

Key Points
Variable regions of CD20 IgA antibodies affected effector mechanisms and efficacy in vitro and in vivo.
Disruption of CD47-SIRPα interactions improved ADCP and ADCC by CD20 IgA antibodies.

Abstract
Blockade of the CD47-SIRPα axis improves lymphoma cell killing by myeloid effector cells, which is an important effector mechanism for CD20 antibodies in vivo. The approved CD20 antibodies rituximab, ofatumumab, and obinutuzumab are of human immunoglobulin G1 (IgG1) isotype. We investigated the impact of the variable regions of these 3 CD20 antibodies when expressed as human IgA2 isotype variants. All 3 IgA2 antibodies mediated antibody-dependent cellular phagocytosis (ADCP) by macrophages and antibody-dependent cellular cytotoxicity (ADCC) by polymorphonuclear cells. Both effector mechanisms were significantly enhanced in the presence of a CD47-blocking antibody or by glutaminyl cyclase inhibition to interfere with CD47-SIRPα interactions. Interestingly, an IgA2 variant of obinutuzumab (OBI-IgA2) was consistently more potent than an IgA2 variant of rituximab (RTX-IgA2) or an IgA2 variant of ofatumumab (OFA-IgA2) in triggering ADCC. Furthermore, we observed more effective direct tumor cell killing by OBI-IgA2 compared with RTX-IgA2 and OFA-IgA2, which was caspase independent and required a functional cytoskeleton. IgA2 variants of all 3 antibodies triggered complement-dependent cytotoxicity, with OBI-IgA2 being less effective than RTX-IgA2 and OFA-IgA2. When we investigated the therapeutic efficacy of the CD20 IgA2 antibodies in different in vivo models, OBI-IgA2 was therapeutically more effective than RTX-IgA2 or OFA-IgA2. In vivo efficacy required the presence of a functional IgA receptor on effector cells and was independent of complement activation or direct lymphoma cell killing. These data characterize the functional activities of human IgA2 antibodies against CD20, which were affected by the selection of the respective variable regions. OBI-IgA2 proved particularly effective in vitro and in vivo, which may be relevant in the context of CD47-SIRPα blockade.
Introduction
Myeloid cells often constitute a relevant population of the tumor immune cell infiltrate,1 where their presence is commonly associated with a poor prognosis for patients.2 On the other hand, macrophages were identified as the predominant effector cell population for the CD20 antibody rituximab (RTX) in mice.3,4 Several approaches have been identified to improve the tumor cell–killing capacity of myeloid cells, including Fc engineering of tumor-directed antibodies5 and blockade of myeloid checkpoint molecules.6 Among the latter, blockade of CD47-SIRPα interactions by the CD47 antibody Hu5F9-G4 (magrolimab) is currently the most advanced in clinical development.7 CD47 is a broadly expressed transmembrane protein that provides a “don’t eat me signal” to phagocytes by binding to the α- isoform of the signaling inhibitory receptor protein (SIRPα; CD172a). SIRPα binding triggers immunoreceptor tyrosine-based inhibitory motif–dependent signaling in phagocytes and, thereby, inhibits their phagocytic and tumor cell killing activity.8 Blockade of CD47-SIRPα interactions has been demonstrated to enhance the therapeutic activity of several tumor-directed antibodies in preclinical models.9,10 Recently, Advani et al were the first to report on a phase 1 clinical study in which the CD47 antibody magrolimab was combined with RTX in lymphoma patients who had relapsed after or were refractory to RTX. Importantly, impressive clinical response rates were observed in heavily pretreated patients with diffuse large B-cell lymphoma (DLBCL) or follicular lymphoma.11
All currently approved and clinically developed CD20 antibodies are of the human immunoglobulin G1 (IgG1) isotype, because IgG1 effectively recruits natural killer (NK) cells for antibody-dependent cellular cytotoxicity (ADCC), triggers antibody-dependent phagocytosis (ADCP) by macrophages, activates complement via the classical pathway, and has a long serum half-life by binding to FcRn. However, human IgG1 is suboptimal for the induction of tumor cell killing by human polymorphonuclear cells (PMNs), the most numerous myeloid effector cell population in blood.12,13 Several studies have shown that PMN-mediated tumor cell killing is triggered more effectively by human IgA antibodies carrying the same variable regions than by their parental IgG1 antibodies.14-19 Human IgA antibodies have not been evaluated clinically, but progress has been made in developing IgA molecules with improved biopharmaceutical and pharmacokinetic properties.20
CD20 antibodies can be classified according to their ability to segregate CD20 in lipid rafts as type I (lipid raft formation) or type II (no lipid raft formation).21 CD20 antibodies can mediate direct cell killing, lymphoma cell phagocytosis by macrophages, trogocytosis by PMNs, and ADCC by NK cells; Fc receptor–mediated functions are enhanced for obinutuzumab (OBI) compared with RTX, presumably as a result of its glycoengineered Fc portion.22-24 However, CD20 antibodies of the human IgG1 isotype did not trigger ADCC by human PMN, whereas IgA2 variants with the same variable regions proved effective.13,15 To understand whether CD47-SIRPα blockade could overcome IgG1’s inefficacy in triggering ADCC by PMNs, we tested RTX, ofatumumab (OFA), and OBI in the absence or presence of CD47 blockade. In addition, we generated IgA2 variants of these 3 CD20 antibodies (RTX-IgA2, OBI-IgA2, and OFA-IgA2) and investigated their modes of lymphoma cell killing in detail. Interestingly, OBI-IgA2 was more effective than RTX-IgA2 and OFA-IgA2 in vitro and in vivo, indicating that the variable regions of IgA CD20 antibodies can critically determine their therapeutic efficacy. To the best of our knowledge, differences in effector cell recruitment that depend on the variable regions have not been described for CD20 antibodies and may prove relevant in the context of myeloid checkpoint blockade.
Materials and methods
Experiments with human material were approved by the Ethical Committees of the participating institutions in accordance with the Declaration of Helsinki. Healthy volunteers and patients gave written informed consent before analyses.
Cell lines and primary patient samples
Cell lines were obtained from the German Collection of Microorganisms and Cell Cultures (Braunschweig, Germany) or from the American Type Culture Collection (Manassas, VA). Generation of human CD20-transgenic (Tg) CHO-K1 cells was described previously.15 EL4 cells stably transduced with human CD20 were described previously.25,26 All cells were grown in media, as recommended by the suppliers, and were obtained between 2011 and 2016. Primary tumor cell samples from patients with Waldenström disease or chronic lymphocytic leukemia (CLL) were prepared from peripheral blood by density gradient centrifugation using Ficoll (GE Healthcare, Chicago, IL).
Antibodies
The approved CD20 antibodies RTX (human IgG1; 2B8; Rituxan) and OBI (afucosylated human IgG1; GA101; GAZYVA) were from Roche (Basel, Switzerland). Ofatumumab (human IgG1; 2F2; Arzerra) was obtained from Novartis (Basel, Switzerland). The EGFR antibody cetuximab (human IgG1, 225; Erbitux) and the C5 antibody eculizumab (human IgG2/IgG4 chimera; 5G1.1; SOLIRIS) were purchased from Merck (Kenilworth, NJ) and Alexion (New Haven, CT), respectively. For murine antibodies and newly generated antibody variants see supplemental Materials and methods.
Direct Fab-mediated cell death analyses
To analyze direct Fab-mediated cell death of lymphoma cells, 1.4 × 105 target cells were incubated with CD20 or isotype control antibodies (all at 10 μg/mL) at 37°C and 5% CO2, in the presence or absence of 10 µM Latrunculin B (LatB; Cayman Chemical Company, Ann Arbor, MI) or 50 µM carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]-fluoromethylketon (Z-VAD-FMK; APExBIO Technology, Houston, TX). Photographs were taken 24 hours later using an Axiovert 40C microscope and an Axiocam ERc 5s camera (both from Zeiss, Oberkochen, Germany). Next, cells were stained using an FITC Annexin V Apoptosis Detection Kit + 7-aminoactinomycin (Bio Legend, San Diego, CA), according to the manufacturer’s instructions, and analyzed by flow cytometry using a Navios flow cytometer, Brea, Beckman Coulter, CA). To quantify lysosomal cell death or apoptosis, 1 × 105 target cells were incubated with CD20 antibodies (10 µg/mL) at 37°C and 5% CO2. After 5 hours of incubation, LysoTracker Red (50 nM; Invitrogen, Carlsbad, CA) was added for 1 hour to determine lysosomal cell death or cells were treated after 6 hours with 10 nM DiOC6 and 20 nM TO-PRO-3 (both from Thermo Fisher, Waltham, MA), following the manufacturer’s instructions. Afterward, cells were analyzed by flow cytometry (BD LSRFortessa; BD Biosciences, Franklin Lakes, NJ). Triton solubility of lipid rafts was determined as described previously.21
Complement binding and deposition
Complement component binding and deposition were characterized by indirect flow cytometry. Target cells (2.5 × 105) were incubated with isotype control or with the indicated CD20 antibodies (all at 10 µg/mL) for 15 minutes at 4°C. Subsequently, a 25% volume-to-volume ratio of human serum was added as a source of complement in the presence of the C5-blocking antibody eculizumab (100 µg/mL) to avoid complement-dependent cytotoxicity (CDC). Samples were stained with polyclonal FITC-conjugated C1q, C4b/c, or C3b/c antibodies (all from Agilent, Santa Clara, CA) and analyzed on a flow cytometer (Navios; Beckman Coulter).
CDC assays
CDC assays were performed as described previously.15 Briefly, target cells were labeled with 200 μCi [51Cr] for 2 hours. A 25% volume-to-volume ratio of freshly drawn human serum was used as a source of complement in the presence of the indicated antibodies (0-50 µg/mL). The percentage of [51Cr] release was calculated using the formula: percentage lysis = (experimental cpm − basal cpm)/(maximal cpm − basal cpm) × 100; maximal [51Cr] release was determined by adding Triton-X (2% final concentration) to target cells, and basal release was measured in the absence of sensitizing antibodies.
Isolation of human effector cells
Human effector cells were isolated from peripheral blood (PMNs) of healthy volunteers or were generated from leukoreduction system chambers (macrophages) from blood donors, using PolymorphoPrep (PROGEN, Heidelberg, Germany) or Ficoll (GE Healthcare), as previously described.12 In brief, monocyte-derived macrophages were generated using monocyte-attachment medium (PromoCell, Heidelberg, Germany), according to the manufacturer’s instructions. For macrophage generation, 50 ng/mL macrophage colony-stimulating factor (PeproTech, Rocky Hill, NJ) was added for ≥7 days.
ADCC assays
Analysis of ADCC was performed using [51Cr] release assays, as described previously.27 granulocyte-macrophage colony-stimulating factor (50 U/mL; CellGenix, Freiburg, Germany)–stimulated PMNs, antibodies at various concentrations, and medium were added to round-bottom microtiter plates (Nunc, Rochester, NY). Assays were started by the addition of effector and target cells at the indicated E:T ratios. After 3 h at 37°C, [51Cr] release from triplicate samples was measured. The percentage of cellular cytotoxicity was calculated using the same formula as for CDC assays.
ADCP assays
ADCP experiments were performed as described.28 In brief, lymphoma cells were washed in phosphate buffered saline (PBS) and labeled with 7.5 µM carboxyfluorescein succinimidyl ester (BioLegend, San Diego, CA). Twenty thousand macrophages were seeded per well in µ- slide 8-well chambered coverslips (ibidi, Gräfelfing, Germany) and allowed to adhere overnight. Forty thousand carboxyfluorescein succinimidyl ester–labeled tumor cells were added. Antibodies were applied at a final concentration of 10 µg/mL. Cells were incubated for 2 hours at 37°C. ADCP was determined by fluorescence microscopy (Nikon, Tokyo, Japan). ADCP was calculated as phagocytic index using the formula: ADCP [phagocytic index] = (number of engulfed target cells/number of macrophages) × 100.
In vivo experiments
All experiments were performed in accordance with international guidelines and approved by the national Central Authority for Scientific Procedures on Animals and the local experimental animal welfare body (AVD115002016410). A detailed description is available in supplemental Materials and methods.
Data processing and statistical analyses
Data were generated from ≥3 independent experiments using blood from different healthy donors. Graphical and statistical analyses were performed using GraphPad Prism 5.0 or 8.0 (GraphPad Software, San Diego, CA). Group data are reported as mean ± standard error of the mean (SEM) or standard deviation; half-maximal effective concentrations were calculated from dose-response curves and also reported as mean ± SEM. Significance was determined by 1- or 2-way analysis of variance (ANOVA) repeated-measures test with the Bonferroni post hoc correction.
Results
Blockade of CD47-SIRPα interactions improves CD20 IgG1 antibody-mediated ADCP by macrophages but does not trigger ADCC by PMNs
First, we investigated myeloid effector cell recruitment by the 3 clinically approved CD20 antibodies RTX, OFA, and OBI in the presence or absence of CD47-SIRPα inhibition. To disrupt CD47-SIRPα interaction, assays were performed using an Fc silent IgG2σ variant of the CD47 antibody 5F9 (Figure 1) or by treating target cells with a glutaminyl cyclase inhibitor, SEN177 (supplemental Figure 1A). Recent results by Logtenberg et al29 demonstrated that SEN177 inhibited a functionally important posttranslational modification of CD47 (pyroglutamate formation) and, thereby, impaired SIRPα binding. As expected, all 3 CD20 antibodies mediated ADCP by macrophages, which was significantly improved by CD47-SIRPα inhibition for a mantle cell lymphoma cell line (Mino) and a Burkitt lymphoma cell line (Raji) (Figure 1A; supplemental Figure 1B). Next, we used neutrophils in ADCC assays with the same 3 CD20 antibodies. As expected from previous studies,13,15,30 none of them triggered ADCC by PMNs (Figure 1B; supplemental Figure 1C). Interestingly, this inefficacy of CD20 IgG1 antibodies in PMN-mediated ADCC could not be overcome by disruption of CD47-SIRPα interactions, as demonstrated (Figure 1B; supplemental Figure 1C). Together, these results demonstrate that CD47 blockade cannot overcome the previously reported inefficacy of CD20 antibodies of the human IgG1 isotype to recruit PMNs for ADCC against lymphoma cells.
![Blockade of CD47-SIRPα interactions improves CD20 IgG1 antibody–mediated ADCP by macrophages but does not trigger ADCC by PMNs. Macrophage-mediated ADCP was assessed by fluorescence microscopy (A), whereas ADCC by PMNs was investigated in 3-hour [51Cr] release assays (B). An IgG2σ version of the CD47 antibody 5F9 (10 µg/mL) was used to block CD47-SIRPα interactions. Mino (mantle cell lymphoma) and Raji (Burkitt lymphoma) cells served as target cells (left and right panels, respectively). The CD20 antibody RTX, OFA, or OBI was used at 10 µg/mL; E:T ratios were 80:1 in ADCC assays and 1:2 in ADCP assays. Results are mean ± SEM from ≥3 experiments with effector cells from different donors. *Significant ADCP by CD20 antibodies vs control (ctrl.) antibody. XSignificant differences in the absence or presence of CD47 blockade. Significant differences (P < .05) were calculated using ANOVA. n.s., not significant; PI, phagocytic index.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/5/19/10.1182_bloodadvances.2021004598/4/m_advancesadv2021004598f1.png?Expires=1769243926&Signature=NW2b~vn1KZdIjimVGJb5SnVc5qvs62zo9SzJ6g1pa2wW5ZJK~CToxXcNkK8-v4gY9b8aRBaKpH4Lx~WcJaXNPzOvNz-XbgeTbxqVy4bbTCjpLVC5VAlWeq6Sa3fLJwzwfPW8p0ge0ZXMsJ-mGn9iYFBL9vK2eZd8v0z3pTPsBMGxe1UzKcGzqs32skAI6WchJ5eBEf5VJSApiCrtka3tsIqJWvKmkPBqWF~7XQcmuwUz8ZDvHgb2J6lGQ52CxvoHK2S3BybGyp04BOfyY0lqs7wR6Ps699xcMZfFEVuUz8xzZhzH7TKEO0C3C0Te~3oJWH2tY74yWGiUL7WF~F3aeA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Blockade of CD47-SIRPα interactions improves CD20 IgG1 antibody–mediated ADCP by macrophages but does not trigger ADCC by PMNs. Macrophage-mediated ADCP was assessed by fluorescence microscopy (A), whereas ADCC by PMNs was investigated in 3-hour [51Cr] release assays (B). An IgG2σ version of the CD47 antibody 5F9 (10 µg/mL) was used to block CD47-SIRPα interactions. Mino (mantle cell lymphoma) and Raji (Burkitt lymphoma) cells served as target cells (left and right panels, respectively). The CD20 antibody RTX, OFA, or OBI was used at 10 µg/mL; E:T ratios were 80:1 in ADCC assays and 1:2 in ADCP assays. Results are mean ± SEM from ≥3 experiments with effector cells from different donors. *Significant ADCP by CD20 antibodies vs control (ctrl.) antibody. XSignificant differences in the absence or presence of CD47 blockade. Significant differences (P < .05) were calculated using ANOVA. n.s., not significant; PI, phagocytic index.
Blockade of CD47-SIRPα interactions improves CD20 IgG1 antibody–mediated ADCP by macrophages but does not trigger ADCC by PMNs. Macrophage-mediated ADCP was assessed by fluorescence microscopy (A), whereas ADCC by PMNs was investigated in 3-hour [51Cr] release assays (B). An IgG2σ version of the CD47 antibody 5F9 (10 µg/mL) was used to block CD47-SIRPα interactions. Mino (mantle cell lymphoma) and Raji (Burkitt lymphoma) cells served as target cells (left and right panels, respectively). The CD20 antibody RTX, OFA, or OBI was used at 10 µg/mL; E:T ratios were 80:1 in ADCC assays and 1:2 in ADCP assays. Results are mean ± SEM from ≥3 experiments with effector cells from different donors. *Significant ADCP by CD20 antibodies vs control (ctrl.) antibody. XSignificant differences in the absence or presence of CD47 blockade. Significant differences (P < .05) were calculated using ANOVA. n.s., not significant; PI, phagocytic index.
Blockade of the CD47-SIRPα axis improves myeloid effector cell recruitment of the IgA2 isotype by CD20 antibodies
Because CD20 antibodies of the IgG1 isotype did not use neutrophils for ADCC, we generated IgA2.0 isotype switch variants of the 3 approved CD20 antibodies: RTX, OFA, and OBI. These IgA antibodies displayed the expected biochemical properties and binding to CD20 (supplemental Figure 2). Similar to IgG1, CD20 antibodies of the IgA2 isotype also induced macrophage-mediated ADCP of lymphoma cells, which was significantly enhanced by CD47 blockade (Figure 2A), as well as by glutaminyl cyclase inhibition (supplemental Figure 3A). Against lymphoma cell lines that are difficult to kill for PMNs, such as Mino and Raji, CD20 IgA2 antibodies alone triggered low levels of ADCC (Figure 2B). However, PMN-mediated ADCC was significantly enhanced for all 3 CD20 IgA2 antibodies in the presence of a CD47-blocking antibody (Figure 2B) or by glutaminyl cyclase inhibition (supplemental Figure 3B).
![Blockade of the CD47-SIRPα axis improves myeloid effector cell recruitment by CD20 antibodies of the IgA2 isotype. Macrophage-mediated ADCP by CD20 antibodies of the IgA2 isotype was determined by fluorescence microscopy (A), whereas ADCC by PMNs was investigated in 3-hour [51Cr] release assays (B). An IgG2σ version of the CD47 antibody 5F9 (10 µg/mL) was used to block CD47-SIRPα interactions. The CD20 antibodies RTX, OFA, and OBI were used at 10 µg/mL; E:T ratios were 80:1 in ADCC assays and 1:2 in ADCP assays. (C) Increasing concentrations of IgA2 variants of RTX, OFA, and OBI (left panel) or increasing E:T ratios (right panel) were compared in 3-hour [51Cr] release ADCC assays using PMNs as effector cells. Target cells were Mino cells (mantle cell lymphoma) and Raji cells (Burkitt lymphoma) (left and right panels, respectively) in (A) and (B) and SU-DHL-4 cells (DLBCL) in (C). CD20 antibodies are depicted as colored circles. Results are mean ± SEM from ≥3 experiments with effector cells from different donors. *Significant difference vs control (ctrl.) antibody. XSignificant difference in the absence and presence of CD47 blockade. §Significant difference between OBI-IgA2 and RTX-IgA2 or OFA-IgA2. Significant differences (P < .05) were calculated using ANOVA. conc., concentration; mAb, monoclonal antibody; n.s., not significant.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/5/19/10.1182_bloodadvances.2021004598/4/m_advancesadv2021004598f2.png?Expires=1769243926&Signature=OgJ-mtGQUNcIGwNv~v5fL~74R80x0l7ikm8QwlkvDrPI7O3TgQT-r-FFaxTrdRAWLSpDQew4JhSYGQzDpbDI0BmGxAL4dbiJAcPgYJ3Fb2V2bDXPBNRLE7PMVASo1ssmFz7V2dBSaGtZi7RFtHCJHgqHumCXCxkN3iBY2pHUVqAILGO-AIsuvNPkR5xBpRW2LYqi~s34FhVmIj9Aooq828pLPAfi2yN5NPYoLRX711zX2JxKtxSmayFnDTtpo6NrI3L9mjuLTdwf~ZOK8F3RdfT95E5HIltG21izK0bOBwWoGKZYq5larlgKR4n~FgpRZNxqb-6rahTAf8WFHiFA~Q__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Blockade of the CD47-SIRPα axis improves myeloid effector cell recruitment by CD20 antibodies of the IgA2 isotype. Macrophage-mediated ADCP by CD20 antibodies of the IgA2 isotype was determined by fluorescence microscopy (A), whereas ADCC by PMNs was investigated in 3-hour [51Cr] release assays (B). An IgG2σ version of the CD47 antibody 5F9 (10 µg/mL) was used to block CD47-SIRPα interactions. The CD20 antibodies RTX, OFA, and OBI were used at 10 µg/mL; E:T ratios were 80:1 in ADCC assays and 1:2 in ADCP assays. (C) Increasing concentrations of IgA2 variants of RTX, OFA, and OBI (left panel) or increasing E:T ratios (right panel) were compared in 3-hour [51Cr] release ADCC assays using PMNs as effector cells. Target cells were Mino cells (mantle cell lymphoma) and Raji cells (Burkitt lymphoma) (left and right panels, respectively) in (A) and (B) and SU-DHL-4 cells (DLBCL) in (C). CD20 antibodies are depicted as colored circles. Results are mean ± SEM from ≥3 experiments with effector cells from different donors. *Significant difference vs control (ctrl.) antibody. XSignificant difference in the absence and presence of CD47 blockade. §Significant difference between OBI-IgA2 and RTX-IgA2 or OFA-IgA2. Significant differences (P < .05) were calculated using ANOVA. conc., concentration; mAb, monoclonal antibody; n.s., not significant.
Blockade of the CD47-SIRPα axis improves myeloid effector cell recruitment by CD20 antibodies of the IgA2 isotype. Macrophage-mediated ADCP by CD20 antibodies of the IgA2 isotype was determined by fluorescence microscopy (A), whereas ADCC by PMNs was investigated in 3-hour [51Cr] release assays (B). An IgG2σ version of the CD47 antibody 5F9 (10 µg/mL) was used to block CD47-SIRPα interactions. The CD20 antibodies RTX, OFA, and OBI were used at 10 µg/mL; E:T ratios were 80:1 in ADCC assays and 1:2 in ADCP assays. (C) Increasing concentrations of IgA2 variants of RTX, OFA, and OBI (left panel) or increasing E:T ratios (right panel) were compared in 3-hour [51Cr] release ADCC assays using PMNs as effector cells. Target cells were Mino cells (mantle cell lymphoma) and Raji cells (Burkitt lymphoma) (left and right panels, respectively) in (A) and (B) and SU-DHL-4 cells (DLBCL) in (C). CD20 antibodies are depicted as colored circles. Results are mean ± SEM from ≥3 experiments with effector cells from different donors. *Significant difference vs control (ctrl.) antibody. XSignificant difference in the absence and presence of CD47 blockade. §Significant difference between OBI-IgA2 and RTX-IgA2 or OFA-IgA2. Significant differences (P < .05) were calculated using ANOVA. conc., concentration; mAb, monoclonal antibody; n.s., not significant.
When more susceptible DLBCL lymphoma cells (SU-DHL-4) served as targets in PMN-mediated ADCC assays, IgA2 variants of all 3 CD20 antibodies alone triggered significant ADCC by PMNs in an antibody concentration–dependent manner or at different E:T ratios (Figure 2C). Interestingly, OBI-IgA2 was significantly more effective than RTX-IgA2 or OFA-IgA2 in both ADCC setups. Thus, we investigated whether this also held true for other B-cell lines and primary lymphoma cells.
OBI-IgA2 is more effective than RTX-IgA2 at triggering ADCC by PMNs
The glycoengineered CD20 antibody OBI is known to trigger improved ADCC by NK cells compared with other CD20 antibodies, which is explained by its enhanced FcγRIIIa binding on NK cells due to its lower level of Fc fucosylation.31 Unexpectedly, OBI-IgA2 also appeared to be more effective than RTX-IgA2 in triggering ADCC by PMNs. This observation cannot be explained by differences in fucosylation, because the glycosylation profiles of all 3 CD20 IgA antibodies were similar (supplemental Figure 2C), and glycosylation of IgA was demonstrated not to contribute to FcαRI (CD89) binding.32 Next, we compared PMN-mediated ADCC by OBI-IgA2 vs RTX-IgA2 against a broader panel of CD20+ lymphoma cell lines, reflecting different biological entities of B-cell lymphomas. As demonstrated, OBI-IgA2 triggered significant ADCC by PMNs against all tested cell lines and was often significantly more potent than the respective RTX-IgA2 variant (Figure 3A). Importantly, OBI-IgA2 also triggered significant ADCC by PMNs against isolated primary lymphoma cells from patients with Waldenström disease or CLL, as well as against isolated primary leukemia cells form patients with CLL (Figure 3B). Here, OBI-IgA2 was significantly more potent than the respective approved CD20 antibody of the IgG1 isotype.
![OBI-IgA2 is more effective than RTX-IgA2 at triggering ADCC by PMNs. (A) Cell lines representing different B-cell lymphoma entities (Daudi (Burkitt lymphoma); Oci-Ly7, CARNAVAL, and Oci-Ly18 (DLBCL); GRANTA-519 and MINO (mantle cell lymphoma); MEC-2 (CLL)) served as target cells in [51Cr] release assays. (B) ADCC was investigated in [51Cr] release assays against freshly isolated tumor cells from patients with Waldenström disease (left panel) or CLL (right panel). PMNs served as effector cells at an E:T ratio of 80:1; antibody concentrations were 10 µg/mL. Results are mean ± SEM from ≥2 experiments with effector cells from different donors. *Significant difference specific antibody vs isotype control. §Significant difference RTX-IgA2 vs OBI-lgA2. #Significant difference lgG1 vs lgA2. Significant differences (P < .05) were calculated using ANOVA. ctrl., control; n.s., not significant; OBI, OBI-IgA2.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/5/19/10.1182_bloodadvances.2021004598/4/m_advancesadv2021004598f3.png?Expires=1769243926&Signature=hqCExCxrNfZN78ePq2UQZWUdIGlDMzPXkXLvXCnoDp8dJjf6LrHY9RiAJ-5wcR-0cfO1MknDyNA3LK2m1wN89FDmujKAWEc3kjZrdzxYyTAVftQdO9YZLP8RR8x923OhgyM8WXLYaxEjFdbWRcEUzdv~rKOddJovX5MlSfeyVrEVhRY5-hor5v4lP2ppFZPjyVbx3HK-LDGuY3PDMH51b5SDnGcUcQw5vEHwjnasaNIllVTjbeosXq6DNteANno~d5~N9X7-e4HUqhoTVzCzEW9L3e87qdBKXuUkYv-cc-NrQUw-i8ClZxqiRWYzYlf29YWrnwUb9ym2CvmpvHs6VQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
OBI-IgA2 is more effective than RTX-IgA2 at triggering ADCC by PMNs. (A) Cell lines representing different B-cell lymphoma entities (Daudi (Burkitt lymphoma); Oci-Ly7, CARNAVAL, and Oci-Ly18 (DLBCL); GRANTA-519 and MINO (mantle cell lymphoma); MEC-2 (CLL)) served as target cells in [51Cr] release assays. (B) ADCC was investigated in [51Cr] release assays against freshly isolated tumor cells from patients with Waldenström disease (left panel) or CLL (right panel). PMNs served as effector cells at an E:T ratio of 80:1; antibody concentrations were 10 µg/mL. Results are mean ± SEM from ≥2 experiments with effector cells from different donors. *Significant difference specific antibody vs isotype control. §Significant difference RTX-IgA2 vs OBI-lgA2. #Significant difference lgG1 vs lgA2. Significant differences (P < .05) were calculated using ANOVA. ctrl., control; n.s., not significant; OBI, OBI-IgA2.
OBI-IgA2 is more effective than RTX-IgA2 at triggering ADCC by PMNs. (A) Cell lines representing different B-cell lymphoma entities (Daudi (Burkitt lymphoma); Oci-Ly7, CARNAVAL, and Oci-Ly18 (DLBCL); GRANTA-519 and MINO (mantle cell lymphoma); MEC-2 (CLL)) served as target cells in [51Cr] release assays. (B) ADCC was investigated in [51Cr] release assays against freshly isolated tumor cells from patients with Waldenström disease (left panel) or CLL (right panel). PMNs served as effector cells at an E:T ratio of 80:1; antibody concentrations were 10 µg/mL. Results are mean ± SEM from ≥2 experiments with effector cells from different donors. *Significant difference specific antibody vs isotype control. §Significant difference RTX-IgA2 vs OBI-lgA2. #Significant difference lgG1 vs lgA2. Significant differences (P < .05) were calculated using ANOVA. ctrl., control; n.s., not significant; OBI, OBI-IgA2.
Complement activation by CD20 antibodies of the IgA2 isotype
Another well-documented effector mechanism of type I CD20 antibodies (eg, RTX and OFA) is CDC,33,34 whereas the CDC activity of type II antibodies (eg, OBI) is considerably weaker.35 Interestingly, IgA antibodies against CD20 also demonstrated C1q-dependent CDC activity,15,36 although IgA lacks a C1q binding site.37 Thus, we compared complement activation between the approved IgG1 CD20 antibodies and our novel IgA2 variants. As expected, all 3 IgG1 antibodies mediated significant CDC, with OBI being significantly less effective than the type I antibodies RTX and OFA (P < .05; Figure 4A, left panel). Interestingly, the 3 IgA2 molecules also triggered CDC with OBI-IgA2 being significantly less efficient than RTX-IgA2 and OFA-IgA2 (P < .05, ANOVA; Figure 4 A, right panel). Maximal CDC by all 3 IgA2 variants was significantly lower compared with the respective IgG1 molecules (Figure 4B).
![CD20 antibodies of the IgA isotype differ in their capacity to trigger CDC. (A) The CDC activity of CD20 antibodies of the IgG1 (left panel) or IgA2 (right panel) isotype was analyzed in [51Cr] release assays against SU-DHL-4 cells using increasing concentrations of antibodies. (B) Comparison of the maximal CDC activity of CD20-IgG1 and CD20-IgA2 antibodies, as determined in (A). (C) CD20 antibody–triggered binding (C1q) and deposition of complement components (C4b/c, C3b/c) on SU-DHL-4 cells was analyzed by indirect flow cytometry. CD20 antibodies were added at 10 µg/mL for 60 minutes. Results are mean ± SEM from ≥3 experiments. *Significant difference specific antibody vs isotype control. §Significant difference between OBI-IgA2 and RTX-IgA2 or OFA-IgA2. #Significant difference lgG1 vs lgA2. Significant differences (P < .05) were calculated using ANOVA. conc., concentration; ctrl, control; MFI, mean fluorescence intensity; n.s., not significant.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/5/19/10.1182_bloodadvances.2021004598/4/m_advancesadv2021004598f4.png?Expires=1769243926&Signature=4kx1CcJyKFQlusAtVu8M-Lole3IUHiQ7FC4Cebzb5x3b5JwbH6oZ8UHiAgISWBXK0PAXXFtAsGPeLSNTQA8Tk4LAl2qE0yQT09DxxtB40WOQeqsSjnlFLKsbhAe4N3XOQA-u-Q5ddOgn2yshNKhnUu453DqiSLFUoxL-j6HZ-SmhidOL3Wwrpp98sb9NeUs8dgeVdCjcGBRE17JoIN4rpSJ~bAn~cW0THd~G2T66mmZ7tyjSGPy4~l99~DMsVaQOzwdJFC5Vh8fEfuPpI79muyufIBStgIQQf-7zZqgE-zuVWAtfN6oLcqU9CwmYI1AS0dCOwTLHxs0FkJGEkooLnA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
CD20 antibodies of the IgA isotype differ in their capacity to trigger CDC. (A) The CDC activity of CD20 antibodies of the IgG1 (left panel) or IgA2 (right panel) isotype was analyzed in [51Cr] release assays against SU-DHL-4 cells using increasing concentrations of antibodies. (B) Comparison of the maximal CDC activity of CD20-IgG1 and CD20-IgA2 antibodies, as determined in (A). (C) CD20 antibody–triggered binding (C1q) and deposition of complement components (C4b/c, C3b/c) on SU-DHL-4 cells was analyzed by indirect flow cytometry. CD20 antibodies were added at 10 µg/mL for 60 minutes. Results are mean ± SEM from ≥3 experiments. *Significant difference specific antibody vs isotype control. §Significant difference between OBI-IgA2 and RTX-IgA2 or OFA-IgA2. #Significant difference lgG1 vs lgA2. Significant differences (P < .05) were calculated using ANOVA. conc., concentration; ctrl, control; MFI, mean fluorescence intensity; n.s., not significant.
CD20 antibodies of the IgA isotype differ in their capacity to trigger CDC. (A) The CDC activity of CD20 antibodies of the IgG1 (left panel) or IgA2 (right panel) isotype was analyzed in [51Cr] release assays against SU-DHL-4 cells using increasing concentrations of antibodies. (B) Comparison of the maximal CDC activity of CD20-IgG1 and CD20-IgA2 antibodies, as determined in (A). (C) CD20 antibody–triggered binding (C1q) and deposition of complement components (C4b/c, C3b/c) on SU-DHL-4 cells was analyzed by indirect flow cytometry. CD20 antibodies were added at 10 µg/mL for 60 minutes. Results are mean ± SEM from ≥3 experiments. *Significant difference specific antibody vs isotype control. §Significant difference between OBI-IgA2 and RTX-IgA2 or OFA-IgA2. #Significant difference lgG1 vs lgA2. Significant differences (P < .05) were calculated using ANOVA. conc., concentration; ctrl, control; MFI, mean fluorescence intensity; n.s., not significant.
To better understand CDC triggered by IgA antibodies against CD20, complement binding (C1q) and deposition (C4b/c and C3b/c) on lymphoma cells was measured by flow cytometry (Figure 4C). For CD20 antibodies of the IgG1 isotype, the expected differences in C1q binding efficacy were observed (OFA > RTX > OBI). None of the 3 IgA2 antibodies triggered measurable C1q binding. All 3 IgG1 antibodies showed high levels of C4b/c deposition, whereas C4b/c deposition by IgA2 antibodies was low, but detectable. All antibodies deposited C3b/c on lymphoma cells, indicative of effective C5 convertase formation. All IgA2 variants were significantly less effective than were the respective IgG1 molecules in C4b/c and C3b/c deposition (Figure 4C). Together, these results demonstrate that IgG and IgA antibodies perform CDC in vitro. IgG1 antibodies do this more efficiently; OBI is the least efficient in CDC induction, as IgG1 and IgA2 variants.
OBI-IgA2 is more effective than RTX-IgA2 or OFA-IgA2 in direct lymphoma cell killing
Next, we investigated direct cell death induction by CD20 antibodies of the IgG1 or IgA2 isotype, starting by visualizing homotypic aggregation of lymphoma cells, which has been described to correlate with the induction of direct cell death.38 As IgG1, the type II antibody OBI induced the greatest degree of homotypic aggregation, followed by RTX and OFA (Figure 5A, upper panels). Interestingly, IgA2 variants of RTX and OFA also triggered homotypic cell aggregation to a similar degree as OBI-IgG1 and OBI-IgA2 (Figure 5A, lower panels). Using flow cytometric assessment of apoptosis induction by DioC6 staining, OBI as IgG1 and IgA2, but not the RTX or OFA isotype variants, induced significant direct cell death (Figure 5B). To further investigate the mechanism of cell death induction by CD20 antibodies of the IgA2 isotype, we investigated lysosomal cell death induction by LysoTracker Red staining.21 Again, OBI-IgG1 and OBI-IgA2 were most effective, but RTX-IgA2 also induced significant loss of lysosomal volume (Figure 5C). Next, we compared the capability of IgG1 and IgA2 variants of the 3 CD20 antibodies to form CD20-containing lipid rafts, a functional characteristic of type I antibodies. Interestingly, this activity of CD20 antibodies also remained unaffected by the isotype switch from IgG1 to IgA2, with both OBI variants being significantly less effective than RTX or OFA variants (Figure 5D). We then investigated the mechanism of direct cell death induction by OBI-IgA2 in more detail using a pan-caspase inhibitor (Z-VAD-FMK) or LatB as an actin polymerization inhibitor. As shown for CD20 antibodies of the IgG1 isotype,21 apoptosis induction by OBI-IgA2 was independent of caspases (Figure 5E) but was fully dependent on actin polymerization (Figure 5F). Additionally, a V11L mutation in the constant region of the heavy chain, which was previously described to reduce lysosomal cell death induction by OBI-IgG1,39 also impaired lysosomal cell death induction by OBI-IgA2 (Figure 5G) but did not negatively affect its ADCC capacity by PMNs (supplemental Figure 4A). Together, these data show that CD20-IgA antibodies trigger direct lymphoma cell killing similar to their IgG1 counterparts.
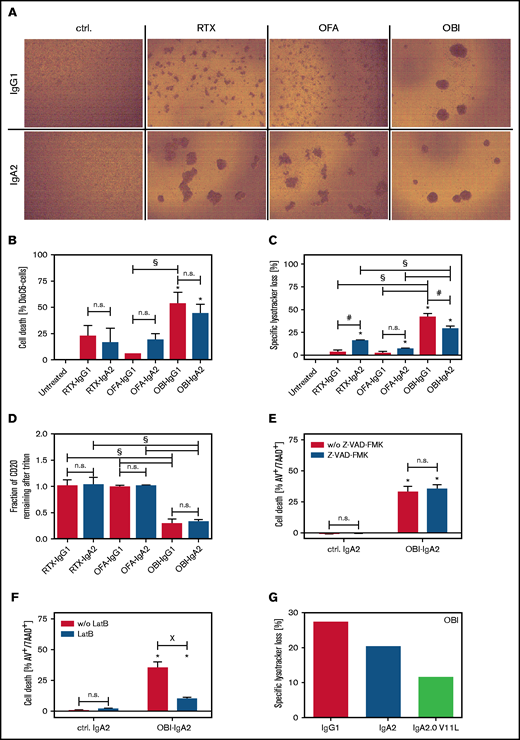
Direct lymphoma cell killing by CD20 antibodies of the IgA2 isotype. (A) Homotypic aggregation by CD20 antibodies was analyzed by phase-contrast microscopy using SU-DHL-4 target cells. Antibodies were added at 10 µg/mL for 24 hours. Representative images of 3 independent experiments are shown. (B) Induction of direct cell death induced by CD20 monoclonal antibody was analyzed by DiOC6 staining. Ramos cells were used as target cells, and antibodies were added at a fixed concentration (10 µg/mL) for 6 hours. (C) Lysosomal cell death was quantified by LysoTracker Red staining on Ramos cells after incubation with CD20 antibodies (10 µg/mL) for 6 hours. (D) Redistribution of CD20 to lipid rafts was analyzed by flow cytometry on Ramos cells. For discrimination between nonraft and lipid raft distribution, cells were treated with Triton X-100 after incubation with 10 µg/mL antibody for 6 hours. Residual fluorescence indicated Triton insolubility of formed lipid rafts. SU-DHL-4 cells were treated with the indicated CD20 antibodies at 10 µg/mL for 24 hours in the presence of the pan caspase inhibitor Z-VAD-FMK (E) or the cytoskeleton inhibitor LatB (F). Subsequently, cells were stained with annexin V and 7-aminoactinomycin to assess cell death by flow cytometry. (F) Lysosomal cell death was quantified by LysoTracker Red staining on Ramos cells after incubation with CD20 antibodies (10 µg/mL) for 6 hours. (G) Three OBI variants (IgG1, IgA2, and IgA2-V11L) were compared with regard to direct cell death induction. Lysosomal cell death was quantified by LysoTracker Red staining of Ramos cells after incubation with the antibodies (10 µg/mL) for 6 hours. Results of 3 independent experiments are shown as mean ± SEM, with the exception of (G), for which n = 1. *Significant difference specific antibody vs untreated/isotype control. #Significant difference lgG1 vs lgA2. XSignificant difference between with vs without inhibitor (Z-VAD-FMK or LatB). Significant differences (P ≤ .05) were calculated using ANOVA. Original mangnification x20 for panel A. AV, annexin V; ctrl., control; n.s., not significant; w/o, without; 7AAD, 7-aminoactinomycin.
Direct lymphoma cell killing by CD20 antibodies of the IgA2 isotype. (A) Homotypic aggregation by CD20 antibodies was analyzed by phase-contrast microscopy using SU-DHL-4 target cells. Antibodies were added at 10 µg/mL for 24 hours. Representative images of 3 independent experiments are shown. (B) Induction of direct cell death induced by CD20 monoclonal antibody was analyzed by DiOC6 staining. Ramos cells were used as target cells, and antibodies were added at a fixed concentration (10 µg/mL) for 6 hours. (C) Lysosomal cell death was quantified by LysoTracker Red staining on Ramos cells after incubation with CD20 antibodies (10 µg/mL) for 6 hours. (D) Redistribution of CD20 to lipid rafts was analyzed by flow cytometry on Ramos cells. For discrimination between nonraft and lipid raft distribution, cells were treated with Triton X-100 after incubation with 10 µg/mL antibody for 6 hours. Residual fluorescence indicated Triton insolubility of formed lipid rafts. SU-DHL-4 cells were treated with the indicated CD20 antibodies at 10 µg/mL for 24 hours in the presence of the pan caspase inhibitor Z-VAD-FMK (E) or the cytoskeleton inhibitor LatB (F). Subsequently, cells were stained with annexin V and 7-aminoactinomycin to assess cell death by flow cytometry. (F) Lysosomal cell death was quantified by LysoTracker Red staining on Ramos cells after incubation with CD20 antibodies (10 µg/mL) for 6 hours. (G) Three OBI variants (IgG1, IgA2, and IgA2-V11L) were compared with regard to direct cell death induction. Lysosomal cell death was quantified by LysoTracker Red staining of Ramos cells after incubation with the antibodies (10 µg/mL) for 6 hours. Results of 3 independent experiments are shown as mean ± SEM, with the exception of (G), for which n = 1. *Significant difference specific antibody vs untreated/isotype control. #Significant difference lgG1 vs lgA2. XSignificant difference between with vs without inhibitor (Z-VAD-FMK or LatB). Significant differences (P ≤ .05) were calculated using ANOVA. Original mangnification x20 for panel A. AV, annexin V; ctrl., control; n.s., not significant; w/o, without; 7AAD, 7-aminoactinomycin.
In vivo efficacy of IgA2 antibodies against CD20
We then investigated our novel IgA2 antibodies against CD20 in 3 in vivo models using human CD89-Tg mice. In an intraperitoneal tumor model, mice were injected with 5 × 106 human CD20-transduced EL4 (EL4-CD20) cells (Figure 6A). Twenty-four hours after monoclonal antibody injection, the remaining EL4-CD20 cells were quantified from peritoneal lavages by flow cytometry. OBI-IgA2, but not RTX-IgA2 or OFA-IgA2, significantly reduced the number of EL4-CD20 cells found in the peritoneal cavity (Figure 6B). Interestingly, all 3 IgA2 antibodies recruited neutrophils to the peritoneal cavity, with OBI-IgA2 causing the greatest neutrophil influx (Figure 6C).
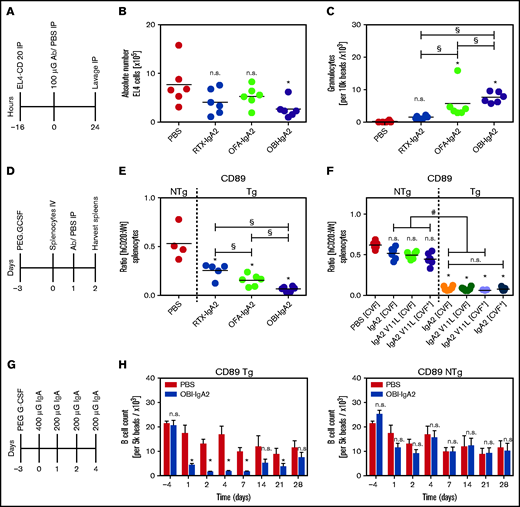
In vivo efficacy of IgA2 antibodies against CD20. (A) Schematic experimental overview of the short intraperitoneal (IP) model. (B) Absolute quantification of EL4-CD20 cells after intraperitoneal injection of the indicated CD20 IgA2 antibodies (100 µg per mouse). (C) Absolute quantification of intraperitoneal Ly6G+ granulocytes after treatment with the indicated CD20 IgA2 antibodies. (D) Schematic overview of the adoptive transfer model. (E) Ratio of labeled CD20-Tg/NTg splenic B cells after treatment of pegylated–granulocyte colony-stimulating factor–primed CD89-Tg or NTg mice with the indicated CD20 IgA antibodies (250 µg per mouse). (F) Ratio of labeled CD20-Tg/NTg B cells using CD89-Tg or NTg mice that did (CVF+) or did not (CVF−) receive intraperitoneal cobra venom factor (CVF) to deplete complement. Mice were treated with an unmodified (IgA2) or a PCD-impaired variant (IgA2 V11L) of OBI-IgA. (G) Schematic experimental overview of a syngeneic human CD20 Tg B-cell depletion model. (H) Quantification of remaining B cells after repeated treatment with OBI-IgA2, as indicated in (G), using CD89-Tg mice (left panel) or NTg mice (right panel). Results are presented as scatter dot blots (A-D) and as mean ± SEM of 6 mice per group (E). Statistical analyses were performed using 1-way ANOVA. *Significant difference vs PBS, unless indicated otherwise. #Significant difference OBI treatment NTg vs OBl treatment Tg. §Significant difference between different IgA2 antibodies. Horizontal line indicates the median. Ab, antibody; G-CSF, granulocyte colony-stimulating factor; h, human; n.s., not significant; Wt, wild-type.
In vivo efficacy of IgA2 antibodies against CD20. (A) Schematic experimental overview of the short intraperitoneal (IP) model. (B) Absolute quantification of EL4-CD20 cells after intraperitoneal injection of the indicated CD20 IgA2 antibodies (100 µg per mouse). (C) Absolute quantification of intraperitoneal Ly6G+ granulocytes after treatment with the indicated CD20 IgA2 antibodies. (D) Schematic overview of the adoptive transfer model. (E) Ratio of labeled CD20-Tg/NTg splenic B cells after treatment of pegylated–granulocyte colony-stimulating factor–primed CD89-Tg or NTg mice with the indicated CD20 IgA antibodies (250 µg per mouse). (F) Ratio of labeled CD20-Tg/NTg B cells using CD89-Tg or NTg mice that did (CVF+) or did not (CVF−) receive intraperitoneal cobra venom factor (CVF) to deplete complement. Mice were treated with an unmodified (IgA2) or a PCD-impaired variant (IgA2 V11L) of OBI-IgA. (G) Schematic experimental overview of a syngeneic human CD20 Tg B-cell depletion model. (H) Quantification of remaining B cells after repeated treatment with OBI-IgA2, as indicated in (G), using CD89-Tg mice (left panel) or NTg mice (right panel). Results are presented as scatter dot blots (A-D) and as mean ± SEM of 6 mice per group (E). Statistical analyses were performed using 1-way ANOVA. *Significant difference vs PBS, unless indicated otherwise. #Significant difference OBI treatment NTg vs OBl treatment Tg. §Significant difference between different IgA2 antibodies. Horizontal line indicates the median. Ab, antibody; G-CSF, granulocyte colony-stimulating factor; h, human; n.s., not significant; Wt, wild-type.
In a second in vivo model, we investigated depletion of syngeneic human CD20-transgenic B cells by IgA2 antibodies (Figure 6D). In this experiment, all 3 CD20 IgA2 antibodies mediated significant B-cell depletion, with OBI-IgA2 causing more effective B-cell killing than RTX-IgA2 or OFA-IgA2 (Figure 6E). Next, we explored the relative importance of potential effector mechanisms of OBI-IgA2 in this model. Thus, we used an OBI-IgA2 variant (V11L) with a reduced capacity to induce lysosomal cell death but preserved ADCC function (see Figure 5G; supplemental Figure 4A), treated mice with cobra venom factor (CVF) to halt complement activation, or used CD89 nontransgenic (NTg) mice to lose the ADCC capacity of IgA antibodies. Interestingly, reduction in direct cell killing capacity, lack of complement activation, or a combination thereof did not have a significant effect on B-cell depletion by OBI-IgA2 in vivo. However, human CD89 expression was required for OBI-IgA2–mediated B-cell depletion, because B-cell depletion in CD89-NTg mice was significantly impaired compared with that in CD89-Tg mice (Figure 6F).
Additionally, we performed in vivo B-cell depletion experiments in human CD89/CD20 double-Tg mice (Figure 6G). Results showed that OBI-IgA2 achieved significant B-cell depletion compared with PBS treatment in CD89-Tg mice during and shortly after antibody injections, but B-cell numbers returned to normal over time (Figure 6H, middle panel). However, when CD89-NTg mice were used in these experiments, OBI-IgA did not significantly deplete B cells (Figure 6H, right panel), again indicating the importance of FcαRI (CD89) and effector cell–mediated tumor cell–killing mechanisms for OBI-IgA2’s therapeutic efficacy in vivo.
In conclusion, our results indicate that investigations into antibodies’ effector mechanisms can be essential for choosing the most appropriate CD20 antibody candidate for particular applications. For the recruitment of myeloid effector cells, and PMNs in particular, the variable regions of OBI proved more effective than did those of RTX or OFA. These observations may be particularly relevant in the therapeutic context of myeloid checkpoint blockade (eg, by CD47-blocking antibodies or by glutaminyl cyclase inhibitors).
Discussion
Here, we demonstrate that OBI-IgA2 was particularly effective in triggering myeloid cell–mediated killing of lymphoma cells in vitro and in vivo. This observation may become relevant in combination with myeloid checkpoint blocking therapy (eg, by magrolimab [Hu5F9-G4]40 or by glutaminyl cyclase inhibitors29 ) because myeloid cells, and PMNs in particular, are effectively activated by human IgA antibodies.
Studies into the mechanisms of action of clinically approved monoclonal antibodies hold promise to identify approaches to improve their therapeutic efficacy.5 In the context of CD20 antibodies, the second-generation antibody OFA is differentiated from RTX by its superior CDC activity.41 OBI as a third-generation antibody is the only approved type II CD20 antibody with enhanced programmed cell death (PCD) and reduced CDC activity compared with the type I antibodies RTX and OFA.39 Additionally, OBI is produced as a low fucosylated IgG1 antibody, which improves FcγRIII (CD16) binding and, thereby, enhances ADCC by NK cells.42,43 However, all 3 CD20 antibodies were not effective in triggering ADCC by PMNs and were similarly effective in recruiting macrophages for engulfment of target cells (Figure 1), with the latter being in agreement with previous observations.44
Importantly, ADCP by macrophages, but not ADCC by PMNs, was enhanced in the presence of CD47 blockade. Because myeloid cells were demonstrated to critically contribute to CD20 antibody efficacy in mice,3,4 are the main recruited effector cell population resulting from myeloid checkpoint blockade with CD47 antibodies,6,8,9 and often are activated more effectively by IgA antibodies than by IgG1 antibodies,14,16,17 we evaluated the functional impact of switching the 3 approved CD20 antibodies from the human IgG1 isotype to the human IgA2 isotype.
The most unexpected finding is the observation that OBI-IgA2 was consistently more effective than RTX-IgA2 or OFA-IgA2 in triggering ADCC by PMNs. Although the underlying mechanisms of tumor cell killing by T or NK cells were intensively investigated and are quite well understood,45 PMN-mediated tumor cell killing was only recently reported to depend on “trogoptosis,” a process that relates to trogocytosis leading to frustrated phagocytosis and, eventually, tumor cell death.46 Macrophage-mediated tumor cell phagocytosis has been demonstrated to be enhanced when tumor-targeting antibodies bind to membrane-distal regions of target antigens, whereas NK cell–mediated ADCC and CDC are more effective when antibodies bind to epitopes that are closer to the tumor cell membrane.47,48 To our knowledge, results from similar studies are not available for PMNs but could not explain the differences among CD20 antibodies, which all bind to partially overlapping epitopes close to the cell membrane.
Tumor cell killing by PMNs was described to critically depend on target antigen density,46 but other less well-defined target antigen characteristics were observed.27,47 The CD20 antigen is a member of the MSA4 protein family with 2 rather small extracellular loops,49,50 which predominantly forms double-barrel dimers.50 The antigen binding sites for the 3 approved CD20 antibodies have been well defined and are partially overlapping.51-53 The enhanced NK cell–mediated ADCC by OBI compared with RTX and OFA has been explained by OBI’s low fucosylated Fc part, which is an established glycomodification used to improve FcγRIII binding for IgG antibodies.42,54 However, binding of IgA antibodies to FcαRI (CD89) is not affected by antibodies’ glycosylation32 ; furthermore, glycoprofiles of our 3 CD20 IgA antibodies were similar (supplemental Figure 2C). Together, these results suggest that an inherent feature of OBI’s variable regions is particularly efficient in ADCC by PMNs. To analyze whether enhanced ADCC by PMNs is a general characteristic of type II CD20 antibodies of the IgA isotype, we generated and tested an IgA2 variant of the type II antibody 11B8. Interestingly, 11B8-IgA2 was also significantly more effective than RTX-IgA2 at triggering ADCC by PMNs (supplemental Figure 4B).
After intensive research by several groups, the fundamental differences between type I and type II CD20 antibodies have been related to differences in clustering of CD20 molecules by CD20 antibodies on the B-cell surface.51,53,55 Recent structural studies elegantly demonstrated that each CD20 dimer is bound by 2 RTX molecules, resulting in cross-linking of CD20 dimers into higher-order assemblies. In contrast to RTX, only 1 OBI molecule can bind to each CD20 dimer, thereby preventing more extensive CD20 cross-linking.51,53 Additional studies revealed that bivalent binding differed between CD20 antibodies: compared with OFA and RTX, OBI displayed the lowest amount of bivalent binding.56 Furthermore, the elbow angle between VH and CH1 of OBI is wider compared with that of RTX, which is reversed by a V11L mutation in OBI.57 Interestingly, V11L impaired direct lymphoma cell killing (Figure 5G) but did not negatively affect PMN-mediated ADCC (supplemental Figure 4A). Thus, type II antibodies of the IgA2 isotype are more effective than type I antibodies in triggering ADCC by PMNs, but their direct lymphoma killing capacity appears to be dispensable for enhanced ADCC by PMNs. In conclusion, differences in antibody orientation after antigen binding or in target antigen clustering on the lymphoma cell membrane appear to affect ADCC by PMNs.
To further improve neutrophil-mediated killing by CD20 antibodies, we explored the effects of CD47 blockade. Previous studies have demonstrated that RTX treatment benefits from blocking the CD47-SIRPα axis in preclinical models,40 which appears to also translate into clinical benefit for lymphoma patients.11 Macrophages were postulated to constitute the predominant effector cell type potentiated by this approach. Nevertheless, it is of interest to understand the impact of CD47 blockade on neutrophil-mediated killing.58 Here, we show that, at least in vitro, IgG1 antibodies do not benefit from CD47 blockade in neutrophil-mediated ADCC. On the other hand, IgA2 antibodies showed significantly increased ADCP with macrophages and increased ADCC with neutrophils after CD47 blockade or glutaminyl cyclase inhibition, suggesting the promise of a combination therapy using IgA CD20 antibodies and myeloid checkpoint inhibition.
In addition to ADCP and ADCC, we investigated complement activation by CD20 antibodies of the IgA2 isotype against lymphoma cell lines as another Fc-dependent effector mechanism. Although IgA antibodies lack a C1q binding site,37 IgA antibodies against CD20 were reported to induce CDC in a C1q-dependent fashion.15,36,59 Similarly, other CD20 antibody molecules lacking a C1q binding site, such as IgG4 and F(ab′)2 fragments, showed activation of the classical complement pathway. Clustering of the B-cell receptor was suggested as a potential mechanism for this so-called “bypass CDC,” which could then provide the required C1q binding moiety.59 However, this explanation is controversial because the same type of complement activation was observed after knockout of the B-cell receptor,60 suggesting that other molecules present within the CD20-containing lipid rafts may provide the required C1q binding site.
These previous results are confirmed by this study. As observed for IgG1 antibodies,61 IgA2 variants of the type I antibodies RTX and OFA were more effective in CDC induction compared with the type II antibody OBI. CDC has been demonstrated to contribute to the therapeutic efficacy of CD20 antibodies in particular mouse models25 and may contribute to the induction of tumor-directed immune responses62,63 ; however, its contribution to the clinical efficacy of CD20 antibodies has remained controversial.34,64,65 On the other hand, uncontrolled complement activation has been suggested to contribute to the first-dose toxicity of CD20 antibodies, which can be mediated by different mechanisms, including cytokine release.66
Another suggested mechanism of action for CD20 antibodies is PCD induction, although its contribution to the clinical efficacy of CD20 antibodies is again controversial.67 Previous studies have reported that an isotype switch of RTX from human IgG1 to IgG2 or IgG4 could affect PCD induction,68 which was supposed to be related to differences in how these antibodies recognize CD20 tetramers on the lymphoma cell membrane.52,57 Within our panel of antibodies, IgG1 and IgA2 versions of the type II antibody OBI were the most effective in inducing direct cell death. As previously shown for OBI,21,69 PCD by OBI-IgA2 was also caspase independent but required a functional cytoskeleton. Interestingly, OBI-IgA2’s PCD activity also appeared to depend on the antibody’s elbow hinge region, because a V11L mutation reduced its PCD activity, as previously described for OBI-IgG1.39 Conversion of type I CD20 antibodies from IgG1 to IgA2 unexpectedly increased homotypic aggregation, whereas lysosomal cell death and lipid raft induction remained mostly unchanged. Thus, overall direct effector mechanisms of the 3 CD20 antibodies were not affected by the isotype switch from IgG1 to IgA2. Importantly, PCD did not appear to contribute to B-cell depletion efficacy in vivo, because the OBI-IgA2 variant with reduced PCD activity in vitro (OBI-IgA2.0–V11L) was as effective as the OBI-IgA2 wild-type antibody in vivo (Figure 5F).
When we elucidated the mechanism of action of OBI-IgA2 in vivo, the presence of the human CD89 transgene proved essential for IgA’s efficacy in vivo (Figure 6F). Together with results obtained after complement depletion by CVF or with the PCD-reduced OBI-IgA2–V11L variant, these observations demonstrated that Fc receptor–dependent mechanisms (ADCC or ADCP) were required, whereas PCD and CDC were dispensable for B-cell depletion by OBI-IgA2 in vivo. This dependency on CD89-dependent mechanisms could help to explain why OBI-IgA2 outperformed RTX-IgA2 and OFA-IgA2 in the same models, because the latter 2 were less effective in ADCC in vitro. Interestingly, OBI-IgA2 also was the most effective anti-CD20 IgA2 at recruiting PMNs into the peritoneal cavity (Figure 6C), the potential effector cell population in this model. Our results showing that IgA antibodies depend on FcαRI (CD89) for in vivo efficacy are in line with results for other CD20 antibodies of the IgA isotype15,36 and match with results for CD20 antibodies of the IgG1 isotype, which require activating FcγRs for their therapeutic activity.3,70 Whether each of these monoclonal antibodies would be more effective in the IgG or IgA2.0 format requires further investigation using complex models encompassing all of the relevant Fc receptors and human target molecules.
Because myeloid checkpoint–blocking therapies (eg, CD47-blocking antibodies, such as magrolimab [Hu5F9-G4]) rely on ADCP or ADCC for their therapeutic efficacy, our results suggest that OBI may be a particularly promising CD20 antibody for this approach. An interesting observation within our in vivo B-cell depletion experiments was the decrease in B-cell numbers in the PBS control groups that were not treated with antibody, which was less pronounced than in OBI-IgA2–treated animals. We attribute these differences to the use of granulocyte colony-stimulating factor before the start of the experiment. Although granulocyte colony-stimulating factor was administered to improve granulocyte numbers and functions in the peripheral blood of mice, it was reported to suppress B lymphopoiesis.71,72
We conclude that OBI-IgA2 is a CD20 antibody with exceptional B-cell killing properties in vitro and in vivo. In particular, OBI-IgA2’s ability to recruit myeloid cells for ADCC and ADCP of lymphoma cells suggests that it could be an interesting molecule to combine with CD47-SIRPα–blocking strategies. Additional preclinical studies are required to assess the clinical potential of this approach.
Acknowledgments
This work was supported by German Research Organization grants DFG Pe 1425/5-1 (M.P.) and DFG Va 124/9-1 (T.V.), the Deutsche Krebshilfe “Mildred Scheel Professorship” program (M.P.), Dutch Cancer Research grant R3695 (M.E., M.J., and J.H.W.L.), Villa Joep grant 17 (M.N.), and grant 227 from the Dutch Kinderen Kankervrij Foundation (T.t.B.).
Authorship
Contribution: M.E., T.R., A.D., J.H.M.J., N.B., T.t.B., M.N., K.E., and K.R. performed experiments and analyzed data; M.E., T.R., J.H.W.L., and T.V. designed experiments and wrote the manuscript; J.H.W.L. and T.V. supervised the project; and all authors critically revised and reviewed the manuscript and approved the final version for submission.
Conflict-of-interest disclosure: J.H.W.L. is a cofounder of and shareholder in TigaTx. T.t.B. performed experiments at TigaTx, which also provided research funding to T.R. and T.V. The remaining authors declare no competing financial interests.
Correspondence: Thomas Valerius, Section for Stem Cell Transplantation and Immunotherapy, Department of Medicine II, Christian Albrechts University and University Medical Center Schleswig-Holstein, Campus Kiel, Arnold Heller Str 3, D-24105 Kiel, Germany; e-mail: t.valerius@med2.uni-kiel.de.

