity for each marker was defined as the percentage of positive cells in the analyzed area. For the 7 selected cases, double-stainings for Foxp3/CCR4 and GATA3/CCR4 were performed. All detailed procedures and manufacturer information are provided in the supplemental Methods.
Statistical analysis
We used χ2, linear-by-linear, and Fisher’s exact tests to compare categorical variables and the Mann-Whitney U test to compare continuous variables, as appropriate. Survival analyses were performed by using the log-rank method, and statistical significance was defined as P < .05. All analyses were performed by using SPSS software (version 25; IBM SPSS Statistics, IBM Corporation, Armonk, NY) and R statistical package 3.6.0 (http://www.r-project.org; R Foundation for Statistical Computing, Vienna, Austria).
Results
Clinicopathological characteristics
The clinicopathological characteristics of 53 patients with SPTCLs are summarized in Table 1 and supplemental Table 1. The median age at diagnosis was 32 years (range, 8-74 years), and 37 (69.8%) patients were women. Fourteen (28.6%) of 49 patients developed HLH/HLH–like systemic illness, and 15 patients (30.6%) experienced disease relapse. Six (11.8%) of 51 patients died due to disease progression or disease-related complications. Treatment information was available for 43 patients: 33 patients (76.7%) received chemotherapy as first-line treatment, and 10 patients (23.3%) were treated with immunosuppressants. Patients with HLH/HLH–like systemic illness were more likely to be treated with immunosuppressants as first-line therapy (P = .017), and there was no differences in the therapeutic approach according to patient age.
Clinicopathological characteristics of SPTCLs according to HAVCR2 genotype
| Characteristic | WT | Y82C | Unknown | Total | P |
|---|---|---|---|---|---|
| Clinical features | |||||
| Age | |||||
| Median (range), y | 40 (16-74) | 26 (8-62) | 52 (35-68) | 32 (8-74) | .002* |
| <30 y | 6 (25.0%) | 18 (72.0%) | 0 (0.0%) | 24 (45.3%) | .001† |
| ≥30 y | 18 (75.0%) | 7 (28.0%) | 4 (100.0%) | 29 (54.7%) | |
| Sex | .404† | ||||
| Male | 6 (25.0%) | 9 (36.0%) | 1 (25.0%) | 16 (30.2%) | |
| Female | 18 (75.0%) | 16 (64.0%) | 3 (75.0%) | 37 (69.8%) | |
| HLH/HLH–like systemic illness | <.001† | ||||
| No | 22 (95.7%) | 11 (45.8%) | 2 (100.0%) | 35 (71.4%) | |
| Yes | 1 (4.3%) | 13 (54.2%) | 0 (0.0%) | 14 (28.6%) | |
| BM involvement | 1.000‡ | ||||
| Absent | 21 (100.0%) | 12 (95.7%) | 0 (0.0%) | 43 (93.5%) | |
| Present | 0 (0.0%) | 1 (4.3%) | 2 (100.0%) | 3 (6.5%) | |
| First-line treatments | .069‡ | ||||
| Chemotherapy | 16 (88.9%) | 14 (63.6%) | 3 (100.0%) | 33 (76.7%) | |
| Immunosuppressive | 2 (11.1%) | 8 (36.4%) | 0 (0.0%) | 10 (23.3%) | |
| HSCT§ | .070‡ | ||||
| No | 8 (80.0%) | 3 (33.3%) | 2 (100.0%) | 13 (61.9%) | |
| Yes | 2 (20.0%) | 6 (66.7%) | 0 (0.0%) | 8 (38.1%) | |
| Relapse | .345† | ||||
| No | 17 (73.9%) | 14 (60.9%) | 3 (100.0%) | 34 (69.4%) | |
| Yes | 6 (26.1%) | 9 (39.1%) | 0 (0.0%) | 15 (30.6%) | |
| 5-y RFS | .001* | ||||
| Median (range), m | 57 (1-60) | 11 (1-60) | 60 (16-60) | 30 (1-60) | |
| Death | 1.000‡ | ||||
| No | 21 (87.5%) | 21 (87.5%) | 3 (100.0%) | 45 (88.2%) | |
| Yes | 3 (12.5%) | 3 (12.5%) | 0 (0.0%) | 6 (11.8%) | |
| 5-y OS | .004* | ||||
| Median (range), m | 60 (2-60) | 25 (1-60) | 60 (16-60) | 39 (1-60) | |
| 5-y DSS | .003* | ||||
| Median (range), m | 59.5 (2-60) | 19 (1-60) | 60 (16-60) | 37 (1-60) | |
| Histopathological features | |||||
| Necrosis | .314† | ||||
| Absent | 14 (66.7%) | 13 (52.0%) | 1 (50.0%) | 28 (58.3%) | |
| Present | 7 (33.3%) | 12 (48.0%) | 1 (50.0%) | 20 (41.7%) | |
| Granulomatous inflammation | .037‡ | ||||
| Absent | 17 (81.0%) | 25 (100.0%) | 2 (100.0%) | 44 (91.7%) | |
| Present | 4 (19.0%) | 0 (0.0%) | 0 (0.0%) | 4 (8.3%) | |
| Lipogranuloma | .293‡ | ||||
| Absent | 16 (76.2%) | 22 (88.0%) | 2 (100.0%) | 40 (83.3%) | |
| Present | 5 (23.8%) | 3 (12.0%) | 0 (0.0%) | 8 (16.7%) | |
| pSTAT3 IHC∥ | .015† | ||||
| Low | 12 (70.6%) | 4 (25.0%) | 0 (0.0%) | 16 (48.5%) | |
| High | 5 (29.4%) | 12 (75.0%) | 0 (0.0%) | 17 (51.5%) | |
| CCR4 IHC‖ | .019† | ||||
| Low | 5 (27.8%) | 12 (66.7%) | 0 (0.0%) | 17 (47.2%) | |
| High | 13 (72.2%) | 6 (33.3%) | 0 (0.0%) | 19 (52.8%) | |
| Total | 24 (45.3%) | 25 (47.2%) | 4 (7.5%) | 53 (100.0%) | |
| Characteristic | WT | Y82C | Unknown | Total | P |
|---|---|---|---|---|---|
| Clinical features | |||||
| Age | |||||
| Median (range), y | 40 (16-74) | 26 (8-62) | 52 (35-68) | 32 (8-74) | .002* |
| <30 y | 6 (25.0%) | 18 (72.0%) | 0 (0.0%) | 24 (45.3%) | .001† |
| ≥30 y | 18 (75.0%) | 7 (28.0%) | 4 (100.0%) | 29 (54.7%) | |
| Sex | .404† | ||||
| Male | 6 (25.0%) | 9 (36.0%) | 1 (25.0%) | 16 (30.2%) | |
| Female | 18 (75.0%) | 16 (64.0%) | 3 (75.0%) | 37 (69.8%) | |
| HLH/HLH–like systemic illness | <.001† | ||||
| No | 22 (95.7%) | 11 (45.8%) | 2 (100.0%) | 35 (71.4%) | |
| Yes | 1 (4.3%) | 13 (54.2%) | 0 (0.0%) | 14 (28.6%) | |
| BM involvement | 1.000‡ | ||||
| Absent | 21 (100.0%) | 12 (95.7%) | 0 (0.0%) | 43 (93.5%) | |
| Present | 0 (0.0%) | 1 (4.3%) | 2 (100.0%) | 3 (6.5%) | |
| First-line treatments | .069‡ | ||||
| Chemotherapy | 16 (88.9%) | 14 (63.6%) | 3 (100.0%) | 33 (76.7%) | |
| Immunosuppressive | 2 (11.1%) | 8 (36.4%) | 0 (0.0%) | 10 (23.3%) | |
| HSCT§ | .070‡ | ||||
| No | 8 (80.0%) | 3 (33.3%) | 2 (100.0%) | 13 (61.9%) | |
| Yes | 2 (20.0%) | 6 (66.7%) | 0 (0.0%) | 8 (38.1%) | |
| Relapse | .345† | ||||
| No | 17 (73.9%) | 14 (60.9%) | 3 (100.0%) | 34 (69.4%) | |
| Yes | 6 (26.1%) | 9 (39.1%) | 0 (0.0%) | 15 (30.6%) | |
| 5-y RFS | .001* | ||||
| Median (range), m | 57 (1-60) | 11 (1-60) | 60 (16-60) | 30 (1-60) | |
| Death | 1.000‡ | ||||
| No | 21 (87.5%) | 21 (87.5%) | 3 (100.0%) | 45 (88.2%) | |
| Yes | 3 (12.5%) | 3 (12.5%) | 0 (0.0%) | 6 (11.8%) | |
| 5-y OS | .004* | ||||
| Median (range), m | 60 (2-60) | 25 (1-60) | 60 (16-60) | 39 (1-60) | |
| 5-y DSS | .003* | ||||
| Median (range), m | 59.5 (2-60) | 19 (1-60) | 60 (16-60) | 37 (1-60) | |
| Histopathological features | |||||
| Necrosis | .314† | ||||
| Absent | 14 (66.7%) | 13 (52.0%) | 1 (50.0%) | 28 (58.3%) | |
| Present | 7 (33.3%) | 12 (48.0%) | 1 (50.0%) | 20 (41.7%) | |
| Granulomatous inflammation | .037‡ | ||||
| Absent | 17 (81.0%) | 25 (100.0%) | 2 (100.0%) | 44 (91.7%) | |
| Present | 4 (19.0%) | 0 (0.0%) | 0 (0.0%) | 4 (8.3%) | |
| Lipogranuloma | .293‡ | ||||
| Absent | 16 (76.2%) | 22 (88.0%) | 2 (100.0%) | 40 (83.3%) | |
| Present | 5 (23.8%) | 3 (12.0%) | 0 (0.0%) | 8 (16.7%) | |
| pSTAT3 IHC∥ | .015† | ||||
| Low | 12 (70.6%) | 4 (25.0%) | 0 (0.0%) | 16 (48.5%) | |
| High | 5 (29.4%) | 12 (75.0%) | 0 (0.0%) | 17 (51.5%) | |
| CCR4 IHC‖ | .019† | ||||
| Low | 5 (27.8%) | 12 (66.7%) | 0 (0.0%) | 17 (47.2%) | |
| High | 13 (72.2%) | 6 (33.3%) | 0 (0.0%) | 19 (52.8%) | |
| Total | 24 (45.3%) | 25 (47.2%) | 4 (7.5%) | 53 (100.0%) | |
BM, bone marrow; DSS, disease-specific survival; HLH, hemophagocytic lymphohistiocytosis; HSCT, hematopoietic stem cell transplantation; OS, overall survival; RFS, relapse-free survival.
Compared between HAVCR2WT and HAVCR2Y82C by using the Mann-Whitney U test.
Compared between HAVCR2WT and HAVCR2Y82C by using the χ2 test.
Compared between HAVCR2WT and HAVCR2Y82C by using Fisher’s exact test.
Either autologous or allogeneic transplantation.
Used median values as cutoffs.
We assessed the mutational status of HAVCR2 using WES, TGS, or direct sequencing (supplemental Tables 1, 8, and 9; Figure 1; supplemental Figure 1). Of 49 patients, 25 had HAVCR2 Y82C mutation (51.0%), of whom 4 patients harbored heterozygous Y82C mutations. Three of them underwent TGS, and heterozygous status was inferred from the presence of WT alleles and the variant allele frequency (VAF) approximating 50.0% (supplemental Table 9); the other patient (SP14) was tested with direct sequencing, in which WT peak on the electropherogram suggested the heterozygous nature of the mutation. The remainder had homozygous Y82C mutations. HAVCR2Y82C SPTCLs were more frequent in patients aged <30 years (P = .001), and 13 of 14 patients who experienced HLH/HLH–like systemic illness harbored HAVCR2Y82C (P < .001) (Table 1). In contrast to patients with homozygous HAVCR2Y82C, none of those with heterozygous HAVCR2Y82C experienced systemic complications (Fisher’s exact test, P = .031).
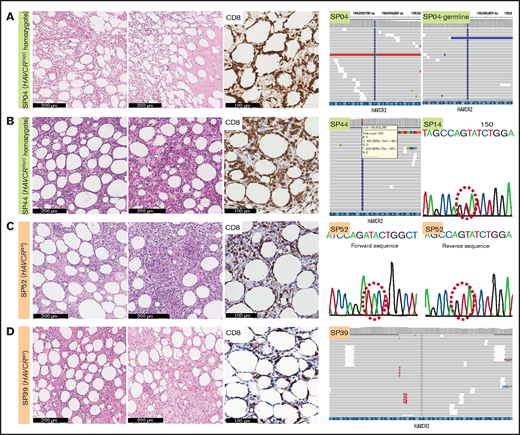
Histopathological features of SPTCLs and detection of HAVCR2Y82C mutations. (A) Excisional biopsy specimen of a 16-year-old female patient with an SPTCL (SP04) exhibited adipocytic rimming by CD8-positive lymphocytes along with prominent necrosis. This patient was confirmed by using WES to have a germline homozygous HAVCR2Y82C mutation. (B) Lipogranulomatous inflammation was observed in a 54-year-old female patient (SP44), and TGS revealed heterozygous HAVCR2Y82C mutations. SP14 harbored heterozygous HAVCR2Y82C mutations, which could be inferred from double peaks on the electropherogram. (C) A 45-year-old female patient (SP52) with the HAVCR2WT genotype had both necrosis and granuloma formation. (D) Lipogranulomatous inflammation was observed in the HAVCR2WT SPTCL of a 53-year-old woman (SP39).
Histopathological features of SPTCLs and detection of HAVCR2Y82C mutations. (A) Excisional biopsy specimen of a 16-year-old female patient with an SPTCL (SP04) exhibited adipocytic rimming by CD8-positive lymphocytes along with prominent necrosis. This patient was confirmed by using WES to have a germline homozygous HAVCR2Y82C mutation. (B) Lipogranulomatous inflammation was observed in a 54-year-old female patient (SP44), and TGS revealed heterozygous HAVCR2Y82C mutations. SP14 harbored heterozygous HAVCR2Y82C mutations, which could be inferred from double peaks on the electropherogram. (C) A 45-year-old female patient (SP52) with the HAVCR2WT genotype had both necrosis and granuloma formation. (D) Lipogranulomatous inflammation was observed in the HAVCR2WT SPTCL of a 53-year-old woman (SP39).
Histopathologically, all cases exhibited adipocytic rimming by CD8-positive T cells, regardless of HAVCR2 status (Figure 1). Tissue necrosis was defined as distinct necrosis with karyorrhectic debris found in at least one high-power field; the necrosis was observed in 41.7% (20 of 48) of patients. Granuloma was noted in 8.3% (4 of 48), and lipogranulomatous inflammation in 16.7% (8 of 48) of patients. Granuloma was more likely to be observed in patients with HAVCR2WT SPTCLs (P = .037).
Mutational profiles in Korean patients with SPTCLs
WES of nine SPTCL samples revealed a total of 399 nonsynonymous mutations in 342 genes (supplemental Table 8); the median number of variants per case was 55 (range, 2-63). No additional recurrent hotspot mutations other than HAVCR2Y82C were detected by using WES (supplemental Figure 2). Altered genes were functionally grouped into different categories: T/natural killer (NK) cell–associated inflammation (HAVCR2, PVRL1, PVRL4, TICAM1, and CD4), epigenetic modifiers (BAZ2A, KMT2D, and SETD1A), and JAK-STAT signaling pathway (IFNL2 and PIAS3). Of note, patient SP04 in the discovery set harbored two point mutations (V272M and K273R) in the same DDX11 allele, which were confirmed somatic (supplemental Figure 3). Subsequent analyses of samples from patient SP03 revealed 56 mutations in the pretreatment sample (SP03-1) and 50 mutations in the recurrence sample (SP03-2), and 36 mutations were identically shared; remaining variants were found in low VAFs from either one of the samples, suggesting that no significant sequential acquisition or dropout of variants occurred during the clinical course (supplemental Figure 2).
To compare mutation profiles between patients with SPTCLs and other diseases resembling panniculitis, we conducted customized TGS. In total, 588 mutations (median, 8; range, 2-193) in 162 genes were detected in patients with SPTCL (supplemental Table 9). None of the tested samples from patients with MF, PCGDTCL, or LP harbored HAVCR2 mutations.
Combined WES and TGS analyses indicated that mutations in genes related to immune responses (ASXL1, JAK3, PIAS3, and PLCG2) and epigenetic modifiers (KMT2D, KMT2C, BAZ2A, and NUP98) were prominent features of SPTCLs (Figure 2). TGS identified DDX11 mutations in 3 additional patients with SPTCLs (SP32, SP47, and SP51) and 1 patient with PCGDTCL (GD3). All SPTCL cases with DDX11 mutations had the HAVCR2Y82C genotype. Notably, patient SP47 was heterozygous for HAVCR2Y82C and harbored the oncogenic hotspot mutation IDH1 R132C. No structural variants were detected by using WES, TGS, or RNA-sequencing.
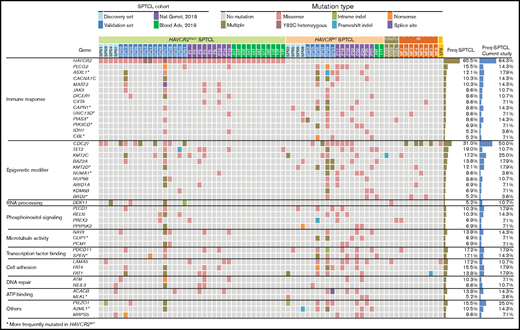
Mutational landscape of SPTCLs and other cutaneous T-cell lymphomas with panniculitic presentation in Korean patients. Integrated mutation map of Korean SPTCLs and previously reported data sets. A subset of genetic alterations was shared with PCGDTCL and MFs; however, the HAVCR2Y82C mutation was seen exclusively in SPTCLs. Genes that were more frequently mutated in HAVCR2WT SPTCLs are indicated with asterisks; statistical significance was determined by using Fisher’s exact test. ATP, adenosine triphosphate.
Mutational landscape of SPTCLs and other cutaneous T-cell lymphomas with panniculitic presentation in Korean patients. Integrated mutation map of Korean SPTCLs and previously reported data sets. A subset of genetic alterations was shared with PCGDTCL and MFs; however, the HAVCR2Y82C mutation was seen exclusively in SPTCLs. Genes that were more frequently mutated in HAVCR2WT SPTCLs are indicated with asterisks; statistical significance was determined by using Fisher’s exact test. ATP, adenosine triphosphate.
To compare the genetic features of HAVCRY82C with those of HAVCR2WT SPTCLs, we extended our data set by integrating 2 previously published WES data sets5,6 (Figure 2). Mutations in ASXL1, CAPN1, UNC13D, PIAS3, PIK3CD, KMT2D, and BRD2 were significantly more frequent in patients with HAVCR2WT SPTCL than in those with HAVCR2 mutations.
Enrichment of inflammation-related pathways in HAVCR2Y82C SPTCLs
We used RNA-sequencing data from the discovery set to conduct GSEA and compare involved pathways between HAVCR2Y82C and HAVCR2WT SPTCLs (Figure 3A-B; supplemental Table 10). Significantly enriched pathways in HAVCR2Y82C SPTCL included tumor necrosis factor-α signaling via NF-kB (normalized enrichment score [NES] = 1.841; FDR q-value = 0.008), hypoxia (NES = 1.860; FDR q-value = 0.009), IL6-JAK-STAT3 signaling (NES = 1.679; FDR q-value = 0.026), apoptosis (NES = 1.437; FDR q-value = 0.121), and MTORC1 signaling (NES = 1.322; FDR q-value = 0.188).
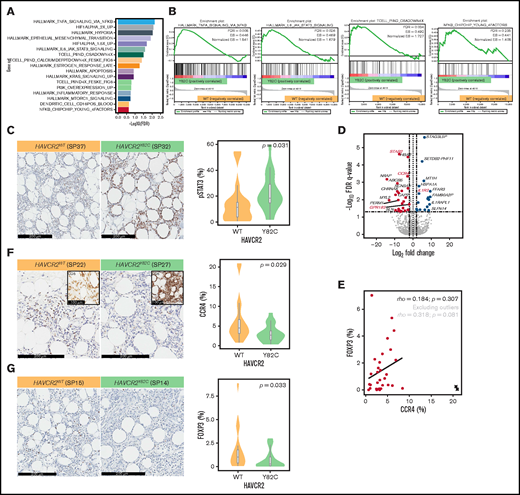
GSEA and DEGs between HAVCR2Y82C and HAVCR2WT SPTCLs. (A) Significantly enriched gene sets in HAVCR2Y82C SPTCLs and their NES are shown. (B) Most enriched gene set in HAVCR2Y82C SPTCLs were associated with increased inflammatory responses. (C) Higher number of pSTAT3-positive cells characterizes HAVCR2Y82C SPTCLs. (D) Volcano plot highlighting 52 genes differentially expressed according to HAVCR2 genotype. (E) CCR4 and Foxp3 positivity was assessed by using IHC. Correlation plot showing percentages of CCR4- and Foxp3-positive cells; circled cross indicates outlier results. (F and G) HAVCR2WT SPTCLs had a significantly higher number of CCR4-positive cells and Foxp3-positive cells compared with HAVCRY82C SPTCLs (Mann-Whitney U test).
GSEA and DEGs between HAVCR2Y82C and HAVCR2WT SPTCLs. (A) Significantly enriched gene sets in HAVCR2Y82C SPTCLs and their NES are shown. (B) Most enriched gene set in HAVCR2Y82C SPTCLs were associated with increased inflammatory responses. (C) Higher number of pSTAT3-positive cells characterizes HAVCR2Y82C SPTCLs. (D) Volcano plot highlighting 52 genes differentially expressed according to HAVCR2 genotype. (E) CCR4 and Foxp3 positivity was assessed by using IHC. Correlation plot showing percentages of CCR4- and Foxp3-positive cells; circled cross indicates outlier results. (F and G) HAVCR2WT SPTCLs had a significantly higher number of CCR4-positive cells and Foxp3-positive cells compared with HAVCRY82C SPTCLs (Mann-Whitney U test).
We performed pSTAT3 IHC, and the number of pSTAT3-positive cells was significantly higher in HAVCR2Y82C SPTCLs compared with HAVCR2WT (P = .031) (Figure 3C; supplemental Table 11); pSTAT3 positivity was observed in reactive cells within the tumor microenvironment as well as adipocyte rimming tumor cells.
Moreover, gene sets associated with T-cell activation mediated by calcium signaling and NFAT nuclear translocation18 were enriched in HAVCR2Y82C SPTCLs (TCELL_PIIND_CSADOWN4X and TCELL_PIIND_CALCIUMDEFPTDOWN4X_FESKE_FIG6; NES = 1.727 and 1.728; FDR q-values = 0.034 and 0.042, respectively). Genes associated with NF-κB subunits on lymphocytic stimulation (NFKB_CHIPCHIP_YOUNG_4FACTORS)19 were significantly enriched in HAVCR2Y82C (NES = 1.408; FDR q-value = 0.235). Taken together, the GSEA results imply that compared with HAVCR2WT SPTCLs, HAVCR2Y82C SPTCLs exhibit enhanced inflammatory responses.
HAVCR2WT SPTCLs are characterized by upregulation of genes involved in lymphocyte homing and immune regulation
To obtain further insight into the role of HAVCR2 mutations in SPTCL pathobiology, we identified DEGs between SPTCL subtypes. A total of 52 DEGs were identified between HAVCR2Y82C and HAVCR2WT SPTCLs (Figure 3D); IL1R2 and 20 other genes were upregulated in HAVCR2Y82C SPTCLs. Genes associated with lymphocyte homing (CCR4 and GPR183) and autoimmunity (STAB2) were among the 31 genes that were significantly upregulated in HAVCR2WT SPTCLs.
CCR4 expression in regulatory T cells (Tregs) residing in nonlymphoid organs has previously been reported20 ; thus, we evaluated differences in CCR4 and Foxp3 expression between HAVCR2Y82C and HAVCR2WT SPTCLs (Figure 3E-G). The number of CCR4-positive cells was significantly higher in HAVCR2WT SPTCLs than in HAVCR2WT SPTCLs (P = .029) (Figure 3F; supplemental Table 11). Microscopic examination revealed that most of the CCR4-positive cells appeared to be distinct from adipocyte-rimming CD8-positive cells (Figure 3F, inlet), implying that these CCR4-positive cells were reactive cells within the microenvironment rather than neoplastic cells.
The percentages of Foxp3-positive cells and CCR4-positive cells showed a trend toward positive correlation (Spearman’s ρ = 0.318 and P = .081 when excluding outlier results) (Figure 3E), and Foxp3-positive Tregs were more abundant in HAVCR2WT SPTCLs (P = .033) (Figure 3G). However, the percentages of Foxp3-positive cells were significantly lower than those of CCR4-positive cells (paired Student t test, P = .001). Double-staining for Foxp3 and CCR4 on selected samples revealed that not all CCR4-positive cells coexpressed Foxp3 (Figure 4). This observation suggests that non-Treg CCR4-positive cells reside in the tumor microenvironment, especially among HAVCR2WT SPTCLs.

Double-staining for Foxp3 and CCR4. Double-staining showed the cells coexpressing Foxp3 and CCR4 (arrow) as well as the cells only positive for CCR4 (arrowhead), suggesting the presence of non-Treg CCR4-positive cells within the tumor microenvironment of SPTCL.
Double-staining for Foxp3 and CCR4. Double-staining showed the cells coexpressing Foxp3 and CCR4 (arrow) as well as the cells only positive for CCR4 (arrowhead), suggesting the presence of non-Treg CCR4-positive cells within the tumor microenvironment of SPTCL.
Clinicopathological risk score for the prognostic stratification of SPTCLs
Patient characteristics and clinical follow-up data are summarized in supplemental Figure 4. RFS analyses according to various clinicopathological variables (Figure 5A-F; supplemental Figure 5) revealed that HAVCR2Y82C mutations and age <30 years were significantly associated with a poor prognosis (P = .023 and P = .033, respectively). In contrast, necrosis, pSTAT3-positivity, CCR4-positivity, HLH/HLH–like systemic illness, and bone marrow involvement had no significant prognostic value. No clinicopathological factors were significantly associated with OS (supplemental Figure 6).
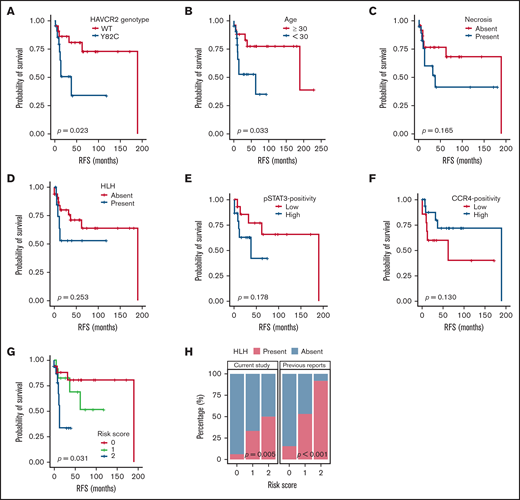
Survival analyses according to clinicopathological factors and development of an SPTCL risk stratification score. (A and B) The presence of the HAVCR2Y82C mutation and age <30 years at diagnosis, respectively, were significant prognostic factors in patients with SPTCLs. (C) A tendency toward poor outcomes was observed in patients with tissue necrosis. (D-F) HLH (or HLH-like systemic illness), pSTAT3 positivity, and lower CCR4 expression were not significantly associated with RFS. (G) The risk score system integrating patients’ age and HAVCR2 status was significantly associated with RFS (P = .031). Patients with score 2 had a shorter RFS compared with patients with score 0 (log-rank test, P = .024). However, there were no significant differences between scores 2 vs 1 and scores 1 vs 2 (log-rank test, P = .068 and 0.354, respectively). (H) Significant correlation between the risk score and event of systemic complication was observed in the current study population (P = .005, linear-by-linear test) as well as in the pooled analysis using the published data from Gayden et al5 and Polprasert et al6 (P < .001, linear-by-linear test).
Survival analyses according to clinicopathological factors and development of an SPTCL risk stratification score. (A and B) The presence of the HAVCR2Y82C mutation and age <30 years at diagnosis, respectively, were significant prognostic factors in patients with SPTCLs. (C) A tendency toward poor outcomes was observed in patients with tissue necrosis. (D-F) HLH (or HLH-like systemic illness), pSTAT3 positivity, and lower CCR4 expression were not significantly associated with RFS. (G) The risk score system integrating patients’ age and HAVCR2 status was significantly associated with RFS (P = .031). Patients with score 2 had a shorter RFS compared with patients with score 0 (log-rank test, P = .024). However, there were no significant differences between scores 2 vs 1 and scores 1 vs 2 (log-rank test, P = .068 and 0.354, respectively). (H) Significant correlation between the risk score and event of systemic complication was observed in the current study population (P = .005, linear-by-linear test) as well as in the pooled analysis using the published data from Gayden et al5 and Polprasert et al6 (P < .001, linear-by-linear test).
We used age <30 years (score 1) and HAVCR2Y82C (score 1) to build a risk scoring system for the prognostic stratification of patients with SPTCLs. The distribution of the 49 patients was as follows: score 0, 18 patients (36.7%; median RFS, 47 months; range, 1-190 months); score 1, 13 patients (26.5%; median RFS, 37 months; range, 6-119 months); and score 2, 18 patients (36.7%; median RFS, 10 months; range, 1-53 months). This risk score was significantly associated with patient outcomes (P = .031) (Figure 5G), and patients with higher score experienced HLH/HLH–like systemic illness more frequently (P = .005) (Figure 5H). We assessed the association between the risk score and events of HLH/HLH–like illness using the previously published clinical data,5,6 which revealed similarly significant results (P < .001).
Discussion
We established a nationwide cohort of patients with SPTCLs and assessed the characteristics of patients with HAVCR2 mutations. We identified differentially enriched cellular pathways and microenvironmental factors according to HAVCR2 genotype by high-throughput sequencing and developed a prognostication score.
To our knowledge, this study is the first to confirm germline HAVCR2Y82C mutations in Korean patients with SPTCLs and is the largest cohort study of East Asian patients with SPTCLs. We provide strong evidence that patients with HAVCR2Y82C SPTCLs exhibit unique clinical features, including younger age, frequent HLH/HLH–like systemic illness, and shorter RFS; some of these findings were previously reported5,7 or failed to be confirmed.6,7 By performing TGS in patients with MF, PCGDTCL, and LP, we showed that the HAVCR2Y82C mutation might be a unique feature of SPTCL.
Four (8.2%) of 49 patients were identified with heterozygous HAVCR2Y82C mutations. One patient with heterozygous HAVCR2I97M was previously reported by Gayden et al,5 and compound heterozygotes with HAVCR2Y82C/I97M and HAVCR2Y82C/T101I have been described.5,6 However, heterozygous HAVCR2Y82C mutations have not been previously reported. A careful review of the histopathological features and IHC results revealed that there were no significant differences between these four patients and patients with homozygous HAVCR2Y82C mutations. Regarding clinical features, heterozygous HAVCR2Y82C patients were less likely complicated by HLH/HLH–like systemic illness, which is compatible with the previous finding that tumor cells of heterozygous HAVCR2Y82C patients exhibited intermediate membranous TIM-3 expression.5 However, survival analyses of various clinicopathological factors after excluding these 4 heterozygous HAVCR2Y82C patients revealed no significant differences in patients’ prognosis compared with our original analyses on the whole study population (data not shown). Considering the minor allele frequency of HAVCRY82C in East Asian subjects is reportedly as high as 0.0036,6 we consider that monoallelic HAVCRY82C alterations alone are not sufficient to cause SPTCL. We sought to identify additional factors contributing to disease presentation; however, no recurrent genetic alterations were found in this subset. Further studies on a larger cohort of patients harboring heterozygous HAVCRY82C mutations are required to elucidate the possible genetic or epigenetic events that contribute to SPTCL development and the clinicopathological implication of heterozygous HAVCRY82C mutations.
Little is known about the somatic mutational profiles of SPTCLs, especially those with the HAVCR2WT genotype. By combining our data with published data set, we found that certain genetic alterations were significantly more frequent in HAVCR2WT than in HAVCR2MUT SPTCLs. For instance, 14.3% (4 of 28) of the study population harbored mutations in PIAS3, the gene-encoding protein inhibitor of activated STAT3 (PIAS3), the main inhibitor of STAT321 ; among these patients, 3 were HAVCR2WT. In addition to the STAT pathway, NF-κB signaling is also affected by PIAS3.22,23 Although the functional effects of the PIAS3 mutations identified in this study remain to be determined, aberrant PIAS3 function may deregulate immune pathways and contribute to the pathogenesis of SPTCLs in the absence of deleterious HAVCR2 mutations.
UNC13D missense mutations were more frequent in HAVCR2WT SPTCLs. Even though UNC13D mutations have been implicated in atypical familial HLH in some Korean patients,24,25 two women in our cohort (SP08 and SP39) were not complicated by HLH. A recent study of Swedish patients suggested that the haploinsufficiency of UNC13D caused by inversion was associated with an increased risk of lymphoma, especially in women.26 The mechanisms linking UNC13D mutations to the pathogenesis of HAVCR2WT SPTCL remain to be unveiled.
Patient SP04 harbored 2 somatic mutations in the DEAD-domain of DDX11, which encodes an RNA helicase family member involved in a rare congenital disease called Warsaw breakage syndrome.27 We identified DDX11 mutations in four HAVCR2Y82C SPTCLs (4 of 25 [16.0%]) and one PCGDTCL (1 of 3 [33.3%]). Mutations in RNA helicase family members have been extensively studied in extranodal NK/T-cell lymphomas28 ; nevertheless, DDX11 alterations have not been described in extranodal NK/T-cell lymphomas or SPTCLs. Of note, DDX11 rearrangements have been associated with diffuse large B-cell lymphoma–associated HLH.29 Furthermore, B-cell lymphoma cell lines strongly depended on DDX11,30 although its role in T-cell lymphomagenesis remains largely unknown.
Comparative analyses between HAVCR2Y82C and HAVCR2WT SPTCLs using RNA-sequencing revealed profound differences between the 2 SPTCL subsets. HAVCR2Y82C SPTCLs were enriched in inflammation-associated cellular pathways, including IL6-JAK-STAT3, further supported by the higher number of pSTAT3-positive cells among HAVCR2Y82C SPTCLs on IHC; pSTAT3 was positive in adipocytic rimming tumor cells and reactive cells, which suggests that activation of the IL6-JAK-STAT pathway could be attributable to both tumor and microenvironmental factors. Upregulation of NF-κB signaling and hypoxia-related genes was also observed, implying uncontrolled immune activation within the tumor milieu. Of note, enrichment of gene sets regulated by calcium signaling in T-lymphocytes underpins the crucial role of activated T cells in SPTCL pathogenesis.
We observed a marked increase in CCR4 expression levels in HAVCR2WT SPTCLs, and numbers of CCR4-positive cells and Foxp3-positive cells were significantly higher in the tumor microenvironment of HAVCR2WT SPTCLs, consistent with a previous study showing decreased Foxp3+ Tregs in HAVCR2MUT SPTCLs.5 CCR4 is a chemokine receptor that regulates Treg homing in nonlymphoid tissues, including the skin31,32 ; loss of CCR4 expression on the Treg compartment resulted in severe inflammatory disease in mouse skin.20 Therefore, in the tumor microenvironment of HAVCR2WT SPTCLs, cytotoxic T cells may be partly controlled by intact CCR4-mediated Treg activity, whereas lack of local immune regulation in HAVCR2Y82C SPTCLs may result in systemic propagation of severe inflammation.
Numbers of CCR4-positive cells were greater than Foxp3-positive cells, and not all CCR4-positive cells coexpressed Foxp3 according to the double-staining; these findings suggested the presence of a non-Treg CCR4-positive cell population. Considering that CCR4 is also a chemoattractant receptor on the T helper 2 cells,33 we performed double-staining for GATA3 and CCR4, in which we observed that some cells were positive for both, whereas others were not (data not shown). Taken together, these findings suggested that a subset of non-Treg CCR4-positive cells could be attributed to the T helper 2 category.
Previous studies have shown HLH to be the most important factor indicating a poor prognosis in SPTCL, yet robust prognostic factors in SPTCL are lacking.3,34 Although HLH alone tended to correlate with poor RFS in the study population, it did not reach statistical significance. Notably, HAVCR2Y82C mutations and younger age were significantly associated with a poor prognosis. We established a risk score system by integrating these 2 factors, which robustly predicted shorter RFS and more frequent events of systemic complication. Although this system showed no significant prognostic impact on patients’ OS, significant association of RFS, events of systemic complication according to our risk score, imply that this system could aid in clinical management and proper triage of the patients with SPTCL. However, because the 2 factors accounting for this score system are closely associated, additional validation in an independent cohort is required to confirm the prognostic value and validity.
Most of the mutational analyses in this study were based on tumor-only sequencing; therefore, copy number analyses were not feasible, and we could not precisely confirm the germline HAVCR2 genotype in most patients. However, we could infer the germline nature of HAVCR2 mutation based on the patterns of VAFs, which approximated to either 50.0% or 100.0%, whereas VAFs of other mutations varied widely. On targeted sequencing, some samples showed an exceptionally high number of variants, implying the possibility of false-positive findings. However, we implemented a thorough variant-filtering process to reduce false-positive findings, as detailed in the supplemental Methods. In addition, not all samples in this cohort were suitable for high-quality next-generation sequencing, and therefore limited genetic information was available from those who underwent direct sequencing of HAVCR2 exon 2 only. Nevertheless, by integrating a previously published data set, we sought to provide a novel insight into the genetics of SPTCLs. Although RNA-sequencing was performed on only a limited number of samples, we performed comparative analyses of SPTCLs according to HAVCR2 genotype and validated the findings by IHC in extended samples, thereby gaining more insight into the biology of this rare disease.
In conclusion, using various sequencing strategies, we assessed the epidemiology and clinicopathological implications of HAVCR2 mutations in a nationwide cohort of Korean patients with SPTCL. Differential distribution of somatic mutations and gene expression profiles according to HAVCR2 genotype were identified. Notably, inflammatory signaling pathways via tumor necrosis factor-α and IL6-JAK-STAT3 axis were enriched in HAVCR2Y82C SPTCLs, whereas a CCR4-rich milieu was observed in in HAVCR2WT SPTCLs, enhancing our current understanding of SPTCL pathogenesis. Additional validation of our proposed risk score may provide a valuable and easy-to-implement tool for the prognostic stratification of patients with SPTCLs.
Acknowledgments
This work was supported by the Basic Science Research through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology Program (grant NRF-2016R1D1A1B01015964), and the Basic Research Program through the NRF funded by the Ministry of Science and ICT (grant 2020R1A4A1017515) and the Seoul National University Hospital Research (grant 0420190470).
Authorship
Contribution: Y.K.J. designed and supervised the project; J.K., I.J., and K.K. performed bioinformatics analyses; J.K. and S.M. performed the experiments; J.K. and Y.K.J. analyzed the results; C.L., H.J.C., Y.H.O., J.-M.K., J.H.H., J.H.P., J.C., Y.H.K., C.-S.P., H.G., J.H., and Y.K.J. contributed to the sample preparation and review of clinical data and pathology; and J.K. and Y.K.J. wrote the manuscript; and all authors read and approved the final manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Yoon Kyung Jeon, Department of Pathology, Seoul National University Hospital, Seoul National University College of Medicine, 101 Daehak-ro, Jongno-gu, Seoul 03080, Republic of Korea; e-mail: ykjeon@snu.ac.kr.

