

Key Points
Pexidartinib is safe and well tolerated in relapsed/refractory AML patients.
Pexidartinib, a type 2 inhibitor of FLT3, including F691 gatekeeper mutations, has clinical activity in FLT3-ITD–mutant AML patients.

Abstract
FMS-like tyrosine kinase 3 (FLT3) tyrosine kinase inhibitors (TKIs) have activity in acute myeloid leukemia (AML) patients with FLT3 internal tandem duplication (ITD) mutations, but efficacy is limited by resistance-conferring kinase domain mutations. This phase 1/2 study evaluated the safety, tolerability, and efficacy of the oral FLT3 inhibitor PLX3397 (pexidartinib), which has activity against the FLT3 TKI–resistant F691L gatekeeper mutation in relapsed/refractory FLT3-ITD–mutant AML. Ninety patients were treated: 34 in dose escalation (part 1) and 56 in dose expansion (part 2). Doses of 800 to 5000 mg per day in divided doses were tested. No maximally tolerated dose was reached. Plasma inhibitory assay demonstrated that patients dosed with ≥3000 mg had sufficient levels of active drug in their trough plasma samples to achieve 95% inhibition of FLT3 phosphorylation in an FLT3-ITD AML cell line. Based on a plateau in drug exposure, the 3000-mg dose was chosen as the recommended phase 2 dose. The most frequently reported treatment-emergent adverse events were diarrhea (50%), fatigue (47%), and nausea (46%). Based on modified response criteria, the overall response rate to pexidartinib among all patients was 21%. Twenty-three percent of patients treated at ≥2000 mg responded. The overall composite complete response rate for the study was 11%. Six patients were successfully bridged to transplantation. Median overall survival (OS) of patients treated in dose expansion was 112 days (90% confidence interval [CI], 77-150 days), and median OS of responders with complete remission with or without recovery of blood counts was 265 days (90% CI, 170-422 days). This trial was registered at www.clinicaltrials.gov as #NCT01349049.
Introduction
Mutations in the FMS-like tyrosine kinase 3 (FLT3) gene are the most frequently found mutations in acute myeloid leukemia (AML).1,2 Constitutively activating internal tandem duplication (ITD) mutations are the most commonly identified mutations in FLT3. ITD mutations occur in ∼20% to 25% of AMLs and confer poor prognosis.3,4 Another 5% to 10% of AMLs are associated with point mutations in the FLT3 tyrosine kinase domain (KD), particularly at the activation loop residue D835.5,6 In recent years, a variety of tyrosine kinase inhibitors (TKIs) of FLT3 have entered clinical development in AML, with variable success. Midostaurin, a multitargeted inhibitor, demonstrated little activity as monotherapy7 but significantly prolonged survival compared with placebo when added to induction chemotherapy in newly diagnosed patients with FLT3 mutations.8 This clinical activity led to US Food and Drug Administration (FDA) approval of midostaurin in newly diagnosed FLT3-mutant AML. More potent and selective next-generation FLT3 inhibitors such as quizartinib9,10 and gilteritinib11 have entered clinical development in AML and, in contrast to midostaurin, have demonstrated high response rates as monotherapy in relapsed/refractory (R/R) patients with FLT3 mutations. Both quizartinib12 and gilteritinib13 have also demonstrated a survival benefit when compared with salvage chemotherapy in R/R FLT3-mutant AML patients, leading to the recent FDA approval of gilteritinib for this indication. Despite this, survival in this R/R group of patients remains limited by the development of therapeutic resistance.
Despite leading to composite complete remission (CRc) rates of 40% to 50%9,10 in R/R FLT3-ITD+ AML, quizartinib is highly vulnerable to acquired resistance-conferring mutations in the FLT3 KD, which occur at the activation loop residue D835 and the kinase gatekeeper residue F691.14 Mutations at these residues have also been implicated in resistance to other FLT3-targeted TKIs, including sorafenib15,16 and sunitinib.15 Type 1 FLT3 inhibitors, such crenolanib and gilteritinib, which bind to the FLT3 active kinase conformation, have been developed to combat quizartinib resistance–conferring D835 mutations and have demonstrated clinical activity against cases with D835 mutations, with and without cooccurring FLT3-ITD mutations.11,17 However, both crenolanib18,19 and gilteritinib20,21 are vulnerable to resistance induced by F691 mutations. Patients treated with either crenolanib or gilteritinib therapy have developed F691L mutations as a cause of clinical resistance.19,21 At this time, limited treatment options exist for patients with FLT3 TKI resistance resulting from F691 mutations. The ABL/FLT3 inhibitor ponatinib, which was rationally designed to inhibit the BCR-ABL gatekeeper mutation T315I, has demonstrated equivalent in vitro activity against the analogous FLT3 F691I mutation but has shown decreased efficacy against the more clinically relevant F691L substitution.22 Ponatinib did induce bone marrow responses in 3 of 12 R/R AML patients in a phase 1 clinical trial23 ; however, no responding patients were known to express FLT3 F691 mutations, and the activity of ponatinib in FLT3-ITD–mutant AML, with or without F691 mutations, has not been confirmed in larger clinical trial experience. Cabozantinib, a multikinase inhibitor of FLT3, MET, AXL, KIT, and VEGFR, has shown in vitro activity against FLT3 F691 mutations, but no bone marrow responses were observed in a dose-escalation clinical trial in AML.24 The common development of resistance-causing FLT3 KD mutations in patients who respond to and relapse on FLT3-targeted therapy underscores the central importance of FLT3 signaling in these patients. However, even with the development of FLT3 TKIs with expanded activity against FLT3 KD mutations, there continues to be no established therapeutic option for patients in whom FLT3 TKI therapy fails because of F691 mutations.
Pexidartinib (formerly PLX3397) is a selective small-molecule kinase inhibitor of CSF1R, KIT, and FLT3-ITD, with a novel chemical structure comprising a pyridine (rather than phenyl) middle ring and a methyl amine (rather than amide or urea) linker, designed to retain binding to FLT3 with a F691L mutation. Based on its activity against CSF1R, pexidartinib was recently approved for the treatment of advanced tenosynovial giant cell tumor, a neoplasm the growth of which is related to CSF1R. In preclinical studies, pexidartinib has demonstrated equipotent activity in cells expressing FLT3-ITD/F691L mutations compared with cells expressing FLT3-ITD alone, although it remains vulnerable to FLT3 D835 mutations.25 To assess the safety and efficacy of pexidartinib in R/R FLT3-ITD–mutant AML, we conducted a phase 1/2 open-label, sequential dose-escalation study followed by a cohort expansion at the recommend phase 2 dose (RP2D) of continuous oral administration of pexidartinib in 28-day cycles.
Patients and methods
Study design
This open-label phase 1/2 study evaluated the safety and tolerability of continuous oral administration of pexidartinib in R/R AML patients with FLT3-ITD mutations. The study consisted of a dose-escalation phase to determine the maximum tolerated dose (MTD)/RP2D, followed by a dose-expansion cohort treated at the RP2D. Patients received pexidartinib orally twice daily in a capsule formulation (100 or 200 mg per capsule) on days 1 through 28 of each 28-day cycle, beginning on cycle 1 day 1 (C1D1). Dose escalation followed a standard 3 + 3 dose-escalation design, based on dose-limiting toxicities (DLTs) during the first 15 days of pexidartinib therapy. A DLT was defined as any adverse event (AE) with a Common Terminology Criteria for Adverse Events (version 4) grade ≥3 severity that was not due to AML and occurred within the first 15 days of treatment initiation. No hematologic DLTs were defined for this protocol. The following doses were administered in part 1 of the study: 800, 1000, 1200, 1400, 2000, 3000, 4000, and 5000 mg per day in split doses. The RP2D was the MTD or the dose level that achieved maximal plasma concentration if an MTD was not reached. Each patient was offered continued dosing with pexidartinib as long as it was well tolerated and the patient was clinically benefitting from treatment. Patients in part 1 and part 2 who were tolerating the study drug but who had evidence of tumor progression at C1D15 remained on study treatment until the C2D1 assessment. Patients continued study drug until unacceptable toxicity or DLT, disease progression or relapse, patient death, or patient decision. Patients who discontinued for reasons other than withdrawal of consent were followed up every 3 months (for up to 2 years) for disease, treatment, and survival status. For part 1, hydroxyurea was permitted at any time, at a dose of ≤5 g per day. For part 2, hydroxyurea was permitted only during the first 2 weeks of treatment, at a dose of ≤5 g per day. For patients in part 1, dose reductions and interruptions were permitted during the first 15 days of cycle 1 only if a patient experienced a DLT. If a patient experienced a DLT, treatment continuation at a lower dose was permitted at the discretion of the investigator and in consultation with the medical monitor. After C1D15, dose reductions or interruptions for AEs could take place at any time. Reduction/interruption of dosing for AEs could take place at any time in part 2.
The study was conducted in compliance with the protocol, Good Clinical Practice, and the applicable regulatory requirements (including International Conference on Harmonisation guidelines) and in accordance with ethical principles founded in the Declaration of Helsinki. All participants gave written informed consent. All authors had access to the data on request.
Study objectives
The primary objective in part 1 was to determine the safety and tolerability of escalating doses of pexidartinib and establish an RP2D in patients with relapsed or refractory FLT3-ITD+ AML. The primary objective in part 2 was to determine the overall response rate (ORR) of pexidartinib at the RP2D.
Study population
Eligibility required a diagnosis of primary AML or secondary AML as per the World Health Organization criteria. Marrow involvement by leukemia was required at screening for enrollment in part 2. Patients were required either to be refractory to initial induction attempt (up to 2 cycles of standard cytotoxic chemotherapy) or to have relapsed after prior CR. The presence of an FLT3-ITD mutation was required from blood or marrow testing at screening using the FLT3-ITD/wild type allelic ratio cutoff defined by local laboratory testing. Patients with history of FLT3-ITD+ leukemia who did not have demonstrable FLT3-ITD at screening could be enrolled after discussion with the study medical monitor. Prior FLT3 inhibitor therapy was allowed, but those with a history of FLT3-TKD mutations at D835 (or for part 2, their presence at screening) were excluded from participation. Patients were required to have resolution of toxicities from prior therapy to grade ≤1, adequate hepatic and renal function at baseline, and Eastern Cooperative Oncology Group performance status of 0 to 2. Patients with prior hematopoietic stem cell transplantation (HSCT) were eligible as long as they were >60 days from transplantation and did not have clinically significant graft-versus-host disease at screening (part 1 only). Patients with diagnosis of acute promyelocytic leukemia, chronic myeloid leukemia, central nervous system leukemia, or active malignancy; with prolonged corrected QT interval by Fredericia (>450 ms for men; >470 ms for women); or who had received treatment with any investigational drug within 28 days were excluded. Patients with refractory nausea/vomiting, gastrointestinal malabsorption, or other serious concurrent uncontrolled illness were also excluded.
Response assessment
Response to treatment was evaluated using a modified version of the International Working Group response criteria.26,27 Responses were defined per standard criteria except that participants achieving a CR with incomplete hematologic recovery (CRi) or a partial response (PR) were not required to be transfusion independent26 (data supplement provides detailed response criteria). The primary efficacy end point was the rate of CRc, which included CR, CRi, and CR with incomplete platelet recovery (CRp) based upon modified International Working Group response criteria, plus the percentage of patients who were successfully bridged to transplantation, defined as any patient who discontinued pexidartinib treatment specifically for the purpose of undergoing HSCT and who subsequently received the planned transplant. Secondary end points included PR, duration of remission, overall survival (OS), and progression-free survival (PFS). PFS was defined as the number of days from the start of therapy to the date of documented disease progression/relapse or death, whichever occurred first.
Results
Patient characteristics and disposition
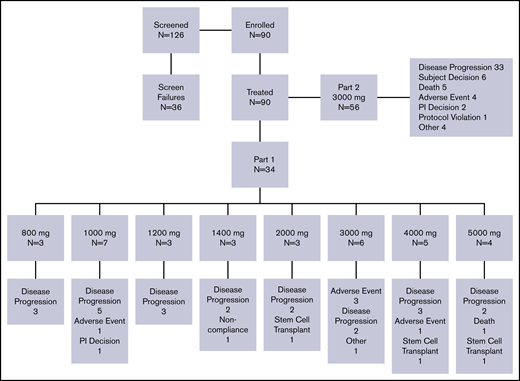
A total of 90 patients with R/R FLT3-ITD+ AML were enrolled in the study (part 1, n = 34; part 2, n = 56) and received at least 1 dose of pexidartinib. Patients were enrolled at 8 sites in the United States. The first patient was enrolled on 21 November 2011, and the data cutoff date was 20 January 2015. Demographics and baseline disease characteristics are shown in Table 1. Median age of patients was 62.5 years (range, 24-82 years) in part 1 and 59.5 years (range, 22-83 years) in part 2. A lower percentage of male patients (38%) were enrolled in part 1, but a higher percentage of men were enrolled in part 2 (59%). Of all 90 patients, 70 had de novo AML and 12 had AML secondary to an antecedent hematologic disorder. In 8 patients, it was not known if AML was de novo or secondary. At screening, 89 (99%) of the enrolled patients were FLT3-ITD mutation positive, and 1 patient with a history of FLT3-ITD was mutation negative. A total of 28 patients (31%) had undergone prior allogeneic transplantation. Median number of prior AML therapies was 4 (range, 1-12; supplemental Figure 1); 27 patients (30%) had received prior FLT3 inhibitor therapy, most commonly sorafenib (n = 23; 26%) and/or quizartinib (n = 5; 6%), and 1 patient had participated in a randomized clinical trial of PKC412; a majority of patients had received 1 prior FLT3 inhibitor, but 2 (7.4%) of 27 patients had received both quizartinib and sorafenib. Patient disposition is shown in Figure 1.
Summary of demographics and AML history
| Characteristic | Part 1 (n = 34) | Part 2 (n = 56) |
|---|---|---|
| Age, y | ||
| Mean | 60.6 | 55.9 |
| Standard deviation | 14.6 | 14.6 |
| Minimum-maximum | 24-82 | 22-83 |
| Sex | ||
| Male | 13 (38) | 33 (59) |
| Female | 21 (62) | 23 (41) |
| Race | ||
| American Indian or Alaskan Native | 1 (3) | 0 |
| Asian | 1 (3) | 3 (5) |
| Black or African American | 7 (21) | 7 (13) |
| White | 21 (62) | 35 (63) |
| Other | 4 (12) | 11 (20) |
| Ethnicity | ||
| Hispanic or Latino | 1 (3) | 5 (9) |
| Not Hispanic or Latino | 33 (97) | 51 (91) |
| ECOG performance status | ||
| Fully active (0) | 6 (18) | 9 (16) |
| Restricted (1) | 17 (50) | 33 (59) |
| Ambulatory (2) | 11 (32) | 14 (25) |
| AML category at diagnosis | ||
| De novo | 26 (76) | 44 (79) |
| Secondary to MDS | 4 (12) | 2 (4) |
| Secondary to chemotherapy for another cancer | 2 (6) | 4 (7) |
| Unknown | 2 (6) | 6 (11) |
| FLT3-ITD+at diagnosis | ||
| Yes | 30 (88) | 45 (80) |
| No | 2 (6) | 4 (7) |
| Unknown | 2 (6) | 7 (13) |
| Prior FLT3 inhibitor therapy | 14 (41) | 12 (21) |
| Prior allogeneic transplantation | ||
| Yes | 7 (21) | 21 (38) |
| No | 27 (79) | 34 (61) |
| Unknown | 0 | 1 (2) |
| No. of prior AML therapies | ||
| Median | 4 | 4 |
| Range | 2-12 | 1-10 |
| Characteristic | Part 1 (n = 34) | Part 2 (n = 56) |
|---|---|---|
| Age, y | ||
| Mean | 60.6 | 55.9 |
| Standard deviation | 14.6 | 14.6 |
| Minimum-maximum | 24-82 | 22-83 |
| Sex | ||
| Male | 13 (38) | 33 (59) |
| Female | 21 (62) | 23 (41) |
| Race | ||
| American Indian or Alaskan Native | 1 (3) | 0 |
| Asian | 1 (3) | 3 (5) |
| Black or African American | 7 (21) | 7 (13) |
| White | 21 (62) | 35 (63) |
| Other | 4 (12) | 11 (20) |
| Ethnicity | ||
| Hispanic or Latino | 1 (3) | 5 (9) |
| Not Hispanic or Latino | 33 (97) | 51 (91) |
| ECOG performance status | ||
| Fully active (0) | 6 (18) | 9 (16) |
| Restricted (1) | 17 (50) | 33 (59) |
| Ambulatory (2) | 11 (32) | 14 (25) |
| AML category at diagnosis | ||
| De novo | 26 (76) | 44 (79) |
| Secondary to MDS | 4 (12) | 2 (4) |
| Secondary to chemotherapy for another cancer | 2 (6) | 4 (7) |
| Unknown | 2 (6) | 6 (11) |
| FLT3-ITD+at diagnosis | ||
| Yes | 30 (88) | 45 (80) |
| No | 2 (6) | 4 (7) |
| Unknown | 2 (6) | 7 (13) |
| Prior FLT3 inhibitor therapy | 14 (41) | 12 (21) |
| Prior allogeneic transplantation | ||
| Yes | 7 (21) | 21 (38) |
| No | 27 (79) | 34 (61) |
| Unknown | 0 | 1 (2) |
| No. of prior AML therapies | ||
| Median | 4 | 4 |
| Range | 2-12 | 1-10 |
Values are n (%) unless otherwise noted.
ECOG, Eastern Cooperative Oncology Group; MDS, myelodysplastic syndrome.
Safety
In part 1 dose escalation, pexidartinib was investigated at 8 dose levels (800-5000 mg). One patient in the 1000-mg dose group experienced grade 3 bilirubin elevation, which was a DLT but considered unrelated to study treatment. Although this AE was considered unrelated to pexidartinib, additional patients were enrolled in the 1000-mg dose group (n = 7). Patients in the next 2 dose cohorts, 1200 (n = 3) and 1400 mg (n = 3), completed the 15-day observation period without the occurrence of a DLT, and the dose was escalated to 2000 mg. One patient in the 2000-mg dose group experienced a DLT of atrial fibrillation, considered unrelated to study medication. The DLT was not discovered until later medical review of the data, and the 2000-mg dose cohort was not expanded. The dose was escalated 3 additional times, to 3000, 4000, and 5000 mg. No protocol-defined DLTs were reported. Evaluation of available pharmacokinetic (PK) data indicated that pexidartinib plasma exposure levels peaked at ∼3000 mg per day and that higher dose levels did not provide higher plasma levels. After extensive discussion, all investigators and the sponsor agreed that pexidartinib at 3000 mg per day was the appropriate R2PD. This dose was used in all 56 patients enrolled in part 2 dose expansion.
A summary of AEs is shown in supplemental Table 1. All patients (n = 90; 100%) who received study medication during this study experienced at least 1 AE. The most frequent treatment-emergent AEs (TEAEs) of pexidartinib are summarized in Table 2. A total of 81 patients (90%) experienced an AE that was Common Terminology Criteria for Adverse Events grade ≥3 (supplemental Table 2). A summary of all TEAEs occurring in the first 30 days of treatment is shown in supplemental Table 3. The most frequently reported TEAE was diarrhea (50% of all patients), followed by fatigue (47%), nausea (46%), and febrile neutropenia (42%). The most frequently reported TEAE that led to changes in study medication administration was febrile neutropenia (12%), followed by sepsis (6%) and aspartate aminotransferase increase (6%). Of note, 4 patients, all treated at 3000 mg, experienced a change in hair color, an associated AE of potent inhibition of c-KIT,28 a known target of pexidartinib. The most frequent grade ≥3 TEAE was febrile neutropenia (42%). Twelve patients (13%) experienced fatal AEs during this study: 3 during part 1 and 9 during part 2. Causes of death were sepsis (n = 5), pneumonia (n = 2), pneumonia aspiration (n = 1), respiratory failure (n = 1), cardiac arrest (n = 1), cytokine release syndrome (differentiation syndrome; n = 1), and cerebral hemorrhage (n = 1). In 11 of 12 patients, study drug had been discontinued before death. All AEs that resulted in death were considered unrelated to study medication, with the exception of 1 patient in part 2 who experienced grade 5 cytokine release syndrome as a result of treatment-induced differentiation syndrome, a known potential complication of FLT3 inhibitor response.29
Summary of all TEAEs that occurred in ≥20% of patients
| Preferred term | Grade 1-2 | Grade ≥3 | All grades |
|---|---|---|---|
| Diarrhea | 44 (49) | 1 (1) | 45 (50) |
| Fatigue | 33 (37) | 9 (10) | 42 (47) |
| Nausea | 40 (44) | 1 (1) | 41 (46) |
| Febrile neutropenia | 0 (0) | 38 (42) | 38 (42) |
| Decreased appetite | 31 (34) | 2 (2) | 33 (37) |
| Vomiting | 32 (36) | 1 (1) | 33 (37) |
| Cough | 26 (29) | 0 (0) | 26 (29) |
| Anemia | 9 (10) | 15 (17) | 24 (27) |
| Hypokalemia | 16 (18) | 6 (7) | 22 (24) |
| Aspartate aminotransferase increased | 14 (16) | 5 (6) | 19 (21) |
| Dyspnea | 15 (17) | 3 (3) | 18 (20) |
| Preferred term | Grade 1-2 | Grade ≥3 | All grades |
|---|---|---|---|
| Diarrhea | 44 (49) | 1 (1) | 45 (50) |
| Fatigue | 33 (37) | 9 (10) | 42 (47) |
| Nausea | 40 (44) | 1 (1) | 41 (46) |
| Febrile neutropenia | 0 (0) | 38 (42) | 38 (42) |
| Decreased appetite | 31 (34) | 2 (2) | 33 (37) |
| Vomiting | 32 (36) | 1 (1) | 33 (37) |
| Cough | 26 (29) | 0 (0) | 26 (29) |
| Anemia | 9 (10) | 15 (17) | 24 (27) |
| Hypokalemia | 16 (18) | 6 (7) | 22 (24) |
| Aspartate aminotransferase increased | 14 (16) | 5 (6) | 19 (21) |
| Dyspnea | 15 (17) | 3 (3) | 18 (20) |
Values are n (%). TEAE defined as AE with start date on or after C1D1.
Patients were withdrawn from study because of disease progression (n = 55), AE (n = 9), death (n = 6), patient decision (n = 6), other (n = 5), investigator decision (n = 3), elective HSCT (n = 3), protocol violation (n = 1), and noncompliance (n = 1). There were no apparent trends in changes in chemistry or hematology parameters throughout the study. No clinically significant electrocardiogram abnormalities were observed at the end of treatment assessment. Echocardiogram/multigated acquisition scan evaluation of ejection fraction (EF) was not originally included in the study protocol but was added because of grade 3 decreased EF noted in 1 patient with unknown baseline EF treated at 5000 mg. Echocardiogram/multigated acquisition assessment was performed for 3 patients in part 1 and for 19 patients in part 2. Two of these patients in part 2 had >10% decrease in EF (grade 2, n = 1; grade 3, n = 1). Assessment of lung diffusion capacity and lung computed tomography scan were also added later in the study because of 2 early reports of pneumonitis (grades 1 and 2) and 1 report of grade 3 dyspnea with bilateral opacities on chest radiograph thought to be possibly drug related. Results of the diffusion capacity assessment demonstrated impaired lung function in some patients but did not change with drug treatment. Clinically significant abnormal computed tomography scans were noted in 9 patients; in 6 of these 9 patients, abnormalities were considered to have resulted from infection; in 3 of 9, no potential etiology was given for diffuse ground glass opacities (n = 1), small pulmonary nodules (n = 1), or pleural effusion (n = 1).
PKs and pharmacodynamics
The PK population of study PLX108-05 consisted of 90 patients in part 1 and part 2. The C1D1 and C1D15 PK parameters are summarized in Table 3. The C1D15 exposures, as assessed by maximum serum concentration, AUC0-4, and AUC0-6, were more than twofold higher than C1D1 exposures at all 8 dose levels (Table 3), indicating significant accumulation at steady state. Nearly dose-proportional increase in steady-state (C1D15) exposure was observed from the 800- to 3000-mg daily dose (AUC0-6, 20 147-58 329 ng × h/mL) in part 1 patients (Table 3). When the relationship between dose and steady-state (C1D15) exposure was investigated using the power model, log (AUC0-6) = a + (b × log [dose]), the intercept a and slope b were determined to be 1.96 and 0.8146, respectively, with an R2 of 0.7665 (Figure 2A). Further increase in exposure was not observed at doses >3000 mg. At steady state, the geometric mean AUC0-6 values at 3000-, 4000-, and 5000-mg doses (n = 4 for each dose) were 58 329, 51 415, and 56 460 ng × h/mL, respectively (Table 3), with significantly overlapping 90% confidence intervals (CIs; 34 256-99 317, 30 300-87 245, and 34 318-92 888 ng × h/mL, respectively). The relatively small sample size used in this study, however, did not allow a definitive conclusion on the plateauing of PKs at 3000 mg. It is possible that AUCs might be higher with 4000- and 5000-mg doses. However, PK improvement, if any, from a dose >3000 mg is likely to be offset by the challenges of greater pill burden. The 3000-mg dose was therefore chosen as the RP2D and administered in part 2 of the study. The geometric mean steady-state exposure (AUC0-6, 62 451 ng × h/mL; 90% CI, 58 280-66 919 ng × h/mL) in part 2 patients (n = 47) was similar to the mean exposure in part 1 patients at the RP2D (AUC0-6, 58 329 ng × h/mL; Table 3). Note that this study was not powered to determine the effects of dose-independent factors (eg, food, hepatic or renal impairment, drug-drug interactions) on the exposure of pexidartinib.
Mean plasma PK parameters of pexidartinib in AML patients after oral administration for 1 day (C1D1) and 15 days (C1D15)
| Daily dose, mg (cohort) | C1D1 | C1D15 | ||||
|---|---|---|---|---|---|---|
| Cmax, ng/mL | AUC0-4, ng × h/mL | AUC0-6, ng × h/mL | Cmax, ng/mL | AUC0-4, ng × h/mL | AUC0-6, ng × h/mL | |
| 800 (part 1, 1) | ||||||
| n | 3 | 3 | 3 | 3 | 3 | 3 |
| G-mean | 1148 | 3300 | 5184 | 4134 | 12 340 | 20 147 |
| CV, % | 133 | 121 | 111 | 47.4 | 78.9 | 64.2 |
| 1000 (part 1, 2) | ||||||
| n | 6 | 6 | 6 | 5 | 5 | 5 |
| G-mean | 2163 | 4566 | 8008 | 7294 | 22 949 | 34 013 |
| CV, % | 44.6 | 63.8 | 48.7 | 25.7 | 31.6 | 37.2 |
| 1200 (part 1, 3) | ||||||
| n | 3 | 3 | 3 | 3 | 3 | 3 |
| G-mean | 2016 | 5392 | 8171 | 4291 | 14 870 | 21 595 |
| CV, % | 92.5 | 92.2 | 93.7 | 110 | 103 | 112 |
| 1400 (part 1, 4) | ||||||
| n | 3 | 3 | 3 | 3 | 3 | 2 |
| G-mean | 2534 | 5510 | 8868 | 7593 | 25 121 | 30 857 |
| CV, % | 96.3 | 139 | 101 | 40 | 37.4 | 36.3 |
| 2000 (part 1, 5) | ||||||
| n | 3 | 3 | 3 | 3 | 3 | 3 |
| G-mean | 3493 | 7159 | 12 133 | 10 633 | 37 214 | 54 464 |
| CV, % | 117 | 153 | 97.5 | 61.2 | 53.6 | 48 |
| 3000 (part 1, 6) | ||||||
| n | 6 | 6 | 6 | 5 | 5 | 4 |
| G-mean | 6132 | 16 550 | 24 986 | 12 124 | 38 834 | 58 329 |
| CV, % | 51.6 | 58.6 | 67.5 | 45.1 | 42 | 47.6 |
| 4000 (part 1, 7) | ||||||
| n | 5 | 5 | 5 | 4 | 4 | 4 |
| G-mean | 3669 | 6530 | 12 766 | 10 406 | 34 446 | 51 415 |
| CV, % | 57.6 | 166 | 101 | 45.8 | 54.3 | 47.3 |
| 5000 (part 1, 8) | ||||||
| n | 4 | 4 | 4 | 4 | 4 | 4 |
| G-mean | 4714 | 11 701 | 18 881 | 11 987 | 39 477 | 56 460 |
| CV, % | 35.8 | 27.6 | 35.6 | 35.6 | 43.2 | 44.3 |
| 3000 (part 2) | ||||||
| n | 56 | 56 | 55 | 48 | 48 | 47 |
| G-mean | 4816 | 10 910 | 18 495 | 12 794 | 43 067 | 62 451 |
| CV, % | 66 | 62.6 | 60.9 | 30.8 | 28 | 28.8 |
| Daily dose, mg (cohort) | C1D1 | C1D15 | ||||
|---|---|---|---|---|---|---|
| Cmax, ng/mL | AUC0-4, ng × h/mL | AUC0-6, ng × h/mL | Cmax, ng/mL | AUC0-4, ng × h/mL | AUC0-6, ng × h/mL | |
| 800 (part 1, 1) | ||||||
| n | 3 | 3 | 3 | 3 | 3 | 3 |
| G-mean | 1148 | 3300 | 5184 | 4134 | 12 340 | 20 147 |
| CV, % | 133 | 121 | 111 | 47.4 | 78.9 | 64.2 |
| 1000 (part 1, 2) | ||||||
| n | 6 | 6 | 6 | 5 | 5 | 5 |
| G-mean | 2163 | 4566 | 8008 | 7294 | 22 949 | 34 013 |
| CV, % | 44.6 | 63.8 | 48.7 | 25.7 | 31.6 | 37.2 |
| 1200 (part 1, 3) | ||||||
| n | 3 | 3 | 3 | 3 | 3 | 3 |
| G-mean | 2016 | 5392 | 8171 | 4291 | 14 870 | 21 595 |
| CV, % | 92.5 | 92.2 | 93.7 | 110 | 103 | 112 |
| 1400 (part 1, 4) | ||||||
| n | 3 | 3 | 3 | 3 | 3 | 2 |
| G-mean | 2534 | 5510 | 8868 | 7593 | 25 121 | 30 857 |
| CV, % | 96.3 | 139 | 101 | 40 | 37.4 | 36.3 |
| 2000 (part 1, 5) | ||||||
| n | 3 | 3 | 3 | 3 | 3 | 3 |
| G-mean | 3493 | 7159 | 12 133 | 10 633 | 37 214 | 54 464 |
| CV, % | 117 | 153 | 97.5 | 61.2 | 53.6 | 48 |
| 3000 (part 1, 6) | ||||||
| n | 6 | 6 | 6 | 5 | 5 | 4 |
| G-mean | 6132 | 16 550 | 24 986 | 12 124 | 38 834 | 58 329 |
| CV, % | 51.6 | 58.6 | 67.5 | 45.1 | 42 | 47.6 |
| 4000 (part 1, 7) | ||||||
| n | 5 | 5 | 5 | 4 | 4 | 4 |
| G-mean | 3669 | 6530 | 12 766 | 10 406 | 34 446 | 51 415 |
| CV, % | 57.6 | 166 | 101 | 45.8 | 54.3 | 47.3 |
| 5000 (part 1, 8) | ||||||
| n | 4 | 4 | 4 | 4 | 4 | 4 |
| G-mean | 4714 | 11 701 | 18 881 | 11 987 | 39 477 | 56 460 |
| CV, % | 35.8 | 27.6 | 35.6 | 35.6 | 43.2 | 44.3 |
| 3000 (part 2) | ||||||
| n | 56 | 56 | 55 | 48 | 48 | 47 |
| G-mean | 4816 | 10 910 | 18 495 | 12 794 | 43 067 | 62 451 |
| CV, % | 66 | 62.6 | 60.9 | 30.8 | 28 | 28.8 |
AUC0-4, area under the curve from d 0 to d 4; AUC0-6, AUC from d 0 to d 6; Cmax, maximum serum concentration; CV, coefficient of variation; G-mean, geometric mean.

PKs and plasma inhibitory activity (PIA). (A) Dose proportionality in the steady-state plasma exposures of PLX3397 (pexidartinib) in AML patients. (B-C) In part 1 of the study, plasma samples from 29 patients across 8 dosing cohorts were collected and evaluated for PIA in the FLT3-ITD–containing human AML cell line Molm14. Predose samples were collected on day 1, day 2, and at steady state (day 15 or 29). (B) Inhibition of pFLT3 by PIA is concentration dependent with an EC95 of 6530 ng/mL. (C) Inhibition of phosphorylated (phospho) FLT3 by PIA increases with dose and is ≥95% at ≥3000 mg for both day 2 and steady-state samples.
PKs and plasma inhibitory activity (PIA). (A) Dose proportionality in the steady-state plasma exposures of PLX3397 (pexidartinib) in AML patients. (B-C) In part 1 of the study, plasma samples from 29 patients across 8 dosing cohorts were collected and evaluated for PIA in the FLT3-ITD–containing human AML cell line Molm14. Predose samples were collected on day 1, day 2, and at steady state (day 15 or 29). (B) Inhibition of pFLT3 by PIA is concentration dependent with an EC95 of 6530 ng/mL. (C) Inhibition of phosphorylated (phospho) FLT3 by PIA increases with dose and is ≥95% at ≥3000 mg for both day 2 and steady-state samples.
Efficacy
Based on modified response criteria, the ORR to pexidartinib (CR, CRi, CRp, and PR) for all treated patients was 21% (n = 19 of 90). The overall CRc (CR, CRi, and CRp) rate for the study was 11% (n = 10 of 90). Of patients treated at ≥2000 mg, 23% (n = 17 of 74) responded (CRc, n = 9 [12.2%] of 74; Figure 3A). One patient who received 2000 mg in part 1 and 1 patient in part 2 achieved CR. Two patients in part 1 (1400- and 2000-mg dose groups) achieved CRi. Six patients in part 2 achieved CRi or CRp. Four patients in part 1 (1000, 2000, 4000, and 5000 mg) achieved PR, and 5 patients in part 2 achieved PR. Six patients underwent HSCT (3 of 6 after response; PR, n = 1; CRi, n = 1; CRp, n = 1). After a protocol amendment to allow maintenance after HSCT, 3 patients bridged to transplantation resumed maintenance therapy with pexidartinib. Of the 3 patients who received pexidartinib maintenance, 2 discontinued maintenance (1 of these patients relapsed 6 months after transplantation, and 1 relapsed 2 years after transplantation), and 1 patient (05_017) was still alive on treatment without relapse at the time of database lock. Of the remaining 3 patients, 2 relapsed <100 days posttransplantation, and 1 was lost to follow-up. A majority of evaluable patients had some reduction in their marrow blast counts (n = 31 [74%] of 42; Figure 3B). Looking at the best percentage change in blasts by steady-state exposure showed a weak but significant exposure-response relationship (Pearson’s 1-sided correlation test r = −3.06; P = .037; supplemental Figure 2). However, no significant difference in steady-state exposure was noted between responders (achieving CRc) and nonresponders (supplemental Figure 2).
![Best clinical response, treatment duration, and survival. (A) Number and proportion of best response (modified criteria) per patient by cohort. Includes all patients with a best response of at least PR. Bars are overlaid with the geometric mean of steady-state AUC0-6 (ng × h/mL) for each cohort. The ORR for the study was 19 (21%) of 90 and CRc rate is 10 (11%) of 90. (B) Best percentage change in blasts is shown for individual patients, and bars are blue gradient–filled with steady-state AUC0-6. Gray-filled bars indicate that AUC0-6 was not determined. Patients who were successfully bridged to transplantation are shown with solid triangles. Best response by modified criteria is indicated at the top of each bar. (C) Treatment duration is represented by a gray line drawn from C1D1 to the day of last known dose. If last dose day is unknown, gray line extends to the day of study discontinuation or database lock. Response assessments by modified criteria are shown with color-coded boxes. Empty boxes indicate that a patient was not assessable (NA). The day of disease progression or death (end PFS), whichever came first, is marked with a red "×." In part 1 of the study, 2 patients were successfully bridged to transplantation (07_002 [2000 mg] and 07_004 [5000 mg]), discontinued the study drug to undergo HSCT, and did not resume pexidartinib treatment. In part 2, 4 patients were successfully bridged to transplantation. Of these, 2 patients (03_009 and 06_013) resumed maintenance treatment with pexidartinib, as provided for by protocol amendment 7, but later discontinued treatment because of an AE or voluntary withdrawal from the study. One patient (05_017) resumed treatment with pexidartinib and was still on treatment at database lock, and 1 patient (06_022) did not resume study treatment. (D) OS stratified by response (≥PR). PD, progressive disease; SD, stable disease.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/4/8/10.1182_bloodadvances.2020001449/2/m_advancesadv2020001449f3.png?Expires=1769157699&Signature=PPH1r6Ix~5Rx05kCZwomcUa4FCJmIw5~4bpNAeHE9aCoRhx767bja-Fjm3oZNfCqqFAHvZtI6MKbW8d0TEOJMoegkGpRG44R5W6GzkplQ~tD-an5wsmbpEpB2M9GCr-QfusseRerGa7wgUOLOkL8ux4IB2ZbkeDrjkO3l3mRDre~y2Ah3sG109fFA9NkWmaU8qxSDaEU8CLHtZIBS0-KcxrglTkYUg11fL8X9cFECvBR6Uz7Ek-VMvr2g6Bc7yoaQ8KtwQ-hCRs56V3flKFcXCYk6VoNAnaUBqOJy6pDp3QakDXhhroHpLKDMOtIxIRRJC8H~9q2hkzE4BwuRH2qgw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Best clinical response, treatment duration, and survival. (A) Number and proportion of best response (modified criteria) per patient by cohort. Includes all patients with a best response of at least PR. Bars are overlaid with the geometric mean of steady-state AUC0-6 (ng × h/mL) for each cohort. The ORR for the study was 19 (21%) of 90 and CRc rate is 10 (11%) of 90. (B) Best percentage change in blasts is shown for individual patients, and bars are blue gradient–filled with steady-state AUC0-6. Gray-filled bars indicate that AUC0-6 was not determined. Patients who were successfully bridged to transplantation are shown with solid triangles. Best response by modified criteria is indicated at the top of each bar. (C) Treatment duration is represented by a gray line drawn from C1D1 to the day of last known dose. If last dose day is unknown, gray line extends to the day of study discontinuation or database lock. Response assessments by modified criteria are shown with color-coded boxes. Empty boxes indicate that a patient was not assessable (NA). The day of disease progression or death (end PFS), whichever came first, is marked with a red "×." In part 1 of the study, 2 patients were successfully bridged to transplantation (07_002 [2000 mg] and 07_004 [5000 mg]), discontinued the study drug to undergo HSCT, and did not resume pexidartinib treatment. In part 2, 4 patients were successfully bridged to transplantation. Of these, 2 patients (03_009 and 06_013) resumed maintenance treatment with pexidartinib, as provided for by protocol amendment 7, but later discontinued treatment because of an AE or voluntary withdrawal from the study. One patient (05_017) resumed treatment with pexidartinib and was still on treatment at database lock, and 1 patient (06_022) did not resume study treatment. (D) OS stratified by response (≥PR). PD, progressive disease; SD, stable disease.
Best clinical response, treatment duration, and survival. (A) Number and proportion of best response (modified criteria) per patient by cohort. Includes all patients with a best response of at least PR. Bars are overlaid with the geometric mean of steady-state AUC0-6 (ng × h/mL) for each cohort. The ORR for the study was 19 (21%) of 90 and CRc rate is 10 (11%) of 90. (B) Best percentage change in blasts is shown for individual patients, and bars are blue gradient–filled with steady-state AUC0-6. Gray-filled bars indicate that AUC0-6 was not determined. Patients who were successfully bridged to transplantation are shown with solid triangles. Best response by modified criteria is indicated at the top of each bar. (C) Treatment duration is represented by a gray line drawn from C1D1 to the day of last known dose. If last dose day is unknown, gray line extends to the day of study discontinuation or database lock. Response assessments by modified criteria are shown with color-coded boxes. Empty boxes indicate that a patient was not assessable (NA). The day of disease progression or death (end PFS), whichever came first, is marked with a red "×." In part 1 of the study, 2 patients were successfully bridged to transplantation (07_002 [2000 mg] and 07_004 [5000 mg]), discontinued the study drug to undergo HSCT, and did not resume pexidartinib treatment. In part 2, 4 patients were successfully bridged to transplantation. Of these, 2 patients (03_009 and 06_013) resumed maintenance treatment with pexidartinib, as provided for by protocol amendment 7, but later discontinued treatment because of an AE or voluntary withdrawal from the study. One patient (05_017) resumed treatment with pexidartinib and was still on treatment at database lock, and 1 patient (06_022) did not resume study treatment. (D) OS stratified by response (≥PR). PD, progressive disease; SD, stable disease.
For patients in part 1, median duration of response was 74 days, median PFS was 57 days, and median OS was 91 days. For patients in part 2, median duration of response was 76 days, median PFS was 48 days, and median OS was 112 days (Figure 3C). Median time to best response was 30 days in both part 1 and part 2. In responders (≥PR), median PFS and OS were extended by 98 and 171 days, respectively, compared with nonresponders (Figures 3D; supplemental Figure 3). Median OS of complete responders (CRi, CRp, or CR) was 265 days (90% CI, 170-422 days; supplemental Figure 4). Median duration of CRc (from 9 responders evaluable for duration of response) was 212 days (90% CI, 37-370 days). Median PFS and OS for patients dosed at ≥3000 mg (RP2D, 3000 mg) were not significantly different than for patients who received lower doses (supplemental Figure 5). We also found that FLT3-ITD length did not affect PFS (Cox proportional hazards hazard ratio, 0.998; 90% CI, 0.9925-1.004; P = .6; supplemental Data; supplemental Figure 6). In this heavily pretreated population, the observed response rate was lower in FLT3 TKI–pretreated patients. Four (14.8%) of 27 patients with FLT3 TKI pretreatment were responders (CR, n = 1; PR, n = 3) compared with 15 (23.8%) of 63 FLT3 TKI–naïve patients (supplemental Figure 7). In all 4 responders, the prior FLT3 TKI treatment was sorafenib. OS did not differ between patients with prior FLT3 inhibitor exposure amd patients who were FLT3 inhibitor naïve (supplemental Figure 7).
To assess the potential clinical activity of pexidartinib in patients expressing FLT3 F691L mutations, we sequenced the FLT3 KD from 42 patients with available blood or bone marrow samples collected before pexidartinib treatment. Four patients had detectable F691L mutations at the time of study drug initiation (supplemental Table 4). All 4 patients were treated below the RP2D of 3000 mg per day (1000 mg, n = 2; 1200 mg, n = 1; 2000 mg, n = 1). Three of the 4 patients had no response to pexidartinib. One patient (5.02), treated at 2000 mg, had an F691L mutation detectable in 2.58% of native FLT3 (non–ITD containing) alleles and achieved a CRi. Of note, at the time of pexidartinib relapse, no F691L mutation was detected, and this patient instead had acquired a new D835Y mutation (data not shown). Additionally, in a previous analysis of 9 patients who achieved response on pexidartinib, no patients had developed new F691L mutations at the time of disease relapse.25
Discussion
Results of this phase 1/2 study demonstrate that pexidartinib was well tolerated and had antileukemic activity in a population of heavily pretreated R/R AML patients with FLT3-ITD mutations. No MTD of pexidartinib was reached in this study. The overall safety profile of pexidartinib in this study was similar to that of other FLT3-targeted inhibitors in clinical development.10,11 As expected in patients with acute leukemia, treatment-emergent infectious complications, such as febrile neutropenia, sepsis, and pneumonia, were common and resulted in death in 8% of patients.
Pexidartinib demonstrated clear antileukemic activity in FLT3-ITD+ AML. The ORR to pexidartinib for all treated patients was 21%, with a CRc rate of 11%. Six patients in the study were successfully bridged to allogeneic stem cell transplantation. Overall, the single-agent response rate for pexidartinib was lower than the ∼40% to 50% CRc rates observed with quizartinib9,10 or gilteritinib11 in similar patient populations. The prior use of other type 2 FLT3 inhibitors, such as sorafenib and quizartinib, in a large proportion of enrolled patients may have contributed to primary resistance (27 [30%] of 90 the patients comprising the overall study population had receiving prior FLT3 TKI treatment). However, the response rate in patients without prior TKI treatment was not substantially different than that of the study population as a whole (15 [23.8%] of 63 patients without prior TKI treatment responded). The lower response rate to pexidartinib may also be attributable to the overall lower potency of pexidartinib compared with either quizartinib or gilteritinib (∼1000 or 100 times less potent, respectively, in cell-based assays).25,30,31 In keeping with this idea, the dose required for >95% median inhibition of FLT3 phosphorylation assessed by plasma inhibitory activity was ≥3000 mg. Our PK data suggest that 3000 mg achieved the maximal possible pexidartinib drug level in patients and that higher doses did not result in increased exposure. Because prolonged and potent inhibition of FLT3 kinase activity is an important prerequisite to achieve clinical response,32 it is possible that inability to achieve sufficient exposure to pexidartinib limited clinical response in a significant proportion of patients, as suggested by the exposure-response correlation shown in supplemental Figure 2. Notably, 4% of patients experienced treatment-emergent changes in hair color, indicative of in vivo kinase inhibition of c-KIT,28 a kinase target against which pexidartinib has activity equivalent to its activity against FLT3-ITD.
Resistance-associated FLT3 KD mutations have been an important cause of clinical resistance to all FLT3 TKIs to date, particularly type 2 inhibitors. Pexidartinib is no exception to this paradigm, and we previously reported the acquisition of polyclonal FLT3-ITD KD mutations, including D835 mutations, at the time of disease progression in 6 of 9 pexidartinib responders.25 Conversely, the development of acquired resistance to pexidartinib as a result of FLT3-ITD KD mutations confirms that the clinical activity of pexidartinib is mediated by inhibition of FLT3 and not by another of its targets, such as KIT or CSF-1R. Although pexidartinib is vulnerable to D835 mutations commonly found in association with clinical resistance to quizartinib and sorafenib, it does have potent activity against the FLT3 F691L gatekeeper mutation also commonly associated with acquired FLT3 TKI resistance. AML patients treated with quizartinib,14 sorafenib,15 sunitinib,15 gilteritinib,21 and crenolanib19 have all developed clinical resistance as a result of FLT3 F691 mutations. FLT3 F691L mutations are the most common KD mutations associated with clinical resistance to type 1 FLT3 TKIs crenolanib and gilteritinib. Given the recent FDA approval of gilteritinib in R/R FLT3-mutant AML, F691L mutations may become a more commonly encountered cause of relapse in patients. Because all currently FDA-approved FLT3 inhibitors except midostaurin have demonstrated in vitro18 and/or clinical15,21 vulnerability to FLT3 F691L mutations, and midostaurin has no single-agent efficacy in this disease,7 pexidartinib may have a potential role in the treatment of patients resistant to gilteritinib (or other FLT3 TKIs) who relapse with F691L mutations. Unfortunately, in this study, only a small minority of patients treated below the R2PD were known to have F691L mutations before pexidartinib initiation, although 1 patient treated at 2000 mg did respond. Additionally, unlike with other FLT3 TKIs,14,15,19,21 the F691L mutation was not associated with pexidartinib relapse in a limited analysis of 9 responding patients.25 However, future studies will be required to determine the true clinical activity of pexidartinib in patients with FLT3 F691L mutations. Recently, translational studies in patients treated with crenolanib19 and gilteritinib21 have also established a variety off-target mechanisms, particularly Ras pathway mutations, as important mediators of FLT3 TKI resistance. Despite activity against FLT3 F691L mutations, it is likely that pexidartinib will also be vulnerable to these non–FLT3-dependent mechanisms of resistance. It is also possible that the unique kinase profile of pexidartinib may contribute to clinical activity in AML in patients without FLT3 mutations. For example, a role for the targeting of CSF1R signaling–dependent supporting cells in AML has also been recently described33 and may be another rationale for the use of pexidartinib in AML patients. Overall, the safety, tolerability, and clinical activity of pexidartinib in the heavily pretreated R/R AML population described in this study are supportive of the further development of pexidartinib in AML.
All data and the full clinical trial protocol will be made available upon request by contacting corresponding author Catherine C. Smith (catherine.smith@ucsf.edu).
Acknowledgment
This study was supported by Plexxikon, a member of the Daiichi Sankyo Group.
Authorship
Contribution: C.C.S., M.J.L., O.F., J.M.P., G.J.R., R.M.S., E.S.W., A.E.P., and S.K. contributed to data acquisition, analysis and interpretation, writing and review of the manuscript, and review and approval of the final manuscript; B.L.W., M.H.L., H.H.H., and C.Z. contributed to study design; M.H.L., C.C.S., A.E.P., H.H.H., P.L.S., B.L., C.Z., S.K., and B.L.W. contributed to data analysis; and G.B. contributed to study design and study oversight.
Conflict-of-interest disclosure: C.C.S. has received research funding from Astellas Pharma, Revolution Medicines, Plexxikon, and FujiFilm and honoraria from Amgen, Astellas Pharma, and Daiichi Sankyo. M.J.L. has received honoraria and research funding from Astellas Pharma, FujiFilm, and Novartis and honoraria from Menarini, Agios, Amgen, and Daiichi Sankyo. A.E.P. has received honoraria from Astellas Pharma, Arog, AbbVie, Actinium Pharmaceuticals, Agios, Jazz, NewLink Genetics, Takeda, and Novartis and research funding from BioMed Valley Discoveries, Daiichi Sankyo, Bayer, and FujiFilm. J.M.P. has received payments for consultancy from Actinium Pharmaceuticals, Gilead, AstraZeneca, and Pharmacyclics. G.J.R. has consulted for and served on advisory committees for AbbVie, Actinium, Agios, Amphivena, Argenx, Array Biopharma, Astex, Astellas, AstraZeneca, Bayer, Celgene, Celltrion, Daiichi Sankyo, Eisai, Epizyme, Helsinn, Janssen, Jasper Therapeutics, Jazz, MEI Pharma (independent data monitoring committee chair), Novartis, Orsenix, Otsuka, Pfizer, Roche/Genentech, Sandoz, Takeda (independent review committee chair), and Trovagene and received research support from Cellectis. E.S.W. has served as an advisor for AbbVie, Kite, Jazz, Astellas, Daiichi Sankyo, Amgen, and Agios and served on speaker’s bureaus and as an advisor for Celyad, Pfizer, and Stemline. M.H.L., H.H.H., P.L.S., C.Z., B.L.W., and G.B. are or were employees of Plexxikon. The remaining authors declare no competing financial interests.
Correspondence: Catherine C. Smith, University of California San Francisco, 505 Parnassus Ave, Suite M1286, Box 1270, San Francisco, CA 94143; e-mail: catherine.smith@ucsf.edu.

