

Key Points
CH is very common among individuals receiving chimeric antigen receptor T-cell therapy.
CH is associated with increased complete response rate and more severe toxicity in patients under 60, but no difference in overall survival.
Abstract
Chimeric antigen receptor (CAR) T-cells have emerged as an efficacious modality in patients with non-Hodgkin lymphoma (NHL) and multiple myeloma (MM). Clonal hematopoiesis of indeterminate potential (CHIP), a state in which mutations in hematopoietic cells give rise to a clonal population of cells, is more common in patients exposed to cytotoxic therapies, has been shown to influence inflammatory immune programs, and is associated with an adverse prognosis in patients with NHL and MM receiving autologous transplantation. We therefore hypothesized that CHIP could influence clinical outcomes in patients receiving CAR T-cell therapy. In a cohort of 154 patients with NHL or MM receiving CAR T-cells, we found that CHIP was present in 48% of patients and associated with increased rates of complete response and cytokine release syndrome severity, but only in patients younger than age 60 years. Despite these differences, CHIP was not associated with a difference in progression-free or overall survival, regardless of age. Our data suggest that CHIP can influence CAR T-cell biology and clinical outcomes, but, in contrast to autologous transplantation, CHIP was not associated with worse survival and should not be a reason to exclude individuals from receiving this potentially life-prolonging treatment.
Introduction
Clonal hematopoiesis (CH) describes an expansion of clonally derived hematopoietic cells in the peripheral blood. CH of indeterminate potential (CHIP), defined by the presence of leukemia-associated mutations with a variant allele fraction (VAF) of at least 2%, is associated with elevated mortality among healthy individuals and patients with cancer.1-3 In patients with non-Hodgkin lymphoma (NHL) or multiple myeloma (MM) who are undergoing autologous transplantation, CHIP is common and associated with inferior outcomes and increased mortality.4,5
Chimeric antigen receptor (CAR) T-cell therapy is a highly efficacious option for patients with relapsed or refractory lymphoid malignancies but is frequently complicated by development of a hyperinflammatory state known as cytokine release syndrome (CRS).6,7 We hypothesized that clonal hematopoiesis would be common in patients receiving CAR T-cell therapy and could influence clinical outcomes through multiple, potentially competing mechanisms. CHIP mutations in myeloid cells have been shown to enhance inflammatory signaling, including via interleukin-6 (IL-6), a key mediator of CRS, and alter interactions between innate and adaptive immune cells.8-11 Moreover, DNMT3A and TET2, genes commonly mutated in CHIP, have been shown to influence CAR T-cell programs.12,13 We therefore sought to understand the frequency and clinical consequences of CH in a cohort of patients receiving CAR T-cell therapy.
Methods
The study was approved by the Dana-Farber institutional review board. We identified all patients with NHL or MM treated with CAR T-cells at our institutions since 2016 for whom frozen blood specimens were available. A preference was made for blood samples obtained on the day of CAR T-cell infusion, followed by samples obtained within 3 months before CAR T-cell infusion, followed by samples as close as possible to CAR T-cell infusion. Next-generation sequencing on a targeted gene panel was performed on genomic DNA isolated from peripheral blood samples as previously described (supplemental Table 1; supplemental Methods).14 All end points were evaluate using a 2-sided type I error of 5%. The primary end points were progression-free survival (PFS) and overall survival (OS). Secondary end points were rate of complete response (CR) and CRS grade ≥2. The statistical methods used can be found in the supplemental Methods.
Results and discussion
Of all patients treated for NHL or MM with CAR T-cells from our institution since 2016, we identified 154 with available blood specimens for genetic analysis (supplemental Table 2; supplemental Figure 1A). Among patients in this cohort, the median age was 63 years old (range, 24-83), 144 patients received a CAR T-cell product targeting CD19 for treatment of NHL, and 10 patients received a CAR T product targeting B-cell maturation antigen for treatment of MM. The median number of prior lines of therapy was 4 (range, 1-10; interquartile range, 2-4), including 41 patients who had received an autologous stem cell transplant and 5 who received an allogeneic stem cell transplant.
CH was detected in 76% of patients with a VAF ≥0.004, and 48% with a VAF ≥0.02 (supplemental Table 3). CH was associated with age, occurring at a VAF >0.01 in 85% of patients older than 70 years (Figure 1A; supplemental Figure 1B). Patients with CH had a median of 2 mutations, with most mutations occurring in PPM1D, DNMT3A, TP53, TET2, and SRCAP, whereas mutations in splicing factors and JAK2 were rare (Figure 1B; supplemental Figure 1B-E).
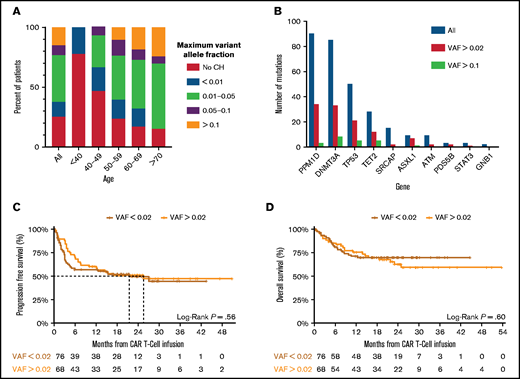
Clonal hematopoiesis in CAR T-cell cohort. (A) The frequency of CH stratified by the size of the clone, as measured by the variant allele fraction across the entire cohort (All) and within particular age groups. (B) The number of mutations (y-axis) identified in each gene (x-axis) at differing VAF across the entire cohort. (C) Progression-free survival of patients with non-Hodgkin lymphoma stratified by absence (red) or presence (orange) of clonal hematopoiesis with a VAF ≥0.02. A log-rank test was performed to compare survival curves. (D) Overall survival of patients with non-Hodgkin lymphoma stratified by absence (red) or presence (orange) of clonal hematopoiesis with a VAF ≥0.02. A log-rank test was performed to compare survival curves.
Clonal hematopoiesis in CAR T-cell cohort. (A) The frequency of CH stratified by the size of the clone, as measured by the variant allele fraction across the entire cohort (All) and within particular age groups. (B) The number of mutations (y-axis) identified in each gene (x-axis) at differing VAF across the entire cohort. (C) Progression-free survival of patients with non-Hodgkin lymphoma stratified by absence (red) or presence (orange) of clonal hematopoiesis with a VAF ≥0.02. A log-rank test was performed to compare survival curves. (D) Overall survival of patients with non-Hodgkin lymphoma stratified by absence (red) or presence (orange) of clonal hematopoiesis with a VAF ≥0.02. A log-rank test was performed to compare survival curves.
Because CHIP (CH with VAF ≥0.02) has previously been associated with inferior outcomes, we adapted this convention for all subsequent analyses.3-5,15 First, we looked at the best overall response and severity of CRS, 2 surrogates of CAR T-cell activity. Among patients with NHL, those with CHIP had a higher likelihood of achieving a CR (77.6% CH vs 57.9% no CH, P = .0133) but only if younger than 60 years of age (88.9% CH vs 46.7% no CH, P = .0067 for younger than 60 years; 73.5% CH vs 66.7% no CH, P = .64 for age 60 years or older) (Table 1A). Similarly, in patients with NHL, CHIP was significantly associated with an increased rate of CRS grade ≥2 only in patients younger than 60 years (77.8% CHIP vs 45.9% no CHIP, P = .042 for younger than 60 years; 62.0% CHIP vs 74.4% no CHIP, P = .26 for 60 years or older) (Table 1B). The same findings held true when including the patients with MM, and we did not detect a significant association between CHIP and evidence of clinical neurotoxicity after CAR T-cell treatment (supplemental Table 4A-B). Although CRS grade was positively associated with maximum detected high-sensitivity C-reactive protein, ferritin, and IL-6 levels, we did not observe an association between the presence of CHIP and levels of these inflammatory markers (supplemental Figure 2).16
Associations between clonal hematopoiesis, best overall response, and cytokine release syndrome
| Association in patients with NHL between the rate of CR and the presence of CH with a VAF ≥ 0.02 compared with all other patients (VAF < 0.02) | |||||
|---|---|---|---|---|---|
| Best overall response | |||||
| CH status | Total patients | CR (%) | No CR (%) | P* | |
| NHL | VAF < 0.02 | 76 | 44 (57.9) | 32 (42.1) | .013 |
| VAF ≥ 0.02 | 67† | 52 (77.6) | 15 (22.4) | ||
| NHL < 60 y | VAF < 0.02 | 37 | 18 (48.6) | 19 (51.4) | .0067 |
| VAF ≥ 0.02 | 18 | 16 (88.9) | 2 (11.1) | ||
| NHL ≥ 60 y | VAF < 0.02 | 39 | 26 (66.7) | 13 (33.3) | .64 |
| VAF ≥ 0.02 | 49† | 36 (73.5) | 13 (26.5) | ||
| Association in all patients between the rate of CRS grade 2 or greater and the presence of CH with a VAF ≥ 0.02 compared with all other patients (VAF < 0.02) | |||||
| ASTCT CRS score | |||||
| CH status | Total patients | Grade < 2 (%) | Grade ≥ 2 (%) | P* | |
| NHL | VAF < 0.02 | 76 | 30 (39.5) | 46 (60.5) | .49 |
| VAF ≥ 0.02 | 68 | 23 (33.8) | 45 (66.2) | ||
| NHL < 60 y | VAF < 0.02 | 37 | 20 (54.1) | 17 (45.9) | .042 |
| VAF ≥ 0.02 | 18 | 4 (22.2) | 14 (77.8) | ||
| NHL ≥ 60 y | VAF < 0.02 | 39 | 10 (25.6) | 29 (74.4) | .26 |
| VAF ≥ 0.02 | 50 | 19 (38.0) | 31 (62.0) | ||
| Association in patients with NHL between the rate of CR and the presence of CH with a VAF ≥ 0.02 compared with all other patients (VAF < 0.02) | |||||
|---|---|---|---|---|---|
| Best overall response | |||||
| CH status | Total patients | CR (%) | No CR (%) | P* | |
| NHL | VAF < 0.02 | 76 | 44 (57.9) | 32 (42.1) | .013 |
| VAF ≥ 0.02 | 67† | 52 (77.6) | 15 (22.4) | ||
| NHL < 60 y | VAF < 0.02 | 37 | 18 (48.6) | 19 (51.4) | .0067 |
| VAF ≥ 0.02 | 18 | 16 (88.9) | 2 (11.1) | ||
| NHL ≥ 60 y | VAF < 0.02 | 39 | 26 (66.7) | 13 (33.3) | .64 |
| VAF ≥ 0.02 | 49† | 36 (73.5) | 13 (26.5) | ||
| Association in all patients between the rate of CRS grade 2 or greater and the presence of CH with a VAF ≥ 0.02 compared with all other patients (VAF < 0.02) | |||||
| ASTCT CRS score | |||||
| CH status | Total patients | Grade < 2 (%) | Grade ≥ 2 (%) | P* | |
| NHL | VAF < 0.02 | 76 | 30 (39.5) | 46 (60.5) | .49 |
| VAF ≥ 0.02 | 68 | 23 (33.8) | 45 (66.2) | ||
| NHL < 60 y | VAF < 0.02 | 37 | 20 (54.1) | 17 (45.9) | .042 |
| VAF ≥ 0.02 | 18 | 4 (22.2) | 14 (77.8) | ||
| NHL ≥ 60 y | VAF < 0.02 | 39 | 10 (25.6) | 29 (74.4) | .26 |
| VAF ≥ 0.02 | 50 | 19 (38.0) | 31 (62.0) | ||
*P values were calculated using a Fisher’s exact test.
†One patient did not have an evaluable best overall response.
Despite differences in response and toxicity, we did not observe any association between CHIP and PFS (median PFS CH 25.6 months, 95% confidence interval [10.1-infinity] vs no CH median 21.5 months, 95% confidence interval [4.5-infinity], log-rank P = .6) or OS (median not achieved in either group, log-rank P = .6) (Figure 1C-D). We did note an early separation in the PFS curves, consistent with the finding that patients with CHIP had higher rates of CR. Stratifying the PFS and OS by age older than or younger than 60 years again highlighted differences in patients younger than 60 with and without CHIP, but the differences in overall PFS and OS outcomes remained nonsignificant and there were no differences noted when restricting to mutations in specific genes (supplemental Figure 3).
We also identified 3 patients who developed a therapy-related myeloid neoplasm during the follow-up period, 2 of whom harbored TP53-mutant CHIP and developed TP53-mutant acute myeloid leukemia.
Even after correcting for differences in sequencing depth and genes sequenced, the prevalence of CHIP with a VAF >0.02 in this cohort was significantly higher than that found in patients treated for solid tumors15 (48% vs 30%, P < .001) and in patients undergoing autologous stem cell transplantation for either NHL4 (48% vs 30%, P < .0001) or for MM5 (48% vs 14%, P < .001). Moreover, as seen in other contexts, prior cytotoxic therapy selected for mutations in the DNA damage response including in TP53.3,4,17,18 Although the presence of CHIP is associated with increased rates of CR and CRS severity, particularly in individuals younger than 60 years, it is not associated with changes in PFS or OS in this cohort. The reasons for this lack of long-term benefit are unknown, and further efforts to understand potential emergent mechanisms of resistance or loss of CAR T-cell activity may further inform the role of CHIP in this setting.
CHIP has the potential to influence CAR T-cell biology and activity through multiple mechanisms. Mutant myeloid cells can influence the activity of CAR T-cells through altered cross-talk between innate immune cells and lymphocytes mediated by changes in secretion of and sensitivity to inflammatory cytokines and chemokines. In particular, changes in the inflammasome, IL-1, IL-6 axis, implicated in both CHIP and CRS after CAR T-cell therapy, could influence CAR T-cell biology in individuals with CHIP.9,10 These alterations likely have a broader impact on human disease, including in states of hyperinflammation, response to immune-targeting therapies, or other autoimmune diseases.11,14,15 Mutations that are present in the CAR T-cell product itself, particularly in TET2 and DNMT3A, can alter T-cell programs and the activity of CAR T-cells.12,13,19,20
Our data support the idea that clonal hematopoietic mutations in blood cells can influence inflammatory pathways through diverse mechanisms and across numerous disease and therapeutic contexts. Our data reveal an association between CHIP and both CR and CRS in patients younger than age 60 years receiving CAR T-cell therapy, but CHIP did not influence long-term outcomes, and patients with CHIP should not be excluded from receiving this potentially lifesaving treatment modality.
Acknowledgments
P.G.M. was supported by a National Institutes of Health National Heart, Lung, and Blood Institute fellowship training grant (4T32HL116324-04), an American Society of Hematology Research Training Award for Fellows, and an Edward P. Evans Young Investigator Award. A.S.S. was supported by a career development award from the National Institutes of Health (K12CA087723). This work received support from the National Institutes of Health, National Heart, Lung, and Blood Institute (R01HL082945 and P50CA206963) and National Cancer Institute (P01CA108631), the Howard Hughes Medical Institute, the Edward P. Evans Foundation, and the Adelson Medical Research Foundation to B.L.E.
Authorship
Contribution: P.G.M., A.S.S., C.J.G., M.B.L., M.J., M.V.M., and B.L.E. initiated the project, designed the research, and wrote the paper with input from other authors; P.G.M., A.S.S., C.J.G., M.B.L., M.J., E.J.B., S.H.G., C.J.W., and C.J. performed the research; and P.G.M., A.S.S., G.G.F., D.S.N., and C.J.G. analyzed the data.
Conflict-of-interest disclosure: B.L.E. has received research funding from Celgene, Deerfield, and Novartis and consulting fees from GRAIL; he serves on the scientific advisory boards and holds equity in Skyhawk Therapeutics, Exo Therapeutics, and Neomorph Therapeutics. P.G.M. has received consulting fees from Foundation Medicine, Inc. C.J.W. holds equity in BioNTech, Inc, and receives research funding from Pharmacyclics. M.V.M. is an inventor on patents related to adoptive cell therapies, held by Massachusetts General Hospital and University of Pennsylvania (some licensed to Novartis), holds equity in TCR2 and Century Therapeutics, and has served as a consultant for multiple companies involved in cell therapies. C.J. is a consultant for Kite/Gilead, Novartis, BMS/Celgene, Nkarta, Precision Biosciences, Lonza, Abbvie, and Bluebird Bio and receives research funding from Kite/Gilead and Pfizer.
Correspondence: Benjamin Ebert, Dana-Farber Cancer Institute, 450 Brookline Ave, D1610A, Boston, MA 02215; e-mail: benjamin_ebert@dfci.harvard.edu

