

Key Points
Survival after HCT vs non-HCT therapeutic options was compared in patients with MF, with results stratified by DIPSS risk.
A long-term survival advantage of HCT was observed in patients with Int-1 or higher risk MF, but at the cost of potential early mortality.

Abstract
Allogeneic hematopoietic cell transplantation (HCT) is the only curative therapy for myelofibrosis (MF). In this large multicenter retrospective study, overall survival (OS) in MF patients treated with allogeneic HCT (551 patients) and without HCT (non-HCT) (1377 patients) was analyzed with Cox proportional hazards model. Survival analysis stratified by the Dynamic International Prognostic Scoring System (DIPSS) revealed that the first year of treatment arm assignment, due to upfront risk of transplant-related mortality (TRM), HCT was associated with inferior OS compared with non-HCT (non-HCT vs HCT: DIPSS intermediate 1 [Int-1]: hazard ratio [HR] = 0.26, P < .0001; DIPSS-Int-2 and higher: HR, 0.39, P < .0001). Similarly, in the DIPSS low-risk MF group, due to upfront TRM risk, OS was superior with non-HCT therapies compared with HCT in the first-year post treatment arm assignment (HR, 0.16, P = .006). However, after 1 year, OS was not significantly different (HR, 1.38, P = .451). Beyond 1 year of treatment arm assignment, an OS advantage with HCT therapy in Int-1 and higher DIPSS score patients was observed (non-HCT vs HCT: DIPSS-Int-1: HR, 2.64, P < .0001; DIPSS-Int-2 and higher: HR, 2.55, P < .0001). In conclusion, long-term OS advantage with HCT was observed for patients with Int-1 or higher risk MF, but at the cost of early TRM. The magnitude of OS benefit with HCT increased as DIPSS risk score increased and became apparent with longer follow-up.
Introduction
Myelofibrosis (MF) is a myeloproliferative neoplasm characterized by clonal myeloid proliferation, extramedullary hematopoiesis, peripheral cytopenias, bone marrow fibrosis, and heterogenous symptom burden.1,2 With the discovery of the JAK2V617F driver mutation in 2005 and subsequent innovation of JAK inhibitors, the therapeutic landscape has evolved and led to significant improvements in the clinical care of MF patients.3-6 Despite improvements in patients’ symptom burden, splenomegaly, and potentially overall survival (OS) with JAK inhibitor therapy, allogeneic hematopoietic cell transplantation (HCT) remains the only curative therapy.7,8 The morbidity and mortality associated with transplant therapy underscores the importance of appropriate patient selection for HCT.9-13
Currently, prognostic stratification is a key clinical feature of transplant assessment in patients with MF. Multiple prognostic scoring systems have assisted clinical decision-making over the years, with more under development.14-20 The International Prognostic Scoring System (IPSS) employs risk factors, including age >65 years, hemoglobin <10 g/dL, white blood cell count >25 × 109/L, circulating blasts ≥1%, and the presence of constitutional symptoms.14 The IPSS highlighted that median survival varies widely from 135 months in those with low risk disease to 27 months in those with high-risk disease. While the IPSS is a useful prognostic estimator at the time of diagnosis, the Dynamic IPSS (DIPSS) is a similar prognostic estimator but may be used at any point during the disease course.15 The DIPSS Plus has added risk factors such as thrombocytopenia, transfusion dependence, and unfavorable karyotype.16 Contemporary prognostic models incorporate clinical, cytogenetic, and mutation data and advance MF prognostication.18,19,21
While emerging prognostic tools and clinical guidelines are useful to facilitate appropriate patient selection for HCT, there is no prospective and randomized data comparing HCT to non-HCT therapies to guide clinical decision-making. This study reports on survival outcomes of HCT vs non-HCT therapy, stratified by DIPSS risk score, in the largest and most inclusive retrospective analysis performed to date.
Methods
Study objective
The primary objective was an OS comparison between HCT and non-HCT therapy in MF when stratified by DIPSS risk status.15 Secondarily, the patients’ disease and treatment-related factors were evaluated for association with differences in survival.
Data source
For the HCT cohort, data were retrospectively obtained from the Center for International Blood and Marrow Transplant Research (CIBMTR) from years 2000 to 2014. The CIBMTR represents an international network of >450 transplant centers that has collected HCT outcomes data for >45 years, resulting in a research database on >475 000 patients. CIBMTR captures data including, but not limited to, baseline recipient and donor characteristics, transplant outcomes, and follow-up data at day +100, day +180, and annually after HCT.
For the non-HCT cohort, data were retrospectively obtained from the Myeloproliferative Neoplasm Research Consortium and its affiliates (a total of 14 academic centers; see supplemental Table 1) between years 2000 and 2014 after approval from institutional review boards of each participating study center. The Myeloproliferative Neoplasm Research Consortium is an interactive group of laboratory and clinical scientists from 11 institutions who work in a coordinated fashion to develop and evaluate therapeutic strategies that will improve the survival of patients with MF.
Study population
In the HCT cohort, patients with MF (either primary, postpolycythemia, or post–essential thrombocythemia MF) who received an HCT were included. Patients receiving HCT from an HLA-identical sibling or well-matched/partially matched unrelated donor after an ablative, nonmyeloablative, or reduced intensity conditioning were included. Patients who were aged ≥20 years and <70 years at the time of transplant were included. Patients who received umbilical cord blood HCT, syngeneic HCT, or haploidentical HCT or patients who have transformed to secondary acute myeloid leukemia prior to HCT were excluded (n = 63).
In the non-HCT cohort, patients with MF (either primary, postpolycythemia, or post–essential thrombocythemia MF) who were ≥20 years and <70 years were included. Those in the non-HCT cohort who were only observed and never on medical treatment were excluded, because they are likely not the ideal comparator for the HCT cohort.
Study variables
Patient-related variables, including age at diagnosis and HCT, race, and Karnofsky performance status (KPS), were collected. Disease-related variables, including diagnosis of primary vs secondary MF, DIPSS at HCT or time of initial treatment/referral to academic center, cytogenetics (favorable: normal karyotype; 13q−, 20q−; unfavorable: complex aberrations or noncomplex abnormalities, including +8, −7/7q−, i(17q), −5/5q−, 12p−, inv(3), or 11q23 rearrangements), and presence/absence of the JAK2V617F were captured. Treatment was recorded, including type and number of medical therapies, HLA matching, graft source, conditioning regimen, total body irradiation delivery, and graft-versus-host disease (GVHD) prophylaxis. Time from diagnosis to HCT or time of initial treatment/referral to academic center, date of death, or date of last contact was collected.
Statistical analysis
A descriptive analysis comparing the HCT and the non-HCT cohorts was performed. Medians, ranges, and percentages of total numbers were evaluated for categorical variables. The hazard ratio (HR) for OS between the HCT and the non-HCT cohorts were compared using the Cox proportional hazards model. The reference time point was DIPSS assessment at time of initiation of medical treatment or time of referral to academic medical center (if time of initiation of medical treatment was unknown) for the non-HCT cohort. The reference time point was DIPSS assessment at the time of transplant for the HCT cohort. The proportional hazards assumption of the Cox model was examined by testing a time-varying effect for each variable. A time-dependent effect was detected for the main effect, and a 12-month breakpoint of posttherapy initiation/referral or transplant was found optimal based on the maximized partial likelihood method to make sure the proportional hazards assumption held within each time period.22 A stepwise model selection approach was then used to identify all significant risk factors at a significance level .01. Potential interactions between the main effect and significant risk factors was tested. Adjusted OS probabilities were calculated to compare the non-HCT cohort and the HCT cohort after adjusting for the significant variables in the final Cox model.23 Adjusted OS was obtained at 5 and 10 years. Multiple imputation with 10 imputations was used to impute missing DIPSS risk scores and then confirmed with the final Cox model using the assumption that missing data are missing at random.24 The variables tested in the multivariable regression analysis included age at DIPSS assessment, race, KPS, primary vs secondary MF, ruxolitinib given as prior treatment, time from diagnosis to DIPSS assessment, and year of DIPSS assessment. Center effect was examined using the score test of homogeneity through the frailty proportional hazards model with random effects.25 The final Cox model was also fitted for each of the DIPSS risk scores as a subset analysis. To investigate whether lead time bias affected the main model given the fact that HCT patients were guaranteed survival up to the point in which they underwent transplant, a sensitivity analysis was performed restricting the time between diagnosis and transplant to 12 months for the HCT cohort. All analyses were performed at a significance level .01 using SAS 9.4 (SAS Institute, Cary, NC).
Results
Patient descriptive analysis
We identified a total of 551 MF patients who underwent HCT and 1377 MF patients treated with non-HCT therapy that met eligibility criteria. See Table 1 for patient characteristics. Diagnosis included primary MF in 463 patients (84%) in HCT group and 894 patients (65%) in non-HCT group. Among the HCT cohort, 74.6% were from the United States, while all the patients in the non-HCT cohort were from US centers. The JAK2V617F mutation was detected in 21% of HCT patients and 55% of non-HCT patients, and JAK2V617F mutation data were unavailable in 64% and 14% of patients, respectively. Other mutational analysis was not available. DIPSS risk at time of transplant was low in 82 patients (15%), intermediate 1 (Int-1) in 248 patients (45%), Int-2/high in 186 patients (34%), and missing in 35 patients (6%). In the non-HCT patients, DIPSS risk was low in 165 patients (12%), Int-1 in 537 patients (39%), Int-2/high in 389 patients (28%), and missing in 286 patients (21%). Among the 1377 non-HCT patients, 88 (6%) reported subsequently proceeding to HCT. The median time from diagnosis to HCT was 19 months (range, 2-360) in the HCT group, and the median time from diagnosis to referral was 2 months (<−383) in the non-HCT group. The median follow-up time of survivors since the reference time point was 72 months (range, 3-193) for HCT patients and 63 months (<1-208) for non-HCT patients.
Descriptive characteristics for patients with MF
| Variable | HCT (n = 551) | Non-HCT (n = 1377) |
|---|---|---|
| Primary MF, % | 84 | 65 |
| Secondary MF, % | 16 | 35 |
| Age at diagnosis, median (range), y | 51 (20-69) | 59 (20-75) |
| Median age at HCT/referral, y | 55 | 61 |
| DIPSS at HCT/referral, % | ||
| Low | 15 | 12 |
| Int-1 | 45 | 39 |
| Int-2/high | 34 | 28 |
| Missing | 6 | 21 |
| Cytogenetics, % | ||
| Normal/favorable | 52 | 73 |
| Unfavorable | 14 | 16 |
| Missing | 34 | 11 |
| Origin, % | ||
| White | 91 | 75 |
| African American | 4 | 6 |
| Asian | 3 | 2 |
| Other | 3 | 16 |
| JAK2V617F mutation, % | ||
| Positive | 21 | 55 |
| Negative | 15 | 32 |
| Not tested or unavailable | 64 | 14 |
| Prior therapy, % | ||
| Ruxolitinib | 10 | 30 |
| Hydroxyurea | 26 | 42 |
| Immunomodulatory | 14 | 26 |
| Splenic irradiation | 2 | 3 |
| Number of prior therapies, % | ||
| 0 | 28 | 16 |
| 1 | 36 | 25 |
| 2 | 17 | 24 |
| ≥3 | 17 | 34 |
| Conditioning intensity, % | ||
| MAC | 51 | — |
| RIC | 41 | — |
| NMA | 7 | — |
| Missing | 2 | — |
| Donor type | ||
| HLA identical | 38 | — |
| Well-matched unrelated | 47 | — |
| Partially matched unrelated | 15 | — |
| Median follow up time of survivors, mo | 72 | 63 |
| Time of diagnosis to treatment, mo | 19 | 2 |
| Number of centers | 138 | 14 |
| Variable | HCT (n = 551) | Non-HCT (n = 1377) |
|---|---|---|
| Primary MF, % | 84 | 65 |
| Secondary MF, % | 16 | 35 |
| Age at diagnosis, median (range), y | 51 (20-69) | 59 (20-75) |
| Median age at HCT/referral, y | 55 | 61 |
| DIPSS at HCT/referral, % | ||
| Low | 15 | 12 |
| Int-1 | 45 | 39 |
| Int-2/high | 34 | 28 |
| Missing | 6 | 21 |
| Cytogenetics, % | ||
| Normal/favorable | 52 | 73 |
| Unfavorable | 14 | 16 |
| Missing | 34 | 11 |
| Origin, % | ||
| White | 91 | 75 |
| African American | 4 | 6 |
| Asian | 3 | 2 |
| Other | 3 | 16 |
| JAK2V617F mutation, % | ||
| Positive | 21 | 55 |
| Negative | 15 | 32 |
| Not tested or unavailable | 64 | 14 |
| Prior therapy, % | ||
| Ruxolitinib | 10 | 30 |
| Hydroxyurea | 26 | 42 |
| Immunomodulatory | 14 | 26 |
| Splenic irradiation | 2 | 3 |
| Number of prior therapies, % | ||
| 0 | 28 | 16 |
| 1 | 36 | 25 |
| 2 | 17 | 24 |
| ≥3 | 17 | 34 |
| Conditioning intensity, % | ||
| MAC | 51 | — |
| RIC | 41 | — |
| NMA | 7 | — |
| Missing | 2 | — |
| Donor type | ||
| HLA identical | 38 | — |
| Well-matched unrelated | 47 | — |
| Partially matched unrelated | 15 | — |
| Median follow up time of survivors, mo | 72 | 63 |
| Time of diagnosis to treatment, mo | 19 | 2 |
| Number of centers | 138 | 14 |
Unless otherwise indicated, the data are presented as percentage of patients.
MAC, myeloablative conditioning; NMA, nonmyeloablative; RIC, reduced intensity conditioning.
Transplant descriptive analysis
Donor types included well-matched unrelated (8/8 match) in 47% of patients, HLA identical sibling/related in 38%, and partially matched unrelated in 15%.26 Peripheral blood stem cell graft source was used in 85% of patients, while bone marrow was used in 15%. The transplant conditioning regimens were myeloablative conditioning in 51% of patients, reduced intensity in 41%, nonmyeloablative in 7%, and unknown in 2%.27 Total body irradiation conditioning was employed in 20% of conditioning regimens. Antithymocyte globulin was used in 36% of patients, and alemtuzumab was used in 3%.
Survival analysis
The probability of survival for the 4 subgroups (DIPSS risk: low, Int-1, Int-2, and high) with HCT vs non-HCT therapy is depicted in Figure 1. In the DIPSS low-risk group, OS was superior with non-HCT therapies compared with HCT in the first-year posttreatment arm assignment, likely due to upfront transplant-related mortality (TRM) risk. However, after 1 year, OS was not significantly different (non-HCT: HR, 0.16; 95% confidence interval [CI], 0.04-0.59; P = .006 at <12 months; and HR, 1.4; 95% CI, 0.60-3.20; P = .45 at > 12 months; Figure 1A). In the DIPSS Int-1 risk group, an OS advantage was present for HCT vs non-HCT therapies, but this OS advantage was only observed beyond 1 year of treatment arm assignment (due to high risk of TRM in the first year post-HCT) (non-HCT vs HCT: HR, 2.64; 95% CI, 1.76-3.98; P < .0001 at <12 months; non-HCT vs HCT: HR, 0.26; 95% CI, 0.17-0.39; P ≤ .001 at >12 months; Figure 1B). Similarly, in those with DIPSS Int-2 and high-risk MF, OS was superior in HCT cohort compared with non-HCT therapies but only observed beyond 1 year of treatment arm assignment (again due to high risk of TRM in the first year post HCT) (non-HCT vs HCT: HR, 2.55; 95% CI, 1.66-3.90; P < .0001 at <12 months; non-HCT vs HCT: HR, 0.39; 95% CI, 0.27-0.57; P < .0001 at >12 months; Figure 1C). Across all risk groups, there was a OS advantage observed with non-HCT therapies in the first year of treatment arm assignment (due to high risk of TRM in the first year post HCT) (non-HCT vs HCT: HR, 0.33; 95% CI, 0.26-0.41; P < .0001); however, OS was improved beyond 1 year of treatment arm assignment with HCT (non-HCT vs HCT: HR, 2.11; 95% CI, 1.66-2.69; P < .0001; Figure 1D).
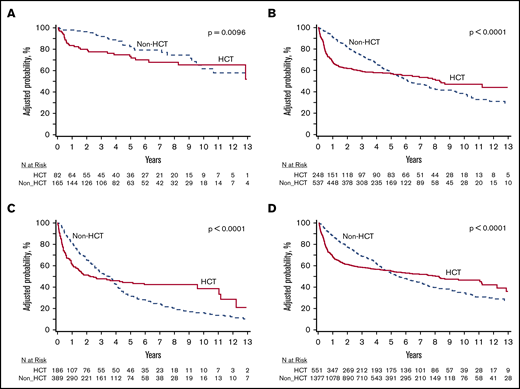
Survival probabilities for the DIPSS risk groups in MF receiving HCT vs non-HCT therapy. (A) DIPSS low risk. (B) DIPSS Int-1. (C) DIPSS Int-2 or higher. (D) Overall (all DIPSS groups). The survival curves presented here, stratified by DIPSS risk score, are a representation of the interventions (ie, HCT vs non-HCT therapy) over a median follow-up of ∼6 years. The curves cross much later in the clinical course than 12 months; however, the slope of the curves changes much earlier (12 months) and then plateaus, indicating the OS benefit associated with HCT begins much earlier than when the curves actually cross. A long-term survival advantage with HCT was observed for patients with Int-1 or higher risk MF, but at the cost of early mortality. The magnitude of OS benefit increased as DIPSS risk score increased.
Survival probabilities for the DIPSS risk groups in MF receiving HCT vs non-HCT therapy. (A) DIPSS low risk. (B) DIPSS Int-1. (C) DIPSS Int-2 or higher. (D) Overall (all DIPSS groups). The survival curves presented here, stratified by DIPSS risk score, are a representation of the interventions (ie, HCT vs non-HCT therapy) over a median follow-up of ∼6 years. The curves cross much later in the clinical course than 12 months; however, the slope of the curves changes much earlier (12 months) and then plateaus, indicating the OS benefit associated with HCT begins much earlier than when the curves actually cross. A long-term survival advantage with HCT was observed for patients with Int-1 or higher risk MF, but at the cost of early mortality. The magnitude of OS benefit increased as DIPSS risk score increased.
By multivariable Cox regression, KPS <90% (HR, 1.71; 95% CI, 1.49-1.98; P < .0001), DIPSS Int-1 or higher (Int-1: HR, 2.24; 95% CI, 1.69-2.97; P < .0001; Int-2: HR, 3.33; 95% CI, 2.50-4.43; P < .0001; high: HR, 5.67; 95% CI, 3.81-8.44; P < .0001), and unfavorable cytogenetics (HR, 1.45; 95% CI, 1.21-1.74; P < .0001) were associated with inferior survival in all patients. Prior ruxolitinib therapy was associated with increased survival (HR, 0.53; 95% CI, 0.44-0.63; P < .0001) (Table 2). The adjusted survival rate at 12 months after adjusting DIPSS, KPS, cytogenetics, and ruxolitinib was 68% (64%-72%) for the HCT group and 87% (85%-89%) for the non-HCT group based on a stratified Cox model.
OS with multivariable regression analysis
| HR | 95% CI | Overall P | |
|---|---|---|---|
| OS (≤12 mo) | <.0001 | ||
| HCT | 1 | ||
| Non-HCT | 0.325 | 0.260, 0.406 | |
| OS (>12 mo) | <.0001 | ||
| HCT | 1 | ||
| Non-HCT | 2.109 | 1.656, 2.685 | |
| DIPSS at HCT/referral | <.0001 | ||
| Low | 1 | ||
| Int-1 | 2.237 | 1.683, 2.973 | |
| Int-2 | 3.327 | 2.496, 4.435 | |
| High | 5.672 | 3.810, 8.443 | |
| KPS at HCT/referral | <.0001 | ||
| ≥90% | 1 | ||
| <90% | 1.713 | 1.485, 1.977 | |
| Cytogenetics | .0004 | ||
| Favorable (normal) | 1 | ||
| Favorable (other) | 1.040 | 0.868, 1.245 | |
| Unfavorable | 1.453 | 1.214, 1.740 | |
| Ruxolitinib | <.0001 | ||
| No | 1 | ||
| Yes | 0.530 | 0.444, 0.633 |
| HR | 95% CI | Overall P | |
|---|---|---|---|
| OS (≤12 mo) | <.0001 | ||
| HCT | 1 | ||
| Non-HCT | 0.325 | 0.260, 0.406 | |
| OS (>12 mo) | <.0001 | ||
| HCT | 1 | ||
| Non-HCT | 2.109 | 1.656, 2.685 | |
| DIPSS at HCT/referral | <.0001 | ||
| Low | 1 | ||
| Int-1 | 2.237 | 1.683, 2.973 | |
| Int-2 | 3.327 | 2.496, 4.435 | |
| High | 5.672 | 3.810, 8.443 | |
| KPS at HCT/referral | <.0001 | ||
| ≥90% | 1 | ||
| <90% | 1.713 | 1.485, 1.977 | |
| Cytogenetics | .0004 | ||
| Favorable (normal) | 1 | ||
| Favorable (other) | 1.040 | 0.868, 1.245 | |
| Unfavorable | 1.453 | 1.214, 1.740 | |
| Ruxolitinib | <.0001 | ||
| No | 1 | ||
| Yes | 0.530 | 0.444, 0.633 |
The impact of HCT vs non-HCT on survival was consistent whether patients received ruxolitinib or not (P value for interaction = .018). Evaluation of the impact of year of referral/HCT on survival (ie, before 2012 vs after 2012, the point at which ruxolitinib was commercially available) revealed no significant association with survival (P = .38). Analysis after excluding partially matched donors revealed no changes to the results (supplemental Table 3). Center effect was not significant (P = .02).
To investigate whether lead time bias affected the main model given the fact that HCT patients were guaranteed survival up to the point in which they underwent transplant, a sensitivity analysis was performed restricting the time between diagnosis and transplant to 12 months for the HCT cohort. This analysis revealed results consistent with main survival results as reported in survival analysis, suggesting minimal impact of lead time bias with chosen reference time point (supplemental Table 4).
Adjusted OS was evaluated at 5 and 10 years (supplemental Table 5). For the entire cohort not stratified by DIPSS, 10-year adjusted OS was 47% (95% CI, 42% to 52%) and 35% (95% CI, 51% to 38%) for HCT and non-HCT (P ≤ .0001), respectively. Cause of death in HCT patients within the first year included GVHD (23.8%), relapse of primary disease (21.1%), organ failure (15.7%), infection (14.1%), acute respiratory distress (8.1%), graft failure (3.8%), secondary malignancy (1.6%), and other causes (10.8).
Discussion
We studied survival outcomes in 1928 MF patients following HCT (551 patients) or non-HCT (1377 patients) in the largest retrospective long-term survival analysis performed in MF to date. This study suggests a long-term survival advantage with HCT for patients with Int-1 or higher risk MF, but with increased early mortality, thus highlighting the need for careful patient selection and continued development of therapies aimed at reducing post-HCT complications.
Long-term survival post-HCT in MF is variable, ranging from 29% to 65% depending on patient and transplant variables, and data are limited.10,12,13,28-45 Non-HCT therapies, including JAK inhibition, are palliative only, and thus, HCT remains the only curative therapy. In 2015, a consensus process guideline was developed by the European Leukemia Net in conjunction with the European Blood and Marrow Transplantation Group guiding allogeneic HCT recommendations in MF.46 In these guidelines, patients with Int-2 or high-risk disease according to IPSS, DIPSS, or DIPSS Plus and age <70 years should be considered potential candidates for HCT. Patients with Int-1 disease and age <65 years were recommended to be considered potential candidates if they present with either transfusion-dependent anemia, percentage of blasts in the peripheral blood >2%, adverse cytogenetics as defined by the DIPSS Plus classification, or high molecular risk (ie, JAK2/CALR/MPL negative, ASXL1 positive).46
The data presented in this study indicate a long-term survival advantage to HCT for patients with DIPSS Int-1 and higher risk MF, independent of transfusion need, blast percentage, or high-risk cytogenetic and molecular features. Clinicians may use the survival curves from this analysis, illustrated in Figure 1, for thorough HCT discussions and joint decision-making. These data propose early consideration for HCT in selected MF patients beginning with Int-1 risk identification and therefore may influence clinical practice.
Although long-term survival was improved with HCT for Int-1 risk and higher disease, this was at the cost of early TRM. The most common causes of nonrelapse early (< 12 months) TRM were GVHD (23.8%), organ failure (15.7%), and infection (14.1%). Although great progress in management of HCT-related morbidity has occurred in recent years, continued advancement of GVHD, infectious, and supportive care therapy is still needed.47
The strength of our study is in the use of a Cox survival analysis that allows for adjustment of covariates that likely govern the decision to proceed to HCT (eg, age, KPS, and DIPSS score). We determined that the ideal time to start the clock is at the time of DIPSS assessment. This was at time of transplant for the HCT group and at time of initiation of medical treatment or referral to academic medical center (if time of initiation of medical treatment is unknown) for the non-HCT arm. With this start time, the question the current study addresses is which intervention (HCT or non-HCT) provides superior OS for a patient with a particular DIPSS risk score that was able to live long enough (and have sufficient resources, etc.) to present to the referral center/HCT center. As such, the results presented here are most clinically applicable to those presenting to a referral center or HCT center and may not be applicable to other settings.
The impact of HCT on survival in patients <65 years with primary MF has previously been evaluated in 438 patients, revealing that the HR of death after HCT vs those treated with non-transplant modalities in low-risk DIPSS was 5.6 (P = .0051); in Int-1 risk, it was 1.6 (P = .19); for Int-2 risk, it was 0.55 (P = .005); and for high-risk, it was 0.37 (P = .0007).10 In this analysis, the authors conclude that patients with Int-2 or high-risk MF clearly benefit from HCT, while those at low risk should receive nontransplant therapy. Individual decision-making was recommended for Int-1–risk disease. In contrast, the current analysis reveals survival benefit for those with Int-1 disease and higher, supporting a stronger recommendation for HCT in this population. While many similarities exist with aforementioned study and the current analysis, the differences may explain the discordant results with respect to the survival benefit observed in the Int-1 patient population. The main differences are that the current analysis includes a much larger sample size (1928 vs 438 patients), all types of MF (including secondary), older patients (<70 vs <65 years), and patients with prior exposure to ruxolitinib (≤30%).
Several factors should be considered when interpreting the results of this analysis. The reference time point of HCT or referral to academic center/start of medical therapy was estimated as the most clinically relevant time point; however, it was recognized a lead time bias was possible with this assumption. To investigate whether lead time bias affected the main model (given the fact that HCT patients were guaranteed survival up to the point in which they underwent transplant), a sensitivity analysis was performed restricting the time between diagnosis and transplant to 12 months for the HCT cohort revealing no significant change in the survival analysis. Thus, lead time bias is estimated to have had little effect on the survival analysis.
Another important consideration is the introduction of JAK inhibitor therapy, which became commercially available in the United States in 2012. We reviewed the impact of ruxolitinib with a multivariate analysis, which revealed decreased risk of death with ruxolitinib therapy (HR, 0.53; 95% CI, 0.44-0.63; P < .0001), consistent with long-term follow up results of the COMFORT analyses.7 However, the impact of HCT vs non HCT on survival was consistent whether patients received ruxolitinib or not (P value for interaction = .018).
Another consideration in the HCT group is the donor source. In our study, 15% of HCT patients received partially mismatched unrelated donor transplants. We performed a sensitivity analysis after excluding the partially matched donors. No major changes were noted in the results, and thus, partially mismatched unrelated donor transplants were included in the analysis (see supplemental Table 3). Cord blood and haploidentical transplants were not included in this analysis due to very low numbers and different outcomes.
Additional HCT factors must also be considered when interpreting the results of the current analyses. Prior studies have suggested the use of MAC rather than reduced intensity (RIC) were associated with higher mortality rates however, optimal conditioning for MF HCT is controversial.32,33,36,39,48,49 In this analysis, MAC was used in 50% of transplant while RIC was used in 41% of patients undergoing HCT. The impact of conditioning regimens was examined and was not found to be significant (P = .142). However, the patient populations were different, and the impact of conditioning on survival was not the purpose of this study.
The significant differences between the clinical-pathologic characteristics between patients in HCT vs non-HCT cohorts must also be considered. The median age of the transplant cohort was younger (55 years) compared with the nontransplant cohort (61 years). There were more white patients in the HCT cohort than the non-HCT cohort; this difference may be partially explained by the exclusion of cord and haploidentical transplant, as ethnic minorities remain underrepresented in donor registries. There was a higher proportion of secondary MF patients in the non-HCT group (35%) vs those receiving transplant (16%); however, the impact of primary vs secondary MF on survival is unknown.50 Unfavorable cytogenetics in the non-HCT group were 16% vs 14% in the HCT group. Therefore, it is recognized the groups are different and survival analysis is potentially impacted. However, we controlled for these observed imbalances in the Cox regression analysis.
Several limitations of the current study are recognized. The current study and all retrospective analyses are burdened with the inherent potential for bias and data-quality issues. A major limitation in this study is lack of clear explanation of why patients were offered non-HCT treatment modalities, including no data regarding the patient’s comorbidities, raising concern of comparison between HCT and non-HCT groups in the absence of this data. DIPSS score was missing in 21% of non-HCT and 6% HCT; thus, more imputation was necessary in the non-HCT arm. The current study is also limited in molecular analysis, a major handicap in the modern era. It is now recognized that the application of molecular and genetic data to prognostic stratification models represents the future of personalized therapy and transplant decision-making for MF.18,19,21 Additionally, there was lack of comparative quality of life data, an important element in transplant consideration. The impact of splenomegaly, splenectomy, or splenic radiation was also not examined in this analysis. The effect of splenomegaly on transplant outcomes is debated, with some studies revealing a negative effect,37,51,52 others finding no effect,13 and others with even a threefold increase in relapse rate after HCT.36 Additionally, the applicability of DIPSS prognostic score, which was developed originally in cohorts of patients with primary MF, is unknown in secondary MF and requires further elucidation.50 New prognostic schemas specifically in secondary MF patients are being described.15,20 The retrospective nature of this study is another limitation, but a randomized study comparing HCT to non-HCT therapies is not likely to be performed in the United States. Thus, this retrospective survival analysis is important for the practicing clinician and may be useful to guide clinical practice.
In conclusion, this multicenter study evaluating survival of 1928 MF patients with HCT vs non-HCT over a median of 6 years follow-up is one of the largest and most inclusive survival analyses performed in MF. Our study suggests consideration of HCT in the setting of DIPSS Int-1–risk MF, supports the accepted recommendation for HCT in DIPSS Int-2/high-risk MF, and highlights the need for improved supportive care strategies in MF in the early post-HCT period.
CIBMTR supports accessibility of research in accord with the NIH Data Sharing Policy and the NCI Cancer Moonshot Public Access and Data Sharing Policy. CIBMTR, in accordance with relevant US and international regulations regarding privacy and confidentiality, releases on its public Web site deidentified analysis data sets and corresponding data dictionaries upon publication without any embargo or no later than after the journal embargo policy of the accepted publication. To adhere to the European Union’s General Data Protection Regulation requirements, CIBMTR does not externally release any European patient-level data. CIBMTR data submitted through its standard reporting structure will be released. Supplemental data may or may not be released depending on the funds used to acquire the data and the contractual agreements in place for those funds and data.
Acknowledgments
The authors thank the CIBMTR, the scientific writing committee, and all data contribution centers for contribution to this paper.
The CIBMTR is supported primarily by Public Health Service grant/cooperative agreement U24CA076518 with the National Institutes of Health (NIH), National Cancer Institute (NCI); National Heart, Lung, and Blood Institute (NHLBI); and National Institute of Allergy and Infectious Diseases (NHLBI and NCI grant/cooperative agreement U24HL138660; NHLBI grants R21HL140314 and U01HL128568; Health Resources and Services Administration [HRSA] contract HHSH250201700006C; Office of Naval Research grants N00014-18-1-2888 and N00014-17-1-2850; HRSA subaward from prime contract award SC1MC31881-01-00; NHLBI subawards from prime grant awards R01HL131731 and R01HL126589; NIH subawards from prime grant awards 5P01CA111412, 5R01HL129472, R01CA152108, 1R01HL131731, 1U01AI126612, and 1R01CA231141) and commercial funds from Actinium Pharmaceuticals, Adaptive Biotechnologies, Allovir, Amgen, and anonymous donation to the Medical College of Wisconsin, Anthem, Astellas Pharma US, Atara Biotherapeutics, BARDA, Be the Match Foundation, bluebird bio, Boston Children’s Hospital, Bristol Myers Squibb, Celgene, Children’s Hospital of Los Angeles, Chimerix, City of Hope Medical Center, CSL Behring, CytoSen Therapeutics, Inc., Daiichi Sankyo, Dana Farber Cancer Institute, Enterprise Science and Computing, Fred Hutchinson Cancer Research Center, Gamida-Cell, Genzyme, Gilead Sciences, GlaxoSmithKline, HistoGenetics, Immucor, Incyte Corporation, Janssen Biotech, Janssen Pharmaceuticals, Janssen Research & Development, Janssen Scientific Affairs, Japan Hematopoietic Cell Transplantation Data Center, Jazz Pharmaceuticals, Karius, Karyopharm Therapeutics, Kite (a Gilead Company), Kyowa Kirin, Magenta Therapeutics, Mayo Clinic and Foundation Rochester, Medac GmbH, Mediware, Memorial Sloan Kettering Cancer Center, Merck & Company, Mesoblast, MesoScale Diagnostics, Millennium, the Takeda Oncology Co., Miltenyi Biotec, Mundipharma EDO, National Marrow Donor Program, Novartis Oncology, Novartis Pharmaceuticals Corporation, Omeros Corporation, Oncoimmune, OptumHealth, Orca Biosystems, PCORI, Pfizer, Phamacyclics, PIRCHE AG, Regeneron Pharmaceuticals, REGiMMUNE, Sanofi Genzyme, Seattle Genetics, Shire, Sobi, Spectrum Pharmaceuticals, St. Baldrick’s Foundation, Swedish Orphan Biovitrum, Takeda Oncology, The Medical College of Wisconsin, University of Minnesota, University of Pittsburgh, University of Texas MD Anderson Cancer Center, University of Wisconsin-Madison, Viracor Eurofins, and Xenikos BV.
The views expressed in this article do not reflect the official policy or position of the National Institutes of Health, the Department of the Navy, the Department of Defense, HRSA, or any other agency of the US Government.
Authorship
Contribution: K.G. developed the first draft and edited and wrote the manuscript; K.B., W.S., K.G., and R.M. designed the study, analyzed data, and participated in writing the manuscript; Z.-H.H. and K.W.A. participated in data interpretation and writing the manuscript; and the remaining authors participated with writing and editing the manuscript.
Conflict-of-interest disclosure: A.T.K. received research funding from Incyte, AbbVie, Janssen, and Colgene. A.T.G. received research funding from Celgene, Incyte, CTI Biopharma, and Pfizer. G.H. received research funding from Bayer, Meirck, Incyte, and Celgene. R.F.O. received research funding from AstraZeneca. Z.D. received research funding from Incyte Corp. M.O.A. received research funding from Incyte, Gilead, Samus Therapeautics, CTI Biopharma, and Janssen. B.R.A. contributed to Best Practice guidelines for the British Med J and received research funding from Juno. V.G. received research funding from Novartis, Celgene, Sierra Oncology, and Incyte. L.C.M. received research funding from ASTEX, Bioline, BMS, Celgene, Janssen, Jazz, Macrogenics, Millennium, Novartis, Pfizer, and TG Therapeutics. J.C. received research funding from Inctye, Daichi-Sankyo, and Jaze Pharmaceuticals. K.G. received consultancy fees from and is a member of the scientific advisory board for Incyte Corporation. R.N. received research funding from Alexion, Celgene, Kirin Kyowa, and Merck. M.T. received research funding from Celgene, CTI Biopharma, Imago, and Constellation. S.V. received research funding from Incyte Corporation, Roche, NS Pharma, Celgene, Gilead, Promisor, CTI BioPharma, Genentech, Blueprint Medicines, Novartis, Sierra Oncology, Pharma Essentia, AstraZeneca, Ital Pharma, and Protagonist Therapeutics. M.G. received consultancy fees from Incyte, Amgen, AbbVie, Astellas, BMS/Celgene, Merck, Pfizer, Premier, Trovagene, Daiichi Sankyo, and Cardinal Health and research funding from Incyte, Amgen, Novartis, Janssen, Genentech/Roche, and Forma Therapeutics. T.J. received consultancy fees from Takeda Oncology and is a member of the CareDx advisory board. The remaining authors declare no competing financial interests.
Correspondence: Krisstina Gowin, Division of Hematology and Oncology, University of Arizona, 3838 N Campbell Ave, Tucson, AZ 85719; e-mail: gowink@email.arizona.edu.

