

Key Points
Trauma-induced organ failure is characterized by an ADAMTS13/VWF imbalance, circulating large VWF multimers, and low platelet counts.
rhADAMTS13 and plasma restore this imbalance, reducing endothelial leakage, microthrombi, and organ failure in a trauma-induced shock model.

Abstract
Trauma-induced organ failure is characterized by endothelial dysfunction. The aim of this study was to investigate the role of von Willebrand factor (VWF) and its cleaving enzyme, ADAMTS13 (a disintegrin and metalloproteinase with thrombospondin type 1 motifs, member 13) in the occurrence of endothelial permeability and organ failure in trauma. In an observational study in a level-1 trauma center, 169 adult trauma patients with clinical signs of shock and/or severe injuries were included. Trauma was associated with low ADAMTS13 and high VWF antigen levels, thus generating an imbalance of ADAMTS13 to VWF. Patients who developed organ failure (23%) had greater ADAMTS13-to-VWF imbalances, persistently lower platelet counts, and elevated levels of high-molecular-weight VWF multimers compared with those without organ failure, suggesting microthrombi formation. To investigate the effect of replenishing low ADAMTS13 levels on endothelial permeability and organ failure using either recombinant human ADAMTS13 (rhADAMTS13) or plasma transfusion, a rat model of trauma-induced shock and transfusion was used. Rats in traumatic hemorrhagic shock were randomized to receive crystalloids, crystalloids supplemented with rhADAMTS13, or plasma transfusion. A 70-kDa fluorescein isothiocyanate–labeled dextran was injected to determine endothelial leakage. Additionally, organs were histologically assessed. Both plasma transfusion and rhADAMTS13 were associated with a reduction in pulmonary endothelial permeability and organ injury when compared with resuscitation with crystalloids, but only rhADAMTS13 resulted in significant improvement of a trauma-induced decline in ADAMTS13 levels. We conclude that rhADAMTS13 and plasma transfusion can reduce organ failure following trauma. These findings implicate the ADAMTS13-VWF axis in the pathogenesis of organ failure.
Introduction
In severe trauma, bleeding contributes to early mortality, whereas late morbidity and mortality is mostly due to multiple organ dysfunction syndrome (MODS).1-3 Due to improved transfusion strategies, more patients survive the initial phase of trauma, rendering them prone to develop MODS.4-6 However, the incidence of MODS has not decreased. As current mortality rates of trauma-induced MODS are 30%, there is a need for additional therapies.7 In the development of MODS, endothelial activation with associated endothelial permeability plays a key role.8 In response to traumatic injury, endothelial cells release von Willebrand factor (VWF).9 VWF binds to platelets, which is the initial step in the formation of a platelet plug at the site of injury. VWF is synthesized in endothelial cells as ultra-large VWF multimers, which are prothrombotic. These multimers are cleaved by the VWF-cleaving protease ADAMTS13 (a disintegrin and metalloprotease with thrombospondin type 1 motifs member 13), thereby reducing their size and rendering the adhesive multimers more quiescent.10 VWF release is critical to control bleeding, reflected by the bleeding tendency of VWF-deficient patients. When the amount of VWF released from the activated endothelium supersedes the cleaving capacity of ADAMTS13, an ADAMTS13-to-VWF imbalance occurs, with ensuing circulating ultra-large VWF multimers. In sepsis, this imbalance goes with a consumption coagulopathy, thrombocytopenia, microthrombi formation, and MODS.11–13 A similar ADAMTS13-to-VWF derangement was recently shown in trauma patients, which was associated with circulating markers of endothelial damage.14,15 Thereby, an ADAMTS13-to-VWF imbalance may also contribute to MODS following trauma-induced shock.
Traumatic bleeding is treated with plasma transfusion, with a supposed beneficial effect of replenishing a deficit in coagulation factors.16,17 However, plasma transfusion does not correct the early trauma-induced coagulopathy, nor does it correct coagulopathy in other patient populations.18–20 An alternative hypothesis is that resuscitation of shock using plasma may limit endothelial permeability. In animal models of hemorrhagic shock, plasma transfusion reduced syndecan-1 shedding from the glycocalyx when compared with the use of crystalloid fluids, associated with less endothelial permeability and less organ damage.21,22 However, the specific mechanism is not known. Of note, plasma is lifesaving in thrombotic thrombocytopenic purpura (TTP) by replenishing low ADAMTS13 levels.23,24 Also, we showed that plasma increases ADAMTS13 levels and decreases VWF levels in critically ill patients with a consumption coagulopathy, associated with reduced syndecan-1 levels.20 Therefore, in these studies, we hypothesize that in patients with trauma-induced MODS, a decreased ADAMTS13/VWF ratio with a high load of circulating VWF multimers is present. We also hypothesize that plasma transfusion and recombinant human ADAMTS13 restore a disturbed ADAMTS13-to-VWF ratio, thereby limiting microthrombi formation, which in turn limits microcirculatory obstruction, hypoxia, endothelial cell activation, and permeability with ensuing organ failure.
Methods
Study population
After institutional and ethical review board approval, a clinical study was conducted as an ancillary study of the Activation of Coagulation and Inflammation in Trauma III (ACIT-III) trial (trial number NL58766.018.16). The ACIT-III is a prospective cohort study on the pathophysiology of inflammation and coagulation in adult trauma patients at the Amsterdam University Medical Centers, location AMC, the Netherlands, between 2012 and 2018. When patients met the inclusion criteria (supplemental Table 1), encompassing severely injured adult patients, citrated blood samples were collected at presentation with deferred consent. Informed consent was obtained from patients or relatives as soon as possible. If informed consent was obtained within 24 hours, then consecutive blood samples at 24 hours and 72 hours post-injury were taken. Patients were included in the present study if ≥2 consecutive blood samples were available. After collection into citrated tubes, blood was centrifuged twice (2 × 1750g, 18°C, 10 minutes, Eppendorf 5804R), and plasma was stored at −80°C for further analysis. Patient characteristics were recorded, including injury severity score, presence of traumatic brain injury, and presence of MODS. Traumatic brain injury was defined as an abbreviated injury score of 3 in the head region. MODS was assessed daily and defined as a sequential organ failure assessment (SOFA) score ≥3 in ≥2 organ systems, yielding a minimum SOFA score of 6. Also, ventilator days, intensive care length of stay, hospital length of stay, and early (≤1 day) and late mortality (>1-30 days) were recorded.
Rat trauma transfusion model
Experiments were approved by the national and Institutional Animal Care and Use Committee of the Amsterdam University Medical Centers, location AMC, in 2018. All procedures were in accordance with European (2010/63/EU) and national laws (Wod, 2014). Animals were group housed (2-4 rats per cage) with access to water and food ad libitum ≥7 days before the experiment. Studies were conducted and reported following the ARRIVE guidelines.25
Preparation of rat plasma product
Plasma products were prepared using syngeneic donor rats as described before.26 In short, whole blood was obtained through heart puncture and collected in citrate-phosphate-dextrose solution in a 9:1 ratio. Whole blood was centrifuged once (1892g, 10 minutes, 20°C) to separate the cells and platelets from plasma. Plasma was collected up to 0.5 cm above the buffy coat. One donor was used for the preparation of 1 plasma product (n = 13), frozen in −80°C and, when used, thawed in a warm water bath of 37°C.
Experimental trauma model.
A rat multiple trauma transfusion model was used (supplemental Figure 1).26 In short, male Sprague Dawley rats (n = 13 per group) were sedated and mechanically ventilated. Baseline blood samples (T = 0) were taken. Thereafter, trauma was inflicted through fracture of the right femur, crush injury of the liver and small intestine and by hemorrhage of 40% of the circulating volume. After one hour of traumatic hemorrhagic shock, rats were randomized to receive resuscitation with Ringer’s lactate (Baxter) infusion (crystalloid) or Ringer’s lactate with recombinant human ADAMTS13 (crystalloid + 0.4 µg/g rhADAMTS13; R&D Systems) or single donor plasma. Dose dependency of rhADAMTS13 was evaluated in a human lung endothelial cell permeability model (supplemental Methods). Rats were resuscitated according to a pressure-fixed transfusion strategy by targeting a mean arterial pressure (MAP) of 60 mm Hg. Transfusion/infusion speed was set at 8 mL per hour. After the transfusion target was achieved for 5 minutes, transfusion/infusion was stopped. Before trauma and at termination (6 hours after trauma), syndecan-1, soluble vascular adhesion molecule-1 (sVCAM-1), interleukin-6 (IL-6) VWF antigen, VWF multimers, and ADAMTS13 antigen levels were measured. Additionally, 30 minutes prior to termination of the experiment, a 70-kDa fluorescein isothiocyanate (FITC)–labeled dextran was administered to assess endothelial permeability. After termination, the circulation was flushed with 50 mL 0.9% NaCl against gravitational force. The hilum of the left lung and left kidney were tied off prior to flushing to further assess endothelial leakage by measuring lung and kidney wet/dry ratios. Hematoxylin and eosin slides were scored by a pathologist (J.K.) who was blinded to treatment allocation (supplemental Table 2).
Measurements
Enzyme-linked immunosorbent assays (ELISAs) for human samples
Human VWF propeptide, VWF antigen, ADAMTS13 antigen, and ADAMTS13 activity were performed as described earlier.27–33 For a detailed description, see supplemental Methods. Syndecan-1 was performed according to instructions of the manufacturer using a commercially available kit (R&D Systems).
Human and rat VWF multimers
High-molecular-weight (HMW) VWF multimers were measured as previously described34 in a subset of patients, including 12 patients with MODS and in 8 control patients without MODS.34,35 Rat VWF multimer analysis was performed using similar methods as human VWF multimers. For a detailed description, see supplemental Methods.
ELISAs for animal samples
Rat VWF antigen and rat and human ADAMTS13 antigen levels were performed as described before.36 For a detailed description, see supplemental Methods. Rat syndecan-1 (eLabscience), sVCAM-1 (eLabscience), and IL-6 (R&D Systems) levels were performed using commercially available kits.
Biochemical assessment of organ failure in the animal model
Aspartate and alanine transaminases, creatinine, and total urine protein were measured by standard enzymatic reactions using spectrophotometric, colorimetric, or turbidimetric measurement methods, measured by the clinical laboratory.
Rotational thromboelastometry
Rotational thromboelastometry was used to determine coagulation abnormalities within the animal model. EXTEM was used to evaluate the extrinsic coagulation pathway (eg, adding tissue factor and phospholipids to initiate coagulation). Clotting time (CT) determines time to 2-mm clot formation, clot formation time evaluates the time from CT to 20 mm of clot formation, maximum clot firmness (MCF) represents the strength of the clot, and the 30-minute lysis index (LI30) determines the percentage of amplitude decrease 30 minutes after maximum clot firmness, indicating clot lysis. FIBTEM was used to evaluate the contribution of fibrinogen to clot formation (eg, EXTEM reagents + cytocholasin D (= platelet inhibitor)).
Immunohistochemical analysis of VWF-platelet microthrombi and endothelial leakage
For determination of VWF-platelet microthrombi in the lung, deparaffinized tissue slides were double stained with rabbit anti-VWF/anti-rabbit alkaline phosphatase and vector blue staining method. Slides were again heated in citrate buffer and blocked to facilitate the second staining using rabbit anti-CD41/anti-rabbit horseradish peroxidase NovaRed staining method. Double staining resulted in purple microthrombi within the lung. Other deparaffinized tissue slides were colored using a rabbit anti-FITC/anti-rabbit horseradish peroxidase and NovaRed coloring method. Five different images of each organ, blinded for the treatment allocation to the assessor, were obtained by microscope (200× zoom) and camera (BX51 with UC90, Olympus). Pictures were inverted using ImageJ. Five random inverted pictures were used to set a threshold of positivity for either VWF-platelet complexes and FITC 70-kDa dextran leakage, respectively. Median percentage of area intensity was used as a measure for endothelial leakage.
Sample size calculation animal model
In a previous trauma-induced hemorrhagic shock experiment, a tyrosine kinase inhibitor compared with vehicle next to a balanced transfusion strategy reduced mean lung injury scores (6.2 vs 4.0, with a standard deviation [SD] of 1.8). Therefore, using these data to extrapolate a sample size with 3 groups using a 1-way analysis of variance (V = 1.076), the use of 11 rats has a power of 80% to reach a statistically significant difference in lung injury scores. With an expected mortality of 15% in our model, we added 2 additional rats per group, yielding n = 13 per group.
Statistical analysis
Analyses were done using SPSS Statistics version 26 (IBM). Graphs were made using GraphPad Prism 8 (GraphPad Software). Normality was tested using Kolmogorov-Smirnov test and visual inspection of histograms, and values are represented as either mean (± SD) or as median (Q1-Q3 = interquartile range [IQR]) in tables. In figures, boxplots are shown with full range. Outliers were defined as >2 SDs from mean and were not incorporated in the statistical analysis. Differences in clinical data were tested with Student t test, Mann-Whitney U test, or χ2 test/Fisher’s exact test, depending on distribution and type of data. For clinical data, paired nonparametric data were tested with a Friedman analysis of variance for variance with post-hoc comparisons. Simple linear regression was used for the correlation between Injury Severity Score (ISS) and the ADAMTS13-VWF axis.
For the experimental data, Kruskal-Wallis with post-hoc multiple comparisons were performed. For paired animal data, similar methods as for the clinical data were used. ELISA values below the reference value were set at half of the lowest detection range.37 A P value of < .05 was considered to be statistically significant. A Bonferroni correction was used to correct for multiple testing.
Results
Of the 169 included trauma patients, 105 (62.1%) had polytrauma and 39 (23.1%) developed MODS (Table 1). On presentation, patients who developed MODS had higher base deficits, prolonged prothrombin times, and decreased fibrinogen levels compared with patients without MODS. Overall mortality rate was 11.8%.
Characteristics of trauma patients at Emergency Department presentation and outcome of trauma
| Parameter | All patients (n = 169) | No MODS (n = 130) | MODS (n = 39) | P |
|---|---|---|---|---|
| Demographics | ||||
| Age (y) | 51 (31-62) | 51 (32-60) | 52 (31-67) | .514 |
| Female sex, n (%) | 35 (20.7) | 24 (18.5) | 11 (28.2) | .126 |
| Blunt injury, n (%) | 159 (94.1) | 122 (93.8) | 37 (94.9) | .881 |
| Injury severity score | 20 (9-27) | 16 (9-25) | 27 (22-38) | <.001 |
| TBI, n (%) nonisolated | 73 (43.2) 34 (20.1) | 48 (36.9) 19 (14.6) | 25 (64.1) 15 (38.5) | .001 .002 |
| Vital parameters | ||||
| Heart rate (bpm) | 82 (70-97) | 81 (68-93) | 89 (78-112) | .024 |
| Systolic blood pressure (mmHg) | 130 (28) | 133 (115-147) | 120 (108-142) | .081 |
| Glasgow Coma Scale, TBI patients | 14 (3-15) 5 (3-14) | 14 (6-15) 6 (3-14) | 4 (3-14) 3 (3-9) | <.001 .162 |
| Hemoglobin (g/dL) | 13.4 (12.1-14.5) | 13.5 (12.6-14.8) | 12.0 (10.4-13.3) | <.001 |
| Lactate (mmol/L) | 2.4 (1.6-3.7) | 2.4 (1.6-3.4) | 2.5 (1.6-3.9) | .390 |
| Base excess (mEq/L) | −1.3 (−3.7 to 0.6) | −1.0 (−3.0 to 1.0) | −2.6 (−6.5 to −0.3) | .004 |
| Coagulation | ||||
| Platelet count (×109/L) | 217 (178-256) | 226 (185-257) | 190 (167-250) | .042 |
| PT (s) | 11.9 (11.2-12.7) | 11.7 (11.2-12.5) | 12.6 (11.9-14.1) | .002 |
| aPTT (s) | 23.5 (22.8-26.0) | 23 (22-25) | 26 (23-28) | .002 |
| Fibrinogen (g/L) | 2.2 (1.6-2.5) | 2.2 (1.8-2.6) | 1.8 (1.1-1.9) | .002 |
| D-dimer (mg/L) | 9.3 (2.5-32.0) | 6.7 (2.1-25.8) | 29.0 (14.2-32.0) | .002 |
| Fluids T0-T24 | ||||
| Crystalloids (mL) | 2000 (1000-2900) | 2000 (1000-2750) | 2400 (1050-5200) | .025 |
| Red blood cells (U) | 1.0 (3.4) | 0.5 (2.3) | 2.9 (5.1) | .007 |
| Plasma (U) | 0.7 (2.9) | 0.3 (1.8) | 1.8 (4.7) | .072 |
| Buffy coat platelets (U) | 0.4 (2.1) | 0.1 (0.9) | 1.2 (3.8) | .091 |
| Late complications and mortality | ||||
| MODS, n (%) | 39 (23.1) | 0 (0) | 39 (100) | ND |
| ICU days | 1 (0-5) | 1 (0-2) | 11 (4-18) | <.001 |
| Ventilator days | 0 (0-3) | 0 (0-1) | 7 (4-15) | <.001 |
| Hospital days | 7 (4-17) | 6 (3-11) | 24 (12-33) | <.001 |
| 24-h mortality, n (%) | 11 (6.5) | 11 (8.4) | 0 (0) | .070 |
| >24-h to 30-d mortality, n (%) | 9 (5.3) | 0 (0) | 9 (23.1) | <.001 |
| Parameter | All patients (n = 169) | No MODS (n = 130) | MODS (n = 39) | P |
|---|---|---|---|---|
| Demographics | ||||
| Age (y) | 51 (31-62) | 51 (32-60) | 52 (31-67) | .514 |
| Female sex, n (%) | 35 (20.7) | 24 (18.5) | 11 (28.2) | .126 |
| Blunt injury, n (%) | 159 (94.1) | 122 (93.8) | 37 (94.9) | .881 |
| Injury severity score | 20 (9-27) | 16 (9-25) | 27 (22-38) | <.001 |
| TBI, n (%) nonisolated | 73 (43.2) 34 (20.1) | 48 (36.9) 19 (14.6) | 25 (64.1) 15 (38.5) | .001 .002 |
| Vital parameters | ||||
| Heart rate (bpm) | 82 (70-97) | 81 (68-93) | 89 (78-112) | .024 |
| Systolic blood pressure (mmHg) | 130 (28) | 133 (115-147) | 120 (108-142) | .081 |
| Glasgow Coma Scale, TBI patients | 14 (3-15) 5 (3-14) | 14 (6-15) 6 (3-14) | 4 (3-14) 3 (3-9) | <.001 .162 |
| Hemoglobin (g/dL) | 13.4 (12.1-14.5) | 13.5 (12.6-14.8) | 12.0 (10.4-13.3) | <.001 |
| Lactate (mmol/L) | 2.4 (1.6-3.7) | 2.4 (1.6-3.4) | 2.5 (1.6-3.9) | .390 |
| Base excess (mEq/L) | −1.3 (−3.7 to 0.6) | −1.0 (−3.0 to 1.0) | −2.6 (−6.5 to −0.3) | .004 |
| Coagulation | ||||
| Platelet count (×109/L) | 217 (178-256) | 226 (185-257) | 190 (167-250) | .042 |
| PT (s) | 11.9 (11.2-12.7) | 11.7 (11.2-12.5) | 12.6 (11.9-14.1) | .002 |
| aPTT (s) | 23.5 (22.8-26.0) | 23 (22-25) | 26 (23-28) | .002 |
| Fibrinogen (g/L) | 2.2 (1.6-2.5) | 2.2 (1.8-2.6) | 1.8 (1.1-1.9) | .002 |
| D-dimer (mg/L) | 9.3 (2.5-32.0) | 6.7 (2.1-25.8) | 29.0 (14.2-32.0) | .002 |
| Fluids T0-T24 | ||||
| Crystalloids (mL) | 2000 (1000-2900) | 2000 (1000-2750) | 2400 (1050-5200) | .025 |
| Red blood cells (U) | 1.0 (3.4) | 0.5 (2.3) | 2.9 (5.1) | .007 |
| Plasma (U) | 0.7 (2.9) | 0.3 (1.8) | 1.8 (4.7) | .072 |
| Buffy coat platelets (U) | 0.4 (2.1) | 0.1 (0.9) | 1.2 (3.8) | .091 |
| Late complications and mortality | ||||
| MODS, n (%) | 39 (23.1) | 0 (0) | 39 (100) | ND |
| ICU days | 1 (0-5) | 1 (0-2) | 11 (4-18) | <.001 |
| Ventilator days | 0 (0-3) | 0 (0-1) | 7 (4-15) | <.001 |
| Hospital days | 7 (4-17) | 6 (3-11) | 24 (12-33) | <.001 |
| 24-h mortality, n (%) | 11 (6.5) | 11 (8.4) | 0 (0) | .070 |
| >24-h to 30-d mortality, n (%) | 9 (5.3) | 0 (0) | 9 (23.1) | <.001 |
Data are represented as number (percentage), median (IQR), or mean (SD). Comparisons were made between patients without MODS and patients with MODS. Buffy coat platelets consisted of 5 donors per unit.
aPTT, activated partial thromboplastin time; bpm, beats per minute; ICU, Intensive Care Unit; ND, not determined; PT, prothrombin time; TBI, traumatic brain injury.
The ADAMTS13/VWF ratio is disturbed after traumatic injury, related to injury severity, and associated with changes indicative of microthrombi formation and the development of MODS
Increasing injury severity scores were associated with increased baseline VWF propeptide and VWF antigen levels, while ADAMTS13 antigen levels decreased (Figure 1A-D). The median time to develop MODS was 2 [IQR 2-3] days post-injury. VWF propeptide levels were increased at presentation to the Emergency Department in both patients with and without MODS. These VWF propeptide levels dropped 24 hours after injury in both groups (P < .01), but remained higher in patients with MODS compared with patients without MODS (Figure 2A). Also, higher VWF antigen levels and lower ADAMTS13 antigen levels up to 24 hours after injury were shown in patients developing MODS (Figure 2B,D). ADAMTS13 activity levels were not different between patient groups during the first 72 hours. In a subset of patients who developed MODS, the percentage of high-molecular-weight (HMW) VWF multimers was higher on baseline compared with patients without MODS (51 [40-53] %, vs 42 [39-43] % multimers, P = .006; Figure 2F,I). Two patients had very low (9.1%) or absent HMW VWF multimers on baseline. One patient was hyperfibrinolytic determined by ROTEM, while the other only had increased ADAMTS13 activity (641%). The difference in HMW VWF multimers disappeared 72 hours after trauma (49 [38-50]% vs 44 [42-47]%, P = .063). Median platelet counts at 72 hours post-injury were lower in patients with MODS compared with patients without MODS, which is long after the bleeding had stopped. Also, other markers of consumption coagulopathy were more deranged in patients with MODS vs without MODS, such as increased D-dimer and prolonged PT. In addition, higher median plasma syndecan-1 were present in patients with MODS compared with patients without MODS (353.0 [125.0-1981.5] vs 125.0 [125.0-214.5] pg/mL, P = .001), depicting more endothelial wall shedding.
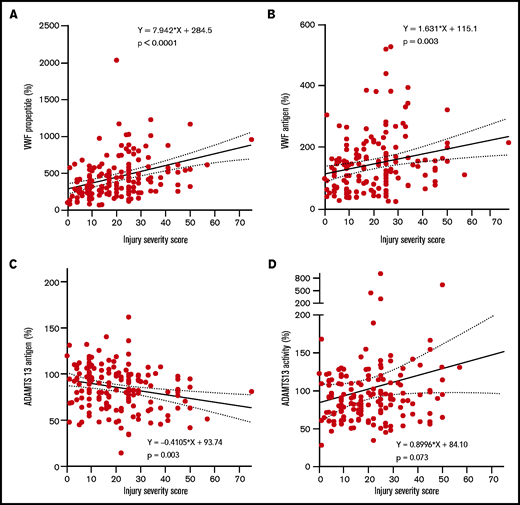
High injury severity scores correlate with high VWF propeptide, high VWF antigen levels, and low ADAMTS13 antigen levels. Line represents a linear regression line. Dotted lines represent the 95% confidence interval. (A) VWF propeptide levels. (B) VWF antigen. (C) ADAMTS13 antigen. (D) ADAMTS13 activity. All measures are calculated to percentage of normal human plasma levels.
High injury severity scores correlate with high VWF propeptide, high VWF antigen levels, and low ADAMTS13 antigen levels. Line represents a linear regression line. Dotted lines represent the 95% confidence interval. (A) VWF propeptide levels. (B) VWF antigen. (C) ADAMTS13 antigen. (D) ADAMTS13 activity. All measures are calculated to percentage of normal human plasma levels.
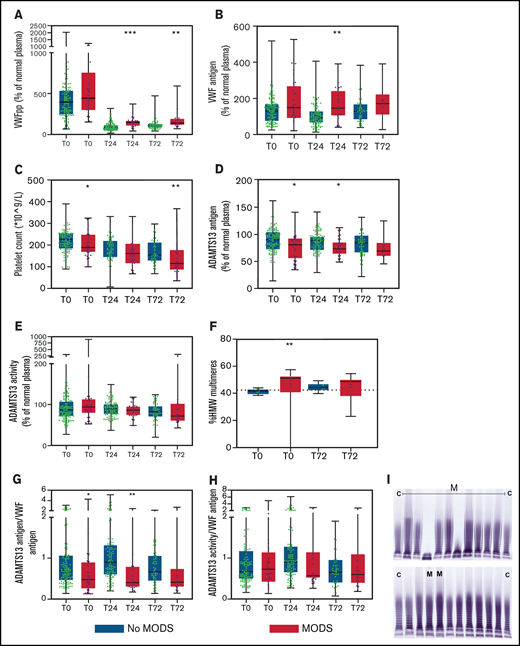
The VWF-ADAMTS13 balance is disturbed in patients who develop MODS and does not normalize over time, associated with platelet consumption and circulation of larger VWF multimers. Data are boxplots with range. (A) VWF propeptide. (B) VWF antigen. (C) Platelet count. (D) ADAMTS13 antigen. (E) ADAMTS13 activity. (F) HMW VWF multimers. (G) ADAMTS13 antigen/VWF antigen ratio. (H) ADAMTS13 activity/VWF antigen ratio. (I) Gel electrophoresis of VWF multimers. Dotted horizontal line in panel F is standard pool plasma level of VWF HMW VWF multimers. Two outliers in the MODS groups had 0% and 9.1% HMW multimers and were not incorporated in the statistical analysis. In panel I, “C” means control (normal human plasma pool) and “M” means patient developing MODS. *P < .05 and **P < .01 between patients without MODS and patients with MODS. VWFpp, human VWF propeptide.
The VWF-ADAMTS13 balance is disturbed in patients who develop MODS and does not normalize over time, associated with platelet consumption and circulation of larger VWF multimers. Data are boxplots with range. (A) VWF propeptide. (B) VWF antigen. (C) Platelet count. (D) ADAMTS13 antigen. (E) ADAMTS13 activity. (F) HMW VWF multimers. (G) ADAMTS13 antigen/VWF antigen ratio. (H) ADAMTS13 activity/VWF antigen ratio. (I) Gel electrophoresis of VWF multimers. Dotted horizontal line in panel F is standard pool plasma level of VWF HMW VWF multimers. Two outliers in the MODS groups had 0% and 9.1% HMW multimers and were not incorporated in the statistical analysis. In panel I, “C” means control (normal human plasma pool) and “M” means patient developing MODS. *P < .05 and **P < .01 between patients without MODS and patients with MODS. VWFpp, human VWF propeptide.
Experimental model of trauma-induced shock and transfusion
To explore whether plasma or rhADAMTS13 protect the endothelium, a rat trauma-induced shock and transfusion model was used (supplemental Figure 1). Baseline parameters are shown in Table 2. Animals were in shock, as depicted by an increase in lactate levels (Figure 3A). Traumatic shock was characterized by a significant decrease in ADAMTS13 antigen levels in the crystalloid-resuscitated animals, whereas VWF levels were not as markedly increased as in trauma patients (Figure 4A). All animals had lost equal amounts of blood. Mortality in this model was 10.3% (4/39), occurring in the crystalloid group (n = 2) or crystalloid + rhADAMTS13 group (n = 2). However, in the crystalloid with rhADAMTS13 group, mortality occurred due to technical difficulties (overdose of ketamine in 1 rat and perforation of the jugular vein in the other rat), whereas in the crystalloid-resuscitated rats, mortality occurred due to an inability to maintain MAP after treatment.
Baseline characteristics and assessments of coagulation and organ failure in an experimental trauma transfusion model
| Parameter | Crystalloid | Crystalloid + rhADAMTS13 | Plasma | |||
|---|---|---|---|---|---|---|
| Before injury | After 6 h | Before injury | After 6 h | Before injury | After 6 h | |
| Weight (g) | 348 (336-369) | ND | 353 (348-379) | ND | 350 (336-380) | ND |
| Arterial blood gas | ||||||
| pH | 7.41 (7.40-7.46) | 7.33 (7.29-7.46) | 7.43 (7.37-7.48) | 7.35 (7.30-7.39) | 7.45 (7.40-7.46) | 7.33 (7.28-7.39) |
| pCO2 (mm Hg) | 34.9 (32.1-41.1) | 22.7 (19.7-31.0) | 34.6 (32.7-44.1) | 31.0 (28.8-33.7)* | 35.1 (31.9-37.5) | 26.6 (24.9-31.5) |
| HCO3− (mmol/L) | 23.5 (21.1-24.9) | 13.2 (11.8-14.9) | 23.6 (22.5-25.0) | 17.6 (16.1-18.2) *,** | 22.5 (21.6-24.0) | 14.1 (11.1-16.2) |
| BE (mEq/L) | −0.1 (−1.9 to 0.4) | −10.3 (−12.8-−9.3) | −0.5 (−0.9 to 0.6) | −7.6 (−8.3 to −5.8) *,** | −1.3 (−1.8 to 0.2) | −10.0 (−14.1 to −8.6) |
| Ca2+ (mmol/L) | 1.1 (1.1-1.1) | 0.9 (0.8-1.1) | 1.1 (1.0-1.2) | 1.0 (1.0-1.1) | 1.1 (1.0-1.1) | 0.9 (0.9-1.1) |
| Hematology | ||||||
| Hb (g/dL) | 15.1 (14.5-15.8) | 9.2 (8.1-10.2) | 15.3 (15.1-16.0) | 9.7 (9.2-10.0) | 15.3 (14.7-15.6) | 9.3 (8.6-9.6) |
| Hematocrit (%) | 44 (43-46) | 27 (26-28) | 46 (43-49) | 29 (27-29) | 45 (44-46) | 27 (26-28) |
| Leukocytes (×109/L) | 5.7 (4.7-6.4) | 3.3 (2.8-5.4) | 6.0 (5.4-6.7) | 5.4 (2.9-6.0) | 5.0 (4.3-6.7) | 4.4 (3.2-5.1) |
| Hepatic | ||||||
| AST (U/L) | 60 (58-69) | 6147 (4019-8179) | 63 (59-67) | 833 (362-1123) *,** | 64 (60-74) | 3354 (2277-5171) |
| ALT (U/L) | 51 (49-56) | 3334 (2307-5680) | 54 (49-57) | 365 (183-553) *,** | 53 (45-58) | 1996 (1451-4609) |
| Renal | ||||||
| Creatinine (μmol/L) | 25 (25-26) | 157 (154-168) | 25 (23-25) | 108 (98-122)*,** | 26 (25-27) | 161 (140-167) |
| Urine protein (g/L) | 0.8 (0.6-1.2) | 2.2 (1.6-2.8) | 0.8 (0.3-1.0)** | 1.9 (1.3-2.0) | 1.3 (0.8-1.6) | 2.1 (1.7-2.6) |
| Coagulation | ||||||
| EXTEM CT (s) | 50 (49-53) | 62 (53-89) | 53 (49-56) | 53 (47-58) | 49 (48-52) | 54 (47-59) |
| EXTEM CFt (s) | 41 (39-44) | 73 (56-122) | 44 (43-49) | 50 (45-58)* | 40 (39-43) | 52 (42-59)* |
| EXTEM MCF (mm) | 71 (70-72) | 60 (51-66) | 70 (69-70) | 67 (64-68) | 71 (69-72) | 67 (64-69) |
| EXTEM Li30 (%) | 100 (100-100) | 100 (100-100) | 100 (100-100) | 100 (100-100) | 100 (100-100) | 100 (100-100) |
| FIBTEM CT (s) | 47 (42-51) | 64 (51-299) | 46 (43-52) | 45 (43-53)* | 48 (45-51) | 47 (45-55) |
| FIBTEM MCF (mm) | 15 (14-17) | 6.5 (3.0-8.3) | 14.5 (13.3-20.5) | 8.0 (6.0-9.0) | 15.0 (14.0-16.5) | 11.0 (10.0-14.0)*,*** |
| Parameter | Crystalloid | Crystalloid + rhADAMTS13 | Plasma | |||
|---|---|---|---|---|---|---|
| Before injury | After 6 h | Before injury | After 6 h | Before injury | After 6 h | |
| Weight (g) | 348 (336-369) | ND | 353 (348-379) | ND | 350 (336-380) | ND |
| Arterial blood gas | ||||||
| pH | 7.41 (7.40-7.46) | 7.33 (7.29-7.46) | 7.43 (7.37-7.48) | 7.35 (7.30-7.39) | 7.45 (7.40-7.46) | 7.33 (7.28-7.39) |
| pCO2 (mm Hg) | 34.9 (32.1-41.1) | 22.7 (19.7-31.0) | 34.6 (32.7-44.1) | 31.0 (28.8-33.7)* | 35.1 (31.9-37.5) | 26.6 (24.9-31.5) |
| HCO3− (mmol/L) | 23.5 (21.1-24.9) | 13.2 (11.8-14.9) | 23.6 (22.5-25.0) | 17.6 (16.1-18.2) *,** | 22.5 (21.6-24.0) | 14.1 (11.1-16.2) |
| BE (mEq/L) | −0.1 (−1.9 to 0.4) | −10.3 (−12.8-−9.3) | −0.5 (−0.9 to 0.6) | −7.6 (−8.3 to −5.8) *,** | −1.3 (−1.8 to 0.2) | −10.0 (−14.1 to −8.6) |
| Ca2+ (mmol/L) | 1.1 (1.1-1.1) | 0.9 (0.8-1.1) | 1.1 (1.0-1.2) | 1.0 (1.0-1.1) | 1.1 (1.0-1.1) | 0.9 (0.9-1.1) |
| Hematology | ||||||
| Hb (g/dL) | 15.1 (14.5-15.8) | 9.2 (8.1-10.2) | 15.3 (15.1-16.0) | 9.7 (9.2-10.0) | 15.3 (14.7-15.6) | 9.3 (8.6-9.6) |
| Hematocrit (%) | 44 (43-46) | 27 (26-28) | 46 (43-49) | 29 (27-29) | 45 (44-46) | 27 (26-28) |
| Leukocytes (×109/L) | 5.7 (4.7-6.4) | 3.3 (2.8-5.4) | 6.0 (5.4-6.7) | 5.4 (2.9-6.0) | 5.0 (4.3-6.7) | 4.4 (3.2-5.1) |
| Hepatic | ||||||
| AST (U/L) | 60 (58-69) | 6147 (4019-8179) | 63 (59-67) | 833 (362-1123) *,** | 64 (60-74) | 3354 (2277-5171) |
| ALT (U/L) | 51 (49-56) | 3334 (2307-5680) | 54 (49-57) | 365 (183-553) *,** | 53 (45-58) | 1996 (1451-4609) |
| Renal | ||||||
| Creatinine (μmol/L) | 25 (25-26) | 157 (154-168) | 25 (23-25) | 108 (98-122)*,** | 26 (25-27) | 161 (140-167) |
| Urine protein (g/L) | 0.8 (0.6-1.2) | 2.2 (1.6-2.8) | 0.8 (0.3-1.0)** | 1.9 (1.3-2.0) | 1.3 (0.8-1.6) | 2.1 (1.7-2.6) |
| Coagulation | ||||||
| EXTEM CT (s) | 50 (49-53) | 62 (53-89) | 53 (49-56) | 53 (47-58) | 49 (48-52) | 54 (47-59) |
| EXTEM CFt (s) | 41 (39-44) | 73 (56-122) | 44 (43-49) | 50 (45-58)* | 40 (39-43) | 52 (42-59)* |
| EXTEM MCF (mm) | 71 (70-72) | 60 (51-66) | 70 (69-70) | 67 (64-68) | 71 (69-72) | 67 (64-69) |
| EXTEM Li30 (%) | 100 (100-100) | 100 (100-100) | 100 (100-100) | 100 (100-100) | 100 (100-100) | 100 (100-100) |
| FIBTEM CT (s) | 47 (42-51) | 64 (51-299) | 46 (43-52) | 45 (43-53)* | 48 (45-51) | 47 (45-55) |
| FIBTEM MCF (mm) | 15 (14-17) | 6.5 (3.0-8.3) | 14.5 (13.3-20.5) | 8.0 (6.0-9.0) | 15.0 (14.0-16.5) | 11.0 (10.0-14.0)*,*** |
Data are presented as median (IQR). Before trauma and before termination, samples were measured for different parameters.
ALT, alanine aminotransferase; AST, aspartate aminotransferase; BE, base excess; CFt, clot formation time; CT, clotting time; Hb, hemoglobin; Li30, lysis index, 30 minutes; MCF, maximum clot firmness; pCO2, partial arterial carbon dioxide pressure.
Bonferroni-adjusted *P < .05 compared with crystalloid, **P < .05 compared with plasma, and ***P < .05 compared with crystalloid + rhADAMTS13.
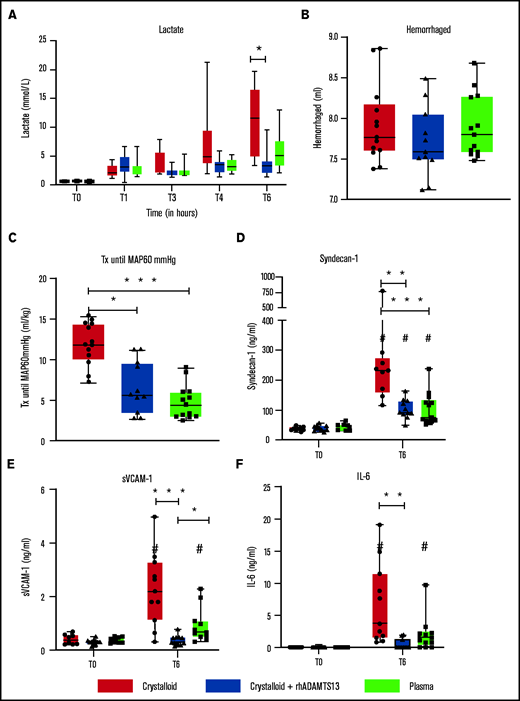
Recombinant human ADAMTS13 and plasma transfusion limit resuscitation fluids and endothelial damage in a rat trauma transfusion model. Data are shown as boxplots with range. (A) Lactate levels over time. After trauma and shock (T = 1 hour), rats were randomized in Ringer’s lactate, Ringer’s lactate + rhADAMTS13, or plasma transfusion. (B) Amount of bleeding during shock. (C) Pressure-fixed transfusion or infusion (Tx) strategy was applied, resulting in differences in the amount of resuscitation fluids needed to reach a MAP of 60 mm Hg. (D-F) Before trauma (T = 0) and 6 hours post-trauma, samples were analyzed for different measures, including syndecan-1 (D), soluble vascular adhesion molecule-1 (E), and IL-6 (F). *P < .05, **P < .01, and ***P < .001 between groups; #P < .05 within groups.
Recombinant human ADAMTS13 and plasma transfusion limit resuscitation fluids and endothelial damage in a rat trauma transfusion model. Data are shown as boxplots with range. (A) Lactate levels over time. After trauma and shock (T = 1 hour), rats were randomized in Ringer’s lactate, Ringer’s lactate + rhADAMTS13, or plasma transfusion. (B) Amount of bleeding during shock. (C) Pressure-fixed transfusion or infusion (Tx) strategy was applied, resulting in differences in the amount of resuscitation fluids needed to reach a MAP of 60 mm Hg. (D-F) Before trauma (T = 0) and 6 hours post-trauma, samples were analyzed for different measures, including syndecan-1 (D), soluble vascular adhesion molecule-1 (E), and IL-6 (F). *P < .05, **P < .01, and ***P < .001 between groups; #P < .05 within groups.
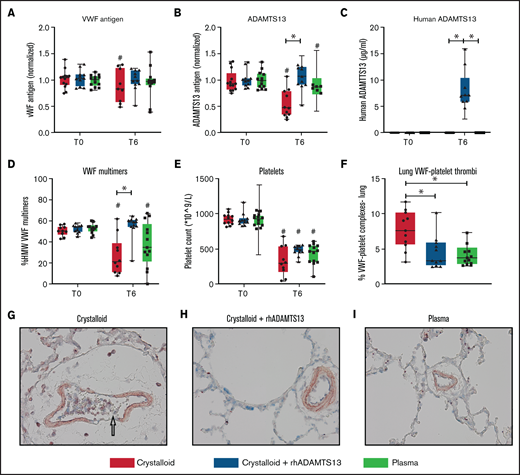
Recombinant human ADAMTS13 and plasma transfusion reduce the deranged ADAMTS13-VWF axis, associated with decreased VWF-platelet thrombi in the lung. Data are shown as boxplots with range. (A-E) Before trauma (T = 0) and 6 hours post-trauma, samples were analyzed for different measures, including VWF antigen (A), rat ADAMTS13 antigen (B), human ADAMTS13 antigen (C), HMW VWF multimers (D), and platelet counts (E). (F) At termination, the lung was assessed for VWF-platelet microthrombi. (G-I) Images (original magnification ×400) of the lung for the assessment of VWF (blue), CD41 + platelets (red), and microthrombi (purple). The arrow in (G) points towards a microthrombus. *P < .05, **P < .01, and ***P < .001 between groups; #P < .05 within groups.
Recombinant human ADAMTS13 and plasma transfusion reduce the deranged ADAMTS13-VWF axis, associated with decreased VWF-platelet thrombi in the lung. Data are shown as boxplots with range. (A-E) Before trauma (T = 0) and 6 hours post-trauma, samples were analyzed for different measures, including VWF antigen (A), rat ADAMTS13 antigen (B), human ADAMTS13 antigen (C), HMW VWF multimers (D), and platelet counts (E). (F) At termination, the lung was assessed for VWF-platelet microthrombi. (G-I) Images (original magnification ×400) of the lung for the assessment of VWF (blue), CD41 + platelets (red), and microthrombi (purple). The arrow in (G) points towards a microthrombus. *P < .05, **P < .01, and ***P < .001 between groups; #P < .05 within groups.
Plasma transfusion reduces vascular leakage and limits lung injury but does not significantly replenish ADAMTS13 levels
Rats resuscitated with plasma needed significantly less volume to reach a MAP of 60 mm Hg (4.5 [3.0-6.0] mL/kg]) compared with rats resuscitated with crystalloids (11.9 [10.1-14.4] mL/kg, P < .001) (Figure 3C). Plasma did not significantly prevent a drop in ADAMTS13 antigen levels (Figure 4B). Also, circulating HMW VWF multimers were lower compared with baseline and not significantly different from the crystalloid-resuscitated animals. Coagulation was less deranged in plasma-resuscitated animals than in both the crystalloid and crystalloid + rhADAMTS13 groups, especially for FIBTEM, which is a marker for the fibrinogen contribution to clot formation (Table 2). Syndecan-1 levels were lower in plasma-resuscitated animals compared with rats receiving crystalloids. Plasma transfusion resulted in less VWF-platelet microthrombi formation, less edema, less vascular leakage and less injury in the lung compared with crystalloids (Figure 5). Other organs scores were not different between groups (Figure 5; supplemental Figure 2).
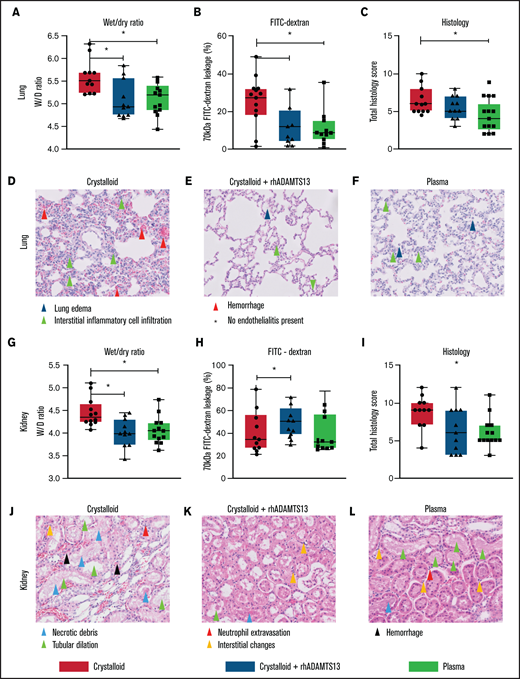
Plasma transfusion, but not crystalloid infusion, limits lung and renal endothelial leakage and organ damage in a rat trauma transfusion model. Data are boxplots with range. (A-C) Lung wet/dry ratio, area of 70-kDa FITC-dextran leakage, and lung total histology scores (supplemental Table 2). (D-F) Images (original magnification ×200) of the lung for all the groups. (G-I) Kidney wet/dry ratio, FITC-dextran leakage, and kidney total histology score. (J-L) Images (original magnification ×200) of the kidney for all groups. *P < .05 between groups.
Plasma transfusion, but not crystalloid infusion, limits lung and renal endothelial leakage and organ damage in a rat trauma transfusion model. Data are boxplots with range. (A-C) Lung wet/dry ratio, area of 70-kDa FITC-dextran leakage, and lung total histology scores (supplemental Table 2). (D-F) Images (original magnification ×200) of the lung for all the groups. (G-I) Kidney wet/dry ratio, FITC-dextran leakage, and kidney total histology score. (J-L) Images (original magnification ×200) of the kidney for all groups. *P < .05 between groups.
Crystalloid resuscitation supplemented with rhADAMTS13 replenishes ADAMTS13 levels, reduces microthrombi formation, reduces vascular leakage, and limits organ injury
To explore whether the observed beneficial effect of plasma was mediated by ADAMTS13, resuscitation was performed by supplementing crystalloids with a single injection of rhADAMTS13. Rats treated with rhADAMTS13 needed less volume (5.6 [3.5-9.6] mL/kg, P < .05) to obtain a MAP of 60 mm Hg compared with rats resuscitated with only crystalloids (Figure 3). In crystalloid-resuscitated rats, there was a decrease in ADAMTS13 levels, which was prevented in rats treated with rhADAMTS13 (Figure 4). Following resuscitation, treatment with rhADAMTS13 resulted in higher circulating levels of HMW VWF multimers compared with crystalloid only. Moreover, rhADAMTS13 treatment reduced sVCAM-1 levels compared with plasma- and crystalloid-resuscitated rats. Also, IL-6 was lower in the rhADAMTS13 group than the crystalloid group (Figure 4). Moreover, there were less deranged ROTEM values in crystalloids with rhADAMTS13-resuscitated rats compared with crystalloid-resuscitated rats (Table 2). Edema in the lung and kidney was reduced following rhADAMTS13 treatment compared with crystalloids, as shown by lower wet/dry ratios. Also, rhADAMTS13 treatment resulted in fewer VWF-platelet microthrombi in the lung compared with crystalloid-treated rats. However, vascular leakage in the kidney was increased in rhADAMTS13-treated rats compared with crystalloid-treated rats. Treatment with rhADAMTS13 resulted in lower aspartate aminotransferase, alanine aminotransferase, and creatinine levels compared with both crystalloid- and plasma-treated rats (Table 2); however, these lower levels were not accompanied by lower organ scores (Figure 5; supplemental Figure 2). Dose dependency of rhADAMTS13 was evaluated in a Transwell system with thrombin-stimulated microvascular lung endothelial cells. Endothelial leakage was reduced in a dose-dependent manner and was associated with less syndecan-1 release (supplemental Figure 3).
Discussion
The findings of this study are that (1) the severity of trauma injury is associated with increased VWF propeptide levels and VWF antigen levels and decreased ADAMTS13 antigen levels; (2) patients who developed MODS have lower ADAMTS13-to-VWF antigen ratios than patients without MODS, associated with lower platelet counts, prolonged prothrombin times, higher D-dimers, and higher percentages of HMW VWF multimers; and (3) plasma transfusion and rhADAMTS13 were associated with less VWF-platelet microthrombi formation in the lung, less vascular leakage, and less organ failure in a rat trauma transfusion model.
Our study is in line with previous studies showing a deranged ADAMTS13-to-VWF ratio in adult and pediatric trauma patients, which was associated with endothelial damage and mortality.14,15 This study extends these findings by showing that in patients developing MODS, increased release of VWF propeptide takes place, an increased amount of VWF multimers are circulating, associated with a decrease in platelet count, which persists long after bleeding had stopped. Collectively, these data underline our hypothesis that severe trauma leads to a rapid release of VWF propeptide and VWF antigen, while ADAMTS13 antigen levels are diminished possibly to a level that is insufficient to deal with large amounts of VWF release, resulting in consumption of platelets that are getting trapped in HMW VWF multimers, leading to microthrombi formation, endothelial activation, obstruction of the microcirculation, and eventually organ failure (supplemental Figure 5). Acknowledging that multiple pathways are involved in the progression of MODS,38,39 the role of an imbalanced ADAMTS13-to-VWF ratio in the development of trauma-induced MODS has only recently been suggested. This pathway shows resemblances with MODS due to septic shock, in which an association between an imbalanced ADAMTS13-to-VWF, consumption coagulopathy and MODS has repeatedly been demonstrated.12,13 However, traumatic shock and septic shock have profoundly different causes of VWF release. In trauma, large amounts of VWF are released due to endothelial activation in response to an injured vessel wall, which supposedly is a short-lived stimulus, whereas in sepsis, VWF is released as part of an ongoing inflammatory host response.12,15,30,40 ADAMTS13 activity levels did not differ in patients with and without MODS. Multiple enzymes affect the activity of ADAMTS13, such as thrombin and plasmin.41 These levels are increased in patients with severe injury and trauma-induced coagulopathy.42 Plasmin and thrombin may in turn also result in degradation of ADAMTS13, resulting in lower antigen levels.41 Alternatively, ADAMTS13 levels may drop due to dilution during the resuscitation of bleeding trauma patients with fluids lacking ADAMTS13. The ADAMTS13 antigen-to-VWF ratio we found in trauma, however, worsens after the bleeding has stopped. Additionally, platelet counts continue to decrease up to 72 hours post-trauma. Thereby, we postulate that after bleeding has stopped following trauma, an unbalanced procoagulant response fueled by an activated endothelium persists. In mice models of renal ischemia-reperfusion and TTP, a low ADAMTS13-to-VWF ratio was associated with more severe endothelial leakage and organ failure.43–45 Thereby, activation of endothelial cells with release of large quantities of VWF and construction of more circulating HMW VWF multimers that supersede ADAMTS13-cleaving capacities may be an important pathway in the progression of MODS in multiple inflammatory shock states.
We found that in rats, plasma transfusion reduced markers of endothelial activation, endothelial leakage, and organ injury compared with crystalloid infusion, associated with a nonsignificant increase in ADAMTS13 levels. Thereby, we propose that the replenishing or at least stabilization of ADAMTS13 levels may be a mechanism by which some of the transfusion trials show beneficial effects of plasma on outcome of trauma.5,46 A comparison with TTP comes to mind, whereby plasma exchange or plasma transfusion to replenish ADAMTS13 levels is lifesaving.47,48 In rats with septic shock, plasma resuscitation improved survival compared with crystalloids.49 Thereby, plasma transfusion may be beneficial in multiple patient groups with severe shock-induced consumption coagulopathy and ADAMTS13-to-VWF imbalance. Alternatively, the benefit of plasma transfusion observed here may be due to other (coagulation) proteins present in plasma.
We found a protective effect of recombinant ADAMTS13 on endothelial barrier function, as shown before in a traumatic brain injury model.50 Previously, we have studied rhADAMTS13 in a similar model comparing 1:1:1 red blood cell/plasma/platelet transfusion ratio with rhADAMTS13 to vehicle.51 We found that rats with rhADAMTS13 compared with vehicle showed reduced organ inflammation but similar levels of endothelial damage. In the current model, we add to these findings, in which the replenishment of low ADAMTS13 levels in the traumatized animals was more effectively done using rhADAMTS13 than plasma. In line, rhADAMTS13 had a more outspoken effect on some markers of inflammation than plasma, including lower liver transaminases and plasma creatinine levels as well as lower circulating sVCAM-1 and IL-6 levels 6 hours after traumatic injury. However, rhADAMTS13 treatment resulted in more vascular leakage in the kidney compared with crystalloid-treated rats. An explanation could be that urine output was higher and plasma creatinine levels were lower in the rhADAMTS13-resuscitated rats compared with crystalloid-treated rats, suggesting higher glomerular filtration rates, resulting in relatively higher amounts of FITC-dextran circulating in the kidney at termination.
Not all beneficial effects were most clear with rhADAMTS13. Plasma transfusion, but not rhADAMTS13 infusion, was associated with reduced pulmonary vascular leakage and organ injury as assessed by histopathology, suggesting that plasma exerts protection also via pathways not involving ADAMTS13. However, both rhADAMTS13 and plasma transfusion reduced VWF-platelet microthrombi in the lung compared with crystalloid. This decrease in microthrombi coincided with higher circulating levels of HMW VWF multimers in the rhADAMTS13 group. An explanation for this result may be consumption of VWF multimers in microthrombi. Alternatively, the decrease in VWF multimers may be a sign of dilutional coagulopathy.
Importantly, although we have provided a rationale as well as circumstantial evidence that plasma is effective by restoring an ADAMTS13-to-VWF antigen imbalance, causality was not determined in our studies. Plasma contains many other proteins that may have an effect on the endothelium.52 Unfortunately, providing direct proof of causality (eg, by using ADAMTS13-knockout animals) was beyond the scope of this study. If this pathway is indeed present in trauma-induced MODS, rhADAMTS13 may be an alternative to plasma transfusion. However, additional (validation) experimental studies are warranted relating to optimal dose and timing as well as testing in an uncontrolled hemorrhage model, because we have administered rhADAMTS13 after 1 hour of shock, when animals were not actively bleeding.
There are limitations to this study. In the clinical study, correction for confounding factors (eg, injury severity score, shock, and trauma-induced coagulopathy) for the development of MODS was not done, which may have influenced our results. In the rat model, we chose a pressure-fixed resuscitation target as this resembles clinical practice. However, this approach may have led to differences in circulating proteins due to different volumes of resuscitation fluids used. On the other hand, volume-fixed transfusion will have resulted in both over- and under transfusion and is not used in clinical practice. Also, as this cohort mainly consisted of blunt trauma patients, results may not apply to trauma patients with penetrating injury. Typically, these patients have a lower incidence of MODS. Furthermore, rats have a different coagulation system (eg, 2-3 times higher platelet counts) than humans. Also, rats did not show the same increase in VWF expression following trauma as found in patients, which may have been due to differences in (timing of) expression, which was not captured with a single measurement in time. Thereby, the rat model was is characterized by endothelial activation, fibrinogen consumption, and organ failure, but differences in coagulation response challenges the translation of the results to humans.
In conclusion, trauma is characterized by a decreased ADAMTS13-to-VWF ratio, associated with injury severity and organ failure. Possibly, a deranged ADAMTS13/VWF ratio is a common pathway of the occurrence of organ injury following shock states. Plasma transfusion and rhADAMTS13 restore this balance in an experimental animal model associated with a reduction in organ failure, at least in part via restoring the ADAMTS13/VWF ratio. Thereby, trauma-induced MODS may be a novel indication for plasma therapy or specific replenishment of ADAMTS13.
Authorship
Contribution: D.J.B.K. drafted the first version of the manuscript; D.J.B.K. and D.D.G.S. analyzed samples for the clinical study. CD, SD analyzed samples for the clinical and animal study; D.J.B.K., D.D.G.S., P.H.S., and M.A.W.M. performed (parts of) the animal studies. D.J.B.K. and PS analyzed data of the animal study; J.K. analyzed the histopathology of the animal study. JM provided reagents for ADAMTS13 activity analysis; J.M., K.B., J.V., K.V., M.W.H., N.P.J. designed and supervised the study; BloodNet commented on all versions of the manuscript; and all authors revised the drafts of the manuscript and approved the final version.
Conflict-of-interest disclosure: J.M. holds a patent “Fluorogenic substrate for ADAMTS-13,” US 8663912, issued to Washington University. The remaining authors declare no competing financial interests.
Correspondence: Derek J. B. Kleinveld, Amsterdam UMC, University of Amsterdam, Intensive Care, Meibergdreef 9, 1105 AZ, Amsterdam, The Netherlands; e-mail: d.j.kleinveld@amsterdamumc.nl.

