

Key Points
- •
High DNMT3A VAF correlates with a more proliferative presentation, granulocytic dysplasia, and inferior outcome.
- •
Presence of 2 concomitant DNMT3A mutations in the same patient is independently associated with shorter EFS and OS.

The clinicopathologic features of DNA methyltransferase 3A (DNMT3A)-mutated de novo acute myeloid leukemia (AML), and the significance of variant type, variant allele frequency (VAF), and multiple concomitant DNMT3A mutations, remain poorly defined. We examined 104 DNMT3A-mutated de novo AML patients from 2 major centers. Most (82%) had normal karyotype (NK); R882H variants were frequent(38%). The most commonly comutated genes included nucleophosmin (NPM1; 53%), Fms-related tyrosine kinase 3 (FLT3)–internal tandem duplication (25%), IDH1 (23%), IDH2 (23%), and TET2 (21%). Patients with high DNMT3A VAF at diagnosis (≥44%; DNMT3AHIGH) had more significant leukocytosis and higher blast counts in peripheral blood and bone marrow. DNMT3AHIGH cases were associated with much shorter event-free survival (EFS; 14.1 vs 56.8 months) and overall survival (OS; 18.3 months vs not reached) compared with cases of patients with low DNMT3A (DNMT3ALOW). Thirteen patients had 2 DNMT3A variants and similar VAFs at diagnosis that tracked together at multiple time points after chemotherapy and/or stem cell transplantation (SCT). In multivariable analyses performed in NK patients who received standard induction chemotherapy, presence of 2 DNMT3A mutations (hazard ratio [HR] = 3.192; P = .038) and SCT in first complete remission (HR = 0.295; P = .001) independently affected EFS; increasing marrow blast percentage (HR = 1.026; P = .025), high DNMT3A VAF (HR = 3.003; P = .010), and 2 DNMT3A mutations (HR = 4.816; P = .020) had independent effects on OS. These data support the adverse prognostic significance of DNMT3AHIGH reveal a novel association between 2 concomitant DNMT3A mutations and inferior outcome in DNMT3A-mutated de novo AML with a NK.
Introduction
Acute myeloid leukemia (AML) is a malignant neoplasm of immature, bone marrow (BM)‐derived myeloid cells. AML has an aggressive clinical course with an overall 5‐year survival rate of ∼25%.1 The majority of AML cases are de novo, without any known history of an antecedent myeloid neoplasm or exposure to cytotoxic therapy.1 De novo AML cases are associated with heterogenous clinical outcomes, necessitating risk stratification at the time of diagnosis by clinicopathologic and genetic criteria. The initial risk stratification of de novo AML cases is based on cytogenetic features, and includes low-, intermediate-, and high-risk categories.1,2 Further risk stratification of the intermediate-risk karyotype group is performed based on the incorporation of pathogenic somatic mutation data.3 The number of recurrent driver mutations encountered in AML is relatively small compared with other tumor types.4,5 These mutations affect genes involved in multiple intracellular processes, resulting in the promotion of cell proliferation and survival (FLT3 and KIT), the prohibition of hematopoietic differentiation and apoptosis (CEBPA, RUNX1, and NPM1), and the abnormal modulation of epigenetic pathways (DNMT3A, TET2, IDH1, and IDH2).5,6 The most common driver mutations in AML involve Fms-related tyrosine kinase 3 (FLT3), nucleophosmin (NPM1), and DNA methyltransferase 3A (DNMT3A).4,5
The latest National Comprehensive Cancer Network (NCCN) and European LeukemiaNet (ELN) guidelines for AML classify non–acute promyelocytic leukemia AML patients into prognostic risk categories based on 3 factors: (1) cytogenetics, (2) genetic mutations (in RUNX1, ASXL1, NPM1, FLT3-internal tandem duplication [FLT3-ITD], TP53, and CEBPA), and (3) variant allele frequency (VAF) of FLT3-ITD.2,7 Biallelic CEBPA mutations have a favorable prognosis in AML. On the other hand, mutations in RUNX1, TP53, and ASXL1 are generally associated with unfavorable outcomes.5 AML cases with mutations in NPM1 and FLT3-ITD have heterogenous clinical outcomes depending on the mutational status of both genes and the allelic ratio of FLT3-ITD.8 In addition, recent work from our group has revealed that the mutant NPM1 VAF at diagnosis may be prognostically significant,9 although additional prospective studies will be important for confirmation of this finding. These data suggest that further exploration into the potential clinical significance of the VAFs of other recurrently mutated genes is warranted.
DNMT3A encodes a DNA methyltransferase that regulates epigenetic modification of gene expression by catalyzing the addition of a methyl group to the cytosine residue of cytosine guanine dinucleotides.10DNMT3A mediates DNA methylation involved in differentiation of the hematopoietic stem cells into predominantly the granulocytic lineage.11DNMT3A mutations are relatively common in myeloid neoplasia and are detected in ∼20% of AML patients.12,13DNMT3A mutation is the most common aberration in clonal hematopoiesis of indeterminate potential, a preleukemic state associated with an increased risk of developing myeloid neoplasia, including AML.14-16 The broad clinical contexts in which DNMT3A mutation occurs have made it difficult to study the prognostic effect of DNMT3A mutations in AML specifically; moreover, numerous studies have revealed varied clinical correlations among DNTM3A-mutated AML patients associated with the specific variant type (eg, R882 vs non-R882).12,13,17-19 Indeed, some DNTM3A-mutated AML patients experience durable remissions despite persistent DNMT3A mutations after treatment.20,21 In this study, we assembled a multi-institutional cohort of DNMT3A-mutated de novo AML patients in order to specifically study associations with VAF, and to identify novel clinicopathologic and genetic features of this AML subtype.
Materials and methods
Case selection
After receiving institutional review board approval from the participating institutions, we identified 104 cases of newly diagnosed intermediate-risk karyotype de novo AML with mutated DNMT3A from the pathology archives of Brigham and Women’s Hospital/Dana-Farber Cancer Institute and Massachusetts General Hospital (2008-2020). We excluded patients who had received prior cytotoxic therapy, had World Health Organization (WHO)-defined recurrent cytogenetic abnormalities (listed after this paragraph), myelodysplasia–related cytogenetic abnormalities (listed after this paragraph), or a history of an antecedent myeloid neoplasm. Cases of AML with myelodysplasia-related changes (AML-MRCs) included in the study had been classified based only on the presence of >50% dysplasia in at least 2 cell lineages, in the absence of NPM1 or biallelic CEBPA mutations. Approximately 89% of patients were treated with standard anthracycline-cytarabine–based induction chemotherapy. All patients received similar induction regimens, and dosages of daunorubicin and cytarabine were not significantly different between the cases of patients with low DNMT3A VAF (DNMT3ALOW) and patients with high DNMT3A VAF (DNMT3AHIGH). Fifty-seven percent of patients received subsequent allogeneic stem cell transplantation (SCT).
AML patients with the following recurrent cytogenetic abnormalities were excluded from the cohort: t(8;21)(q22;q22.1), inv(16)(p13.1q22) or t(16;16)(p13.1;q22), t(15;17)(q22;q11-12), t(9;11)(p21.3;q23.3), t(6;9)(p23;q34.1), inv(3)(q21.3q26.2) or t(3;3)(q21.3;q26.2), and t(1;22)(p13.3;q13.1)
AML patients with the following myelodysplastic syndrome–related cytogenetic abnormalities were excluded from the cohort: −7/del(7q), del(5q), i(17q)/t(17p), −13/del(13q), del(11q), del(12p)/t(12p), del(9q), idic(X)(q13), t(11;16), t(3;21), t(1;3), t(2;11), inv(3)/t(3;3), t(6;9), morphologic criteria and +8, del(20q), or −Y.
NGS studies
Targeted next-generation sequencing (NGS) studies were performed at the time of diagnosis, on fresh BM aspirate or peripheral blood samples. Target regions of 87 genes (hybrid capture system; Agilent Technologies, Santa Clara, CA) were evaluated in 43 patients, 95 genes (rapid heme panel) (TruSeq custom amplicon kit; Illumina, San Diego, CA) in 47 patients, and 54 genes (SNaPshot; TruSeq [Illumina]; Massachusetts General Hospital) in 14 patients. Genes assessed in all platforms included DNMT3A, IDH1, IDH2, TET2, ASXL1, BCOR, BCORL, CREBBP, EZH2, SETBP1, SH2B3, SETD2, CEBPA, ETV6, MLL, NPM1, PHF6, RUNX1, WT1, PDS5B, RAD21, SMC3, STAG2, CBL, FLT3, JAK2, KIT, KRAS, NF1, NRAS, PTPN11, RIT1, PRPF40b, SF3B1, SRSF2, U2AF1, and ZRSR2. Regions analyzed included mutational hotspots in oncogenic driver genes and the coding sequence of tumor-suppressor genes. All cases were evaluated for average read count across all amplicons and for the read count and VAF of DNMT3A. Data for multiple time points (range, 2-8) were available for 7 patients who had 2 DNTM3A variants at diagnosis.
BM morphologic evaluation
BM aspirate smears were evaluated for blast count as well as dysplasia in the erythroid, granulocytic, and megakaryocytic lineages as previously reported.22 Dysplasia was scored in a blinded fashion by 3 board-certified, expert hematopathologists (O.P., R.P.H., and O.K.W.). Each lineage was assessed for individual dysplastic features (erythroid precursors: megaloblastic change, multinucleation, and nuclear irregularities; granulocytic precursors: abnormal nuclear shape and hypogranulation; megakaryocytes: micromegakaryocytes, widely separated nuclear lobes, and hypolobated nuclei). The severity of dysplasia in each lineage was scored on a scale of 0 to 4 (0, no dysplasia; 1, mild dysplasia; 2, moderate dysplasia; 3, significant dysplasia; 4, severe dysplasia), and the percentage of dysplastic cells in each lineage was quantified, as previously described.22
Statistical analyses
Regression analyses were used to assess correlation between 2 variables. Overall survival (OS) and event-free survival (EFS) were evaluated in normal karyotype patients (including isolated −Y) who received induction chemotherapy (n = 76). OS was defined as the time in months from the date of diagnosis to last follow-up or death. EFS was defined as the time in months from the date of diagnosis to relapse, death, or last follow-up (the latter was used as the end point in those who did not relapse).23 EFS and OS were estimated and compared between groups using the log-rank test and/or Cox proportional hazard models. Univariate analyses (UVAs) and multivariable analyses (MVAs) were performed for OS and EFS in normal karyotype patients who received standard induction chemotherapy, as described earlier in this section. Any variables associated with P values of ≤.20 were included in initial multivariable models, followed by backward elimination until only variables associated with P < .05 remained in order to establish the final models. Results were quantified in terms of hazard ratios (HRs) along with the corresponding 95% confidence intervals. Unless otherwise stated, a P value <.05 was used to define statistical significance. Statistical analyses were performed using Prism 8.4 (GraphPad) and XLSTAT (2020.1) software packages.
Results
Study cohort
The study cohort consisted of 104 DNMT3A-mutated (DNMT3AMUT) de novo AML patients who met inclusion criteria. The median patient age was 61 years, male-to-female ratio (M:F) = 0.86, and the median follow-up time was 14.5 months (Table 1). Ninety-three patients (89%) were treated with standard induction chemotherapy, and 59 patients (57%) underwent SCT in first complete remission (CR1) with a median time of 4.5 months from diagnosis to SCT (Table 1). Five patients (10%) with FLT3-ITD mutations received FLT3-inhibitor therapy before any relapse (midostaurin, n = 2; crenolanib, n = 2; gilteritinib, n = 1). Forty-five patients (48%) relapsed, and 55 patients (59%) were alive at last follow-up (Table 1). Eighty-five patients (82%) had a normal karyotype and 19 (18%) had an abnormal karyotype (supplemental Table 1).
Characteristics of patients with de novo AML with DNMT3AMUT (N = 104)
| DNMT3AMUT, N = 104 | DNMT3ALOW, N = 57 | DNMT3AHIGH, N = 47 | P* | |
|---|---|---|---|---|
| Patient characteristics | ||||
| Median age (range), y | 61 (23-87) | 60 (23-87) | 62 (30-87) | >.05 |
| M:F ratio | 0.86 | 0.90 | 0.81 | >.05 |
| Abnormal karyotype, N (%) | 19 (18) | 9 (16) | 10 (21) | >.05 |
| Treatment | ||||
| Induction chemotherapy, N (%) | 93 (89) | 52 (91) | 41 (87) | >.05 |
| FLT3-inhibitor therapy, N (%) | 5 (5) | 5 (9) | 0 | >.05 |
| Allo-SCT, N (%) | 59 (57) | 38 (67) | 21 (45) | .0243 |
| Median time from diagnosis to allo-SCT (range), mo | 4.5 (2-66.5) | 4.4 (2-36.5) | 4.9 (2.1-66.5) | >.05 |
| Outcome | ||||
| Achieved complete remission, N (%) | 83 (89) | 48 (92) | 35 (85) | >.05 |
| Relapsed, N (%) | 45 (48) | 20 (39) | 25 (61) | .0311 |
| Alive at last follow-up, N (%) | 55 (59) | 37 (71) | 18 (44) | .0076 |
| Median EFS (range), mo | 25.2 (0.7-83.3) | 56.8 (1.1-83.3) | 14.1 (0.7-73.8) | .0154 |
| Median OS (range), mo | 44.5 (0.7-83.3) | NR (1.1-83.3) | 18.3 (0.7-73.8) | .0011 |
| DNMT3A mutation | ||||
| Two DNMT3A mutations, N (%) | 13 (14) | 7 (12) | 6 (13) | >.05 |
| Type of DNMT3A mutations, N (%) | ||||
| Missense mutations | 80 (77) | 41 (72) | 39 (83) | >.05 |
| • R882H | 39 (38) | 18 (32) | 21 (45) | >.05 |
| • R882C | 11 (11) | 6 (11) | 5 (11) | >.05 |
| • R882P | 3 (3) | 0 | 3 (6) | |
| • Others | 29 (28) | 17 (30) | 12 (26) | >.05 |
| Nonsense mutations | 9 (9) | 5 (9) | 4 (9) | >.05 |
| Frameshift mutations | 12 (12) | 9 (16) | 3 (6) | >.05 |
| Splice site mutations | 2 (2) | 2 (4) | 0 | >.05 |
| Clinical parameters | ||||
| Median Hb (range), g/dL | 8.8 (3.7-15) | 8.7 (5.7-12.5) | 8.8 (3.7-15) | >.05 |
| Median platelets (range), ×103/µL | 84 (12-543) | 99 (18-543) | 64.5 (12-271) | >.05 |
| Median WBC (range), ×109/L | 13.8 (0.6-220) | 5.6 (0.6-174) | 37 (0.6-220) | <.0001 |
| Median PB blasts (range), % | 16 (0-98) | 11 (0-88) | 24 (0-98) | .0093 |
| Median BM blasts (range), % | 65.5 (20-96) | 60 (20-90) | 70 (22-96) | .0086 |
| WHO classification, N (%) | ||||
| AML, NOS | 34 (33) | 23 (40) | 11 (23) | >.05 |
| AML-MRC | 7 (7) | 4 (7) | 3 (6) | >.05 |
| AML with NPM1 mutation | 54 (52) | 26 (46) | 28 (60) | >.05 |
| AML with RUNX1 mutation | 9 (9) | 4 (7) | 5 (11) | >.05 |
| Morphologic parameters | ||||
| Erythroid dysplasia | ||||
| Mean dysplasia, % | 28 | 25 | 33 | >.05 |
| Mean megaloblastic change | 0.9 | 0.9 | 1.0 | >.05 |
| Mean multinucleation | 0.7 | 0.6 | 0.8 | >.05 |
| Mean nuclear irregularities | 1.5 | 1.3 | 1.7 | >.05 |
| Granulocytic dysplasia | ||||
| Mean dysplasia, % | 27 | 18 | 35 | .0177 |
| Mean abnormal nuclear shape | 1.0 | 0.7 | 1.3 | .0082 |
| Mean hypogranulation | 1.2 | 0.9 | 1.5 | .0201 |
| Megakaryocytic dysplasia | ||||
| Mean dysplasia, % | 44 | 38 | 49 | >.05 |
| Mean micromegakaryocytes | 1.2 | 1.0 | 1.4 | >.05 |
| Mean separated nuclear lobes | 1.2 | 1.2 | 1.2 | >.05 |
| Mean hypolobated nuclei | 1.4 | 1.4 | 1.4 | >.05 |
| DNMT3AMUT, N = 104 | DNMT3ALOW, N = 57 | DNMT3AHIGH, N = 47 | P* | |
|---|---|---|---|---|
| Patient characteristics | ||||
| Median age (range), y | 61 (23-87) | 60 (23-87) | 62 (30-87) | >.05 |
| M:F ratio | 0.86 | 0.90 | 0.81 | >.05 |
| Abnormal karyotype, N (%) | 19 (18) | 9 (16) | 10 (21) | >.05 |
| Treatment | ||||
| Induction chemotherapy, N (%) | 93 (89) | 52 (91) | 41 (87) | >.05 |
| FLT3-inhibitor therapy, N (%) | 5 (5) | 5 (9) | 0 | >.05 |
| Allo-SCT, N (%) | 59 (57) | 38 (67) | 21 (45) | .0243 |
| Median time from diagnosis to allo-SCT (range), mo | 4.5 (2-66.5) | 4.4 (2-36.5) | 4.9 (2.1-66.5) | >.05 |
| Outcome | ||||
| Achieved complete remission, N (%) | 83 (89) | 48 (92) | 35 (85) | >.05 |
| Relapsed, N (%) | 45 (48) | 20 (39) | 25 (61) | .0311 |
| Alive at last follow-up, N (%) | 55 (59) | 37 (71) | 18 (44) | .0076 |
| Median EFS (range), mo | 25.2 (0.7-83.3) | 56.8 (1.1-83.3) | 14.1 (0.7-73.8) | .0154 |
| Median OS (range), mo | 44.5 (0.7-83.3) | NR (1.1-83.3) | 18.3 (0.7-73.8) | .0011 |
| DNMT3A mutation | ||||
| Two DNMT3A mutations, N (%) | 13 (14) | 7 (12) | 6 (13) | >.05 |
| Type of DNMT3A mutations, N (%) | ||||
| Missense mutations | 80 (77) | 41 (72) | 39 (83) | >.05 |
| • R882H | 39 (38) | 18 (32) | 21 (45) | >.05 |
| • R882C | 11 (11) | 6 (11) | 5 (11) | >.05 |
| • R882P | 3 (3) | 0 | 3 (6) | |
| • Others | 29 (28) | 17 (30) | 12 (26) | >.05 |
| Nonsense mutations | 9 (9) | 5 (9) | 4 (9) | >.05 |
| Frameshift mutations | 12 (12) | 9 (16) | 3 (6) | >.05 |
| Splice site mutations | 2 (2) | 2 (4) | 0 | >.05 |
| Clinical parameters | ||||
| Median Hb (range), g/dL | 8.8 (3.7-15) | 8.7 (5.7-12.5) | 8.8 (3.7-15) | >.05 |
| Median platelets (range), ×103/µL | 84 (12-543) | 99 (18-543) | 64.5 (12-271) | >.05 |
| Median WBC (range), ×109/L | 13.8 (0.6-220) | 5.6 (0.6-174) | 37 (0.6-220) | <.0001 |
| Median PB blasts (range), % | 16 (0-98) | 11 (0-88) | 24 (0-98) | .0093 |
| Median BM blasts (range), % | 65.5 (20-96) | 60 (20-90) | 70 (22-96) | .0086 |
| WHO classification, N (%) | ||||
| AML, NOS | 34 (33) | 23 (40) | 11 (23) | >.05 |
| AML-MRC | 7 (7) | 4 (7) | 3 (6) | >.05 |
| AML with NPM1 mutation | 54 (52) | 26 (46) | 28 (60) | >.05 |
| AML with RUNX1 mutation | 9 (9) | 4 (7) | 5 (11) | >.05 |
| Morphologic parameters | ||||
| Erythroid dysplasia | ||||
| Mean dysplasia, % | 28 | 25 | 33 | >.05 |
| Mean megaloblastic change | 0.9 | 0.9 | 1.0 | >.05 |
| Mean multinucleation | 0.7 | 0.6 | 0.8 | >.05 |
| Mean nuclear irregularities | 1.5 | 1.3 | 1.7 | >.05 |
| Granulocytic dysplasia | ||||
| Mean dysplasia, % | 27 | 18 | 35 | .0177 |
| Mean abnormal nuclear shape | 1.0 | 0.7 | 1.3 | .0082 |
| Mean hypogranulation | 1.2 | 0.9 | 1.5 | .0201 |
| Megakaryocytic dysplasia | ||||
| Mean dysplasia, % | 44 | 38 | 49 | >.05 |
| Mean micromegakaryocytes | 1.2 | 1.0 | 1.4 | >.05 |
| Mean separated nuclear lobes | 1.2 | 1.2 | 1.2 | >.05 |
| Mean hypolobated nuclei | 1.4 | 1.4 | 1.4 | >.05 |
Bold P values represent < .05.
allo-SCT, allogeneic SCT; Hb, hemoglobin; NOS, not otherwise specified; NR, not reached; PB, peripheral blood.
P values comparing DNMT3AHIGH and DNMT3ALOW groups.
The median read count in the DNMT3A gene was 387 (range, 17-3658), with a read count of >100 in 87% of cases and a read count of >300 in 54% of cases. DNMT3A VAF ranged from 2% to 96% with a median of 43%. There was no significant difference in DNMT3A VAF measured between the 3 testing platforms (hybrid capture: median, 0.44 [n = 43]; rapid heme panel: median, 0.43 [n = 47]; SNaPshot: median, 0.43 [n = 14]; P = .6553). There was no identifiable relationship between DNMT3A VAF and DNMT3A read count (r = −0.08; P = .4915). A single DNMT3A mutation was detected in the majority of cases (87.5%). Missense mutations were most common (n = 80; 77%), and frequently the R882H variant, as reported previously (Table 1; supplemental Table 2).10,17,19 There was no significant effect of DNMT3A R882 mutation on EFS and OS in normal karyotype patients receiving induction chemotherapy (supplemental Figure 1). Nonsense, frameshift, and/or splice site mutations were identified in 24 patients (23%). Thirteen patients (12.5%) had 2 DNMT3A mutations.
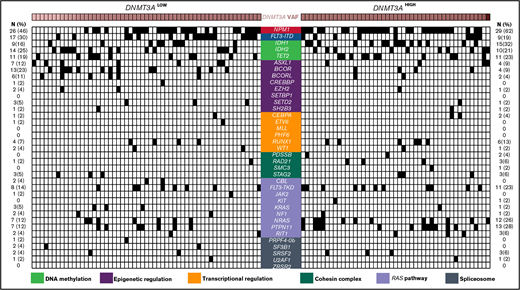
Comparison of comutational profiles for all patients with de novo AML with mutated DNMT3A. All cases of de novo AML with mutated DNMT3A evaluated (n = 104). Each column represents an individual patient. Intensity gradient corresponds to DNMT3A VAF. All comutations are provided in binary format with a black box indicating presence of comutation in that patient. The number and percentage of cases with a specific comutation are listed on the left (for DNMT3ALOW cases) and right (for DNMT3AHIGH cases).
Comparison of comutational profiles for all patients with de novo AML with mutated DNMT3A. All cases of de novo AML with mutated DNMT3A evaluated (n = 104). Each column represents an individual patient. Intensity gradient corresponds to DNMT3A VAF. All comutations are provided in binary format with a black box indicating presence of comutation in that patient. The number and percentage of cases with a specific comutation are listed on the left (for DNMT3ALOW cases) and right (for DNMT3AHIGH cases).
Characteristics of patients with single DNMT3A mutation and double DNMT3A mutations
| Single DNMT3A mutation, N = 91 | Double DNMT3A mutations, N = 13 | P* | |
|---|---|---|---|
| Patient characteristics | |||
| Median age (range), y | 61 (23-87) | 62 (43-83) | >.05 |
| M:F ratio | 0.9 | 0.6 | >.05 |
| Abnormal karyotype, N (%) | 16 (18) | 3 (23) | >.05 |
| Treatment | |||
| Induction chemotherapy, N (%) | 82 (90) | 11 (85) | >.05 |
| FLT3-inhibitor therapy, N (%) | 5 (6) | 0 | >.05 |
| Allo-SCT, N (%) | 54 (59) | 5 (38) | >.05 |
| Median time from diagnosis to allo-SCT (range), mo | 8.4 | 4.1 | >.05 |
| Outcome | |||
| Achieved complete remission, N (%) | 78 (86) | 7 (54) | .0099 |
| Relapsed, N (%) | 44 (48) | 9 (69) | >.05 |
| Alive at last follow-up, N (%) | 53 (58) | 6 (46) | >.05 |
| Median EFS (range), mo | 28.2 (0.7-83.3) | 5.4 (1.3-19.2) | .0190 |
| Median OS (range), mo | 47 (0.7-83.3) | 9.3 (1.7-25.0) | .0156 |
| Type of DNMT3A mutations, N (%) | |||
| Missense mutations | 72 (79) | 17 (65) | >.05 |
| • R882H | 38 (42) | 1 (4) | .0002 |
| • R882C | 11 (12) | 0 | >.05 |
| • R882P | 3 (3) | 0 | >.05 |
| • Others | 20 (22) | 16 (62) | <.0001 |
| Nonsense mutations | 6 (7) | 4 (15) | >.05 |
| Frameshift mutations | 11 (12) | 4 (15) | >.05 |
| Splice site mutations | 2 (2) | 1 (4) | >.05 |
| Clinical parameters | |||
| Median Hb (range), g/dL | 8.8 (3.7-15) | 8.8 (7.6-12.5) | >.05 |
| Median platelets (range), ×103/µL | 88 (12-543) | 63 (18-225) | >.05 |
| Median WBC (range), ×109/L | 15.8 (0.6-220) | 9 (1.1-174) | >.05 |
| Median PB blasts (range), % | 16 (0-98) | 13 (0-93) | >.05 |
| Median BM blasts (range), % | 67 (20-96) | 44 (25-84) | >.05 |
| WHO classification, N (%) | |||
| AML, NOS | 27 (30) | 7 (54) | >.05 |
| AML-MRC | 6 (7) | 1 (8) | >.05 |
| AML with NPM1 mutation | 50 (55) | 4 (31) | >.05 |
| AML with RUNX1 mutation | 8 (9) | 1 (8) | >.05 |
| Morphologic parameters | |||
| Erythroid dysplasia | |||
| Mean dysplasia, % | 28 | 30 | >.05 |
| Mean megaloblastic change | 0.9 | 1.3 | >.05 |
| Mean multinucleation | 0.7 | 0.4 | >.05 |
| Mean nuclear irregularities | 1.5 | 1.4 | >.05 |
| Granulocytic dysplasia | |||
| Mean dysplasia, % | 25 | 53 | >.05 |
| Mean abnormal nuclear shape | 0.9 | 1.9 | >.05 |
| Mean hypogranulation | 1.1 | 2.1 | >.05 |
| Megakaryocytic dysplasia | |||
| Mean dysplasia, % | 42 | 76 | >.05 |
| Mean micromegakaryocytes | 1.2 | 1.4 | >.05 |
| Mean separated nuclear lobes | 1.2 | 2 | >.05 |
| Mean hypolobated nuclei | 1.4 | 1.4 | >.05 |
| Single DNMT3A mutation, N = 91 | Double DNMT3A mutations, N = 13 | P* | |
|---|---|---|---|
| Patient characteristics | |||
| Median age (range), y | 61 (23-87) | 62 (43-83) | >.05 |
| M:F ratio | 0.9 | 0.6 | >.05 |
| Abnormal karyotype, N (%) | 16 (18) | 3 (23) | >.05 |
| Treatment | |||
| Induction chemotherapy, N (%) | 82 (90) | 11 (85) | >.05 |
| FLT3-inhibitor therapy, N (%) | 5 (6) | 0 | >.05 |
| Allo-SCT, N (%) | 54 (59) | 5 (38) | >.05 |
| Median time from diagnosis to allo-SCT (range), mo | 8.4 | 4.1 | >.05 |
| Outcome | |||
| Achieved complete remission, N (%) | 78 (86) | 7 (54) | .0099 |
| Relapsed, N (%) | 44 (48) | 9 (69) | >.05 |
| Alive at last follow-up, N (%) | 53 (58) | 6 (46) | >.05 |
| Median EFS (range), mo | 28.2 (0.7-83.3) | 5.4 (1.3-19.2) | .0190 |
| Median OS (range), mo | 47 (0.7-83.3) | 9.3 (1.7-25.0) | .0156 |
| Type of DNMT3A mutations, N (%) | |||
| Missense mutations | 72 (79) | 17 (65) | >.05 |
| • R882H | 38 (42) | 1 (4) | .0002 |
| • R882C | 11 (12) | 0 | >.05 |
| • R882P | 3 (3) | 0 | >.05 |
| • Others | 20 (22) | 16 (62) | <.0001 |
| Nonsense mutations | 6 (7) | 4 (15) | >.05 |
| Frameshift mutations | 11 (12) | 4 (15) | >.05 |
| Splice site mutations | 2 (2) | 1 (4) | >.05 |
| Clinical parameters | |||
| Median Hb (range), g/dL | 8.8 (3.7-15) | 8.8 (7.6-12.5) | >.05 |
| Median platelets (range), ×103/µL | 88 (12-543) | 63 (18-225) | >.05 |
| Median WBC (range), ×109/L | 15.8 (0.6-220) | 9 (1.1-174) | >.05 |
| Median PB blasts (range), % | 16 (0-98) | 13 (0-93) | >.05 |
| Median BM blasts (range), % | 67 (20-96) | 44 (25-84) | >.05 |
| WHO classification, N (%) | |||
| AML, NOS | 27 (30) | 7 (54) | >.05 |
| AML-MRC | 6 (7) | 1 (8) | >.05 |
| AML with NPM1 mutation | 50 (55) | 4 (31) | >.05 |
| AML with RUNX1 mutation | 8 (9) | 1 (8) | >.05 |
| Morphologic parameters | |||
| Erythroid dysplasia | |||
| Mean dysplasia, % | 28 | 30 | >.05 |
| Mean megaloblastic change | 0.9 | 1.3 | >.05 |
| Mean multinucleation | 0.7 | 0.4 | >.05 |
| Mean nuclear irregularities | 1.5 | 1.4 | >.05 |
| Granulocytic dysplasia | |||
| Mean dysplasia, % | 25 | 53 | >.05 |
| Mean abnormal nuclear shape | 0.9 | 1.9 | >.05 |
| Mean hypogranulation | 1.1 | 2.1 | >.05 |
| Megakaryocytic dysplasia | |||
| Mean dysplasia, % | 42 | 76 | >.05 |
| Mean micromegakaryocytes | 1.2 | 1.4 | >.05 |
| Mean separated nuclear lobes | 1.2 | 2 | >.05 |
| Mean hypolobated nuclei | 1.4 | 1.4 | >.05 |
Bold P values represent < .05.
See Table 1 for expansion of abbreviations.
P values comparing cases with single DNMT3A mutation vs double DNMT3A mutations.
The median number of mutated genes per case, including DNMT3A, was 4 (range, 1-7). DNMT3A was the sole mutation in 4 patients (4%). The most common comutated genes included NPM1 (53%), FLT3-ITD (25%), IDH1 (23%), IDH2 (23%), and TET2 (21%) (Figure 1; supplemental Table 4). DNA methylation pathway gene comutations were overall the most frequent (n = 66; 64%), followed by mutations in genes involved in epigenetic regulation (n = 36; 35%), RAS pathway (n = 35; 34%), transcriptional regulation (n = 13; 13%), the cohesin complex (n = 13; 13%), and the spliceosome apparatus (n = 10; 10%) (supplemental Table 4).
Effects of high DNMT3A VAF
Cases were partitioned into 2 groups using the median DNMT3A VAF: DNMT3ALOW cases (DNMT3A VAF ≤ 43%) and DNMT3AHIGH cases (DNMT3A VAF ≥ 44%). To classify double DNMT3A mutant cases into the DNMT3ALOW or DNMT3AHIGH group, the higher VAF of the 2 mutations was used (supplemental Table 3). Clinical parameters, morphologic variables, comutations, and patient outcomes were compared between the 2 groups (Table 1). The number of patients who received induction chemotherapy were similar in the 2 groups. However, significantly more patients in the DNMT3ALOW group received SCT (Table 1). When comparing patients who received SCT to those who did not, there was no significant difference in patients with an abnormal karyotype (17% vs 20%; P > .05) or FLT3-ITD mutation status (18% vs 31%; P > .05). In addition, when comparing the DNMT3ALOW and DNMT3AHIGH groups, there was no significant difference in the number of cases with abnormal karyotypes or FLT3-ITD mutation (P > .05). Therefore, cytogenetics and FLT3-ITD mutation status were not the primary drivers of the higher SCT rate in DNMT3ALOW patients. Based on a review of the patients’ electronic medical records, many clinical variables were considered prior to SCT, including age, comorbidities, prior treatment history, and availability of a suitable donor.
MVA of factors influencing EFS and OS in NK patients receiving induction chemotherapy (n = 76)
| Variable | P | HR | 95% CI |
|---|---|---|---|
| EFS | |||
| • Two DNMT3A mutations | .038 | 3.192 | 1.068 = 9.542 |
| • Received allo-SCT (in CR1) | .001 | 0.295 | 0-0.595 |
| OS | |||
| • DNMT3AHIGH | .010 | 3.003 | 1.308-6.896 |
| • BM blast % (continuous, per % increase) | .025 | 1.026 | 1.003-1.049 |
| • Two DNMT3A mutations | .020 | 4.816 | 1.284-18.061 |
| Variable | P | HR | 95% CI |
|---|---|---|---|
| EFS | |||
| • Two DNMT3A mutations | .038 | 3.192 | 1.068 = 9.542 |
| • Received allo-SCT (in CR1) | .001 | 0.295 | 0-0.595 |
| OS | |||
| • DNMT3AHIGH | .010 | 3.003 | 1.308-6.896 |
| • BM blast % (continuous, per % increase) | .025 | 1.026 | 1.003-1.049 |
| • Two DNMT3A mutations | .020 | 4.816 | 1.284-18.061 |
CI, confidence interval; NK, normal karyotype.
Morphological and clinical parameters.
DNMT3AHIGH cases were associated with leukocytosis and elevated blast count in the peripheral blood and BM (Table 1). DNMT3A VAF demonstrated statistically significant positive correlations with white blood cell (WBC) count (r = 0.31; P = .0012), peripheral blast percentage (r = 0.24; P = .0158), and BM blast percentage (r = 0.21; P = .0295). DNMT3AHIGH cases were associated with more significant granulocytic dysplasia (ie, more severe individual dysplastic features and elevated mean dysplasia) (Table 1).
CD34 expression on blasts.
We examined the correlation between DNMT3A VAF and the cell type that acquired the first DNMT3A mutation (ie, does a higher DNMT3A VAF correlate with a more stem cell–like AML with expression of immature markers?). CD34 expression in blasts (assessed by flow cytometry or immunoperoxidase studies) was compared between the 2 groups. There was no significant difference in cases with CD34+ blasts between the 2 groups (65% of DNMT3ALOW cases vs 49% of DNMT3AHIGH cases; P = .1194). In addition, there was no significant difference in the DNMT3A VAF in cases with CD34− blasts compared with cases with CD34+ blasts (42% vs 37%; P = .082). Taken together, these data indicate that DNMT3A VAF does not correlate with CD34 expression on the blasts. Interestingly, CD34+ blasts were significantly higher in the double DNMT3A mutant cases compared with single DNMT3A mutant cases (92% vs 52%; P = .0065). Additional studies with single-cell sequencing need to be performed to precisely identify the specific cell in which the first DNMT3A mutation occurs.
Comutational profiles.
There was no association between the different types of DNMT3A mutations and the DNMT3ALOW and DNMT3AHIGH groups (supplemental Table 2). The median VAF and VAF range of NPM1 and FLT3-ITD were not significantly different between the 2 groups (data not shown). AML with NPM1 mutation is associated with granulocytic dysplasia.1 So, we evaluated the contribution of NPM1 comutation to the granulocytic dysplasia seen in the DNMT3AHIGH group. NPM1 VAF did not show a significant correlation with granulocytic dysplasia in both the DNMT3ALOW (r = 0.05; P = .7814) and DNMT3AHIGH (r = −0.16; P = .3883) groups, suggesting that granulocytic dysplasia observed in the DNMT3AHIGH cases is independent of NPM1 comutation.
Clinical outcomes in normal karyotype patients receiving induction chemotherapy.
Among normal karyotype patients receiving induction chemotherapy, high DNMT3A VAF was associated with a significantly higher rate of relapse (EFS, 14.1 vs 56.8 months; P = .0154) and mortality (OS, 18.3 months vs not reached; P = .0011) compared with the DNMT3ALOW cases (Figure 2). Interestingly, this effect was only observed in patients with a single DNMT3A mutation, but not in patients with double mutations, who had short EFS and OS irrespective of the DNMT3A VAF (Figure 2).
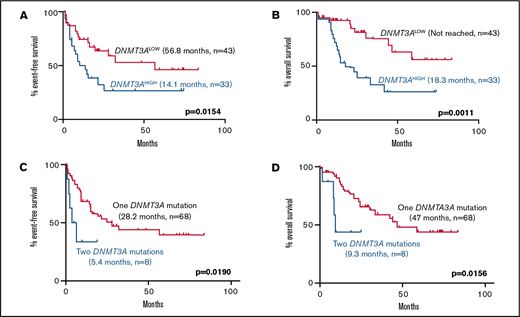
Kaplan-Meier curves showing effect of DNMT3A VAF and double DNMT3A mutations in normal karyotype patients receiving induction chemotherapy. (A-B) Effect of high DNMT3A VAF on (A) EFS (14.1 months, n = 33 vs 56.8 months, n = 43; P = .0154) and (B) OS (18.3 months, n = 33 vs not reached, n = 43; P = .0011). (C-D) Effect of double DNMT3A mutations on (E) EFS (5.4 months, n = 8 vs 28.2 months, n = 68; P = .0190) and (F) OS (9.3 months, n = 8 vs 47.0 months, n = 68; P = .0156) in the entire cohort. Values were generated using the log-rank test.
Kaplan-Meier curves showing effect of DNMT3A VAF and double DNMT3A mutations in normal karyotype patients receiving induction chemotherapy. (A-B) Effect of high DNMT3A VAF on (A) EFS (14.1 months, n = 33 vs 56.8 months, n = 43; P = .0154) and (B) OS (18.3 months, n = 33 vs not reached, n = 43; P = .0011). (C-D) Effect of double DNMT3A mutations on (E) EFS (5.4 months, n = 8 vs 28.2 months, n = 68; P = .0190) and (F) OS (9.3 months, n = 8 vs 47.0 months, n = 68; P = .0156) in the entire cohort. Values were generated using the log-rank test.
To assess the contributions of comutations toward the differences seen in patient outcomes between the DNMT3ALOW and DNMT3AHIGH groups, EFS was examined in genetically defined subpopulations. DNMT3AHIGH cases were associated with shorter EFS in cases with wild-type NPM1 and FLT3-ITD (Figure 3). In cases with mutations in NPM1, or FLT3-ITD, or both, high DNMT3A VAF was associated with a non–statistically significant shorter EFS (Figure 3).
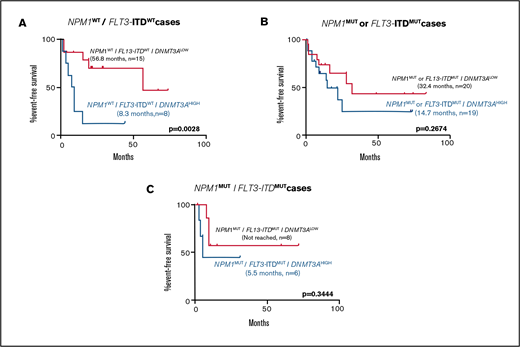
Kaplan-Meier curves showing effect of DNMT3A VAF in normal karyotype patients receiving induction chemotherapy in selected genetically defined subpopulations. Effect of high DNMT3A VAF on EFS in cases with (A) NPM1WT/FLT3-ITDWT (8.3 months, n = 8 vs 56.8 months, n = 15; P = .0028), (B) NPM1MUT or FLT3-ITDMUT (14.7 months, n = 19 vs 32.4 months, n = 20; P = .2674), and (C) NPM1MUT/FLT3-ITDMUT (5.5 months, n = 6 vs not reached, n = 8; P = .3444) subpopulations. Values were generated using the log-rank test.
Kaplan-Meier curves showing effect of DNMT3A VAF in normal karyotype patients receiving induction chemotherapy in selected genetically defined subpopulations. Effect of high DNMT3A VAF on EFS in cases with (A) NPM1WT/FLT3-ITDWT (8.3 months, n = 8 vs 56.8 months, n = 15; P = .0028), (B) NPM1MUT or FLT3-ITDMUT (14.7 months, n = 19 vs 32.4 months, n = 20; P = .2674), and (C) NPM1MUT/FLT3-ITDMUT (5.5 months, n = 6 vs not reached, n = 8; P = .3444) subpopulations. Values were generated using the log-rank test.
Subpopulation with double DNMT3A mutations
Patients with 2 DNMT3A mutations (n = 13) were diagnosed at a median age of 62 years (range, 43-83 years) and had an M:F ratio of 0.6 (Table 2). The different types of DNMT3A mutations and comutations in this cohort are listed in Table 2 and supplemental Tables 3 and 5. One of the 26 total variants was R882 compared with 38 single-mutant cases that harbored the R882 variant (P < .0001). Interestingly, we identified a significant positive correlation between the diagnostic VAF for the 2 DNMT3A variants (supplemental Table 3; supplemental Figure 2) on paired analysis (Spearman r = 0.8; P = .002) with a median absolute difference between values in a single patient of 2.0%. The VAFs for both DNMT3A variants tracked together in several patients with NGS data available at multiple time points (supplemental Figure 2).
Clinical outcome measures in normal karyotype patients receiving induction chemotherapy
UVAs and MVAs for factors affecting EFS in normal karyotype patients receiving induction chemotherapy.
Among patients with a normal karyotype, factors adversely affecting EFS included: 2 DNMT3A mutations (HR = 3.561; P = .021), high DNMT3A VAF (HR = 2.273; P = .010), increasing DNMT3A VAF (continuous) (HR = 1.025; P = .03), increasing WBC (continuous) (HR = 1.007; P = .019), increasing peripheral blast percentage (continuous) (HR = 1.013; P = .009), and increasing marrow blast percentage (continuous) (HR = 1.029; P = .003). SCT in CR1 was the only factor associated with a favorable effect on EFS in this subgroup (HR = 0.289; P = .0005). Additional variables associated with favorable or adverse effects on EFS in this subgroup (P ≤ .020) only included increasing total mutation count (continuous) (HR = 1.191).
In MVAs performed on patients with a normal karyotype, the following factors were independently associated with a significant effect on EFS: 2 DNMT3A mutations (HR = 3.192; P = .038) and SCT in CR1 (HR = 0.295; P = .001) (Table 3).
UVAs and MVAs for factors affecting OS in normal karyotype patients receiving induction chemotherapy.
Among patients with a normal karyotype, factors adversely affecting OS included: 2 DNMT3A mutations (HR = 3.450; P = .049), high DNMT3A VAF (HR = 3.621; P = .001), increasing DNMT3A VAF (continuous) (HR = 1.030; P = .037), increasing peripheral blast percentage (continuous) (HR = 1.015; P = .011), and increasing marrow blast percentage (continuous) (HR = 1.032; P = .008). Only SCT in CR1 was associated with a favorable effect on OS in this subgroup (HR = 0.402; P = .016). Additional variables associated with favorable or adverse effects on OS in this subgroup (P ≤ .020) included increasing WBC (continuous) (HR = 1.006) and FLT3-ITD (HR = 0.533). Neither lineage dysplasia nor NPM1 mutation exhibited any effect on OS in either group in UVA.
In MVAs performed on patients with a normal karyotype, the following factors were independently associated with a significant effect on OS: increasing marrow blast percentage (continuous) (HR = 1.026; P = .025), high DNMT3A VAF (HR = 3.003; P = .010), and 2 DNMT3A mutations (HR = 4.816; P = .020) (Table 3).
Discussion
In this study, we assessed for potential associations between clinical variables, comutations, DNMT3A mutational burden, and DNMT3A mutation count at diagnosis in a uniform cohort of 104 DNMT3A-mutated AML patients. The effects of age, SCT in CR1, abnormal karyotype, and specific comutations on clinical outcome, in patients stratified by DNMT3A VAF, were also examined in defined subpopulations. Salient findings from our analyses include (1) an association between high DNMT3A VAF (VAF ≥ 44%) and a more proliferative presentation and granulocytic dysplasia; (2) an adverse prognostic impact of high DNMT3A VAF, particularly in patients with a normal karyotype and those lacking NPM1 and/or FLT3-ITD comutations; and (3) a negative prognostic effect of 2 concurrent DNMT3A mutations.
For the total study cohort, we stratified patients at the median DNMT3A VAF (43%) into DNMT3ALOW and DNMT3AHIGH groups. The high median VAF of DNMT3A in our cohort, and in others previously reported in the literature,17,21,24 may potentially reflect a proportional dominance of the total marrow cellularity by a mutant clone harboring a heterozygous mutation; however, in this retrospective study, we were unable to perform more sensitive genome-wide cytogenetic assays (eg, single-nucleotide polymorphism array analysis) to exclude the possibility of copy number neutral loss of heterozygosity at the 2p locus affecting the wild-type DNMT3A allele.25 Of note, outcome analyses based on stratification of the cohort into quartiles based on the VAF were also performed in normal karyotype patients receiving induction chemotherapy, and revealed a consistent association between higher VAF and inferior outcome (supplemental Figure 2).
Interestingly, DNMT3AHIGH cases were associated with quantitative and qualitative differences in the granulocytic compartment, but not in the erythroid or megakaryocytic series. We identified no significant differences in median hemoglobin levels or platelet counts between the 2 groups, however, DNMT3AHIGH patients presented with leukocytosis and higher blast counts. Moreover, we observed a statistically significant positive correlation between VAF and WBC as well as both peripheral blood and marrow blast counts. These data suggest that a higher DNMT3A VAF may be associated with a more proliferative phenotype. PTPN11 mutations were more frequent in the DNMT3AHIGH group (P < .05), whereas FLT3-ITD was less frequent but not significantly different (supplemental Table 4). We further identified a significant correlation between higher DNMT3A VAF and morphologic dysplasia, which was notably restricted to the granulocytic lineage. We hypothesize that DNMT3A mutations may affect granulopoiesis in the context of an evolving myeloid neoplasia, including AML.
Our group was recently the first to identify and report on a potentially significant adverse prognostic effect of high VAF in NPM1-mutated de novo AML. Similarly, recent studies by others24,26 have interrogated for a relationship between VAF and clinical outcome in DNMT3A-mutated AML, with conflicting results. Although Yuan et al26 identified an independent prognostic effect of high DNMT3A VAF, the group led by Linch et al24 reported that an adverse effect of increasing DNMT3A VAF in UVAs was lost in multivariable modeling, likely secondary to the favorable effect of NPM1 comutation. In this cohort, we also observed significantly shorter EFS and OS in DNMT3AHIGH patients in UVAs; moreover, in additional subgroup analyses, the negative effect of DNMT3AHIGH on EFS was stronger in wild-type NPM1 (NPM1WT) than NPM1MUT patients. Of note, stratification by either NPM1 or FLT3-ITD status revealed no statistically significant differences in patient outcome, indicating that the prognostic effects of these mutations is affected by the DNMT3A mutation status, as has been previously shown.27,28 No other specific comutational patterns were associated with significant clinical outcome differences in this cohort, although an abnormal karyotype was unfavorable in both UVAs and MVAs for factors affecting EFS and OS.
In multivariable modeling to identify factors independently associated with effects on EFS and/or OS, we found retention of the adverse effect of DNMT3AHIGH only in patients with a normal karyotype, and only with respect to OS but not EFS. Conversely, the presence of 2 concomitant DNMT3A mutations was associated with an independent adverse prognostic effect on both EFS and OS in patients with normal karyotype. To the best of our knowledge, this is the first identification of such an association in DNTM3A-mutated AML patients. The presence of 2 DNMT3A mutations was associated with predominantly normal karyotypes, a lack of “classical” adverse comutations (eg, FLT3-ITD), relative infrequency of the R882 variant, and relative enrichment for IDH2 and BCOR comutations. We found a significant positive correlation between paired diagnostic VAFs and no significant difference between their median values. Further investigation of the subset of patients for whom we had NGS data from multiple time points revealed a close VAF association between both variants across time points. We therefore hypothesize that the majority of these patients harbored monoallelic or biallelic DNMT3A alterations within the same cells, which generated a more robust leukemia-promoting and/or -sustaining clone, potentially as a result of a dosage effect. Whether the apparent enrichment for concomitant IDH2 and/or BCOR mutations reflects a synergistic relationship, or simply an epiphenomenon, cannot be precisely determined within our study design.
We acknowledge that our study is limited by its retrospective nature, relatively small sample size, and the lack of a controlled clinical trial setting; the relatively short median follow-up time may be a consequence of these study characteristics, and validation of our data in larger cohorts with lengthier follow-up will be important for definitive confirmation of these data. However, our findings nonetheless highlight potentially novel aspects of the underlying biology of DNMT3A-mutated AML: we have identified a relationship between increasing DNMT3A VAF and proliferative presentation, granulocytic dysplasia, and inferior patient outcome. Our findings also reveal a novel association between 2 concomitant DNMT3A mutations and shorter EFS and OS. Additional studies in other DNMT3A-mutated myeloid neoplasms are warranted, specifically to further explore the observed associations between DNMT3A VAF and both clinical parameters and dysgranulopoiesis, and to confirm the adverse prognostic effect of 2 concomitant DNMT3A mutations. Prospective studies, ideally within the framework of a clinical trial, will be required to evaluate the potential for these parameters to be useful in guiding patient management and predicting clinical outcome in the context of other known risk factors in AML.
Authorship
Contribution: D.N., S.S.P., and O.K.W. designed research, analyzed data, and wrote the paper; O.P. and R.P.H. performed research and analyzed data; and all authors approved the final manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Olga K. Weinberg, Department of Pathology, UT Southwestern Medical Center, BioCenter, 2230 Inwood Rd, EB03.220G, Dallas, TX 75235; e-mail: olga.weinberg@utsouthwestern.edu.

