

Key Points
Complement and interferon interplay promotes thrombotic microangiopathy and can serve as novel therapeutic target.
Combined interferon and complement blockade might improve clinical outcomes in thrombotic microangiopathy.

Abstract
Transplant-associated thrombotic microangiopathy (TA-TMA) is an important cause of morbidity and mortality after hematopoietic stem cell transplantation (HSCT). The complement inhibitor eculizumab improves TA-TMA, but not all patients respond to therapy, prompting a search for additional targetable pathways of endothelial injury. TA-TMA is relatively common after HSCT and can serve as a model to study mechanisms of tissue injury in other thrombotic microangiopathies. In this work, we performed transcriptome analyses of peripheral blood mononuclear cells collected before HSCT, at onset of TA-TMA, and after resolution of TA-TMA in children with and without TA-TMA after HSCT. We observed significant upregulation of the classical, alternative, and lectin complement pathways during active TA-TMA. Essentially all upregulated genes and pathways returned to baseline expression levels at resolution of TA-TMA after eculizumab therapy, supporting the clinical practice of discontinuing complement blockade after resolution of TA-TMA. Further analysis of the global transcriptional regulatory network showed a notable interferon signature associated with TA-TMA with increased STAT1 and STAT2 signaling that resolved after complement blockade. In summary, we observed activation of multiple complement pathways in TA-TMA, in contrast to atypical hemolytic uremic syndrome (aHUS), where complement activation occurs largely via the alternative pathway. Our data also suggest a key relationship between increased interferon signaling, complement activation, and TA-TMA. We propose a model of an “interferon-complement loop” that can perpetuate endothelial injury and thrombotic microangiopathy. These findings open opportunities to study novel complement blockers and combined anti-complement and anti-interferon therapies in patients with TA-TMA and other microangiopathies like aHUS and lupus-associated TMAs.
Introduction
Transplant-associated thrombotic microangiopathy (TA-TMA) results from acute injury to the vascular endothelium by chemotherapy, radiation, infectious pathogens, and/or immune dysregulation, leading to target organ injury (eg, kidney, gut), or multiorgan dysfunction syndrome.1 Clinical outcomes for untreated TA-TMA with multiorgan dysfunction syndrome after hematopoietic stem cell transplant (HSCT) are very poor.2-7 Complement dysregulation plays an important role in TA-TMA and complement blockade with eculizumab, a monoclonal antibody directed against C5, improves survival after TA-TMA.8-10 However, some patients with TA-TMA do not have a clinical response to eculizumab, suggesting that other mechanisms of endothelial injury may be involved in the pathogenesis of thrombotic microangiopathies and could potentially serve as therapeutic targets.
TMAs have been reported as a treatment-limiting side effect of interferon-α and interferon-β therapy for malignancies and chronic hepatitis.11,12 Interferons have also been implicated in pathogenesis of monogenic lupus, a disease with high levels of inflammation that can present with lupus nephritis and thrombotic microangiopathy.13,14 Therefore, we hypothesized that interferons, in addition to complement, contribute to vascular endothelial injury and TA-TMA.
Here, we present novel evidence using gene expression analysis of complement activation via all complement pathways-alternative, classical, and lectin. In addition, we present a strong downstream interferon signature in TA-TMA. Our data indicate important roles for activation of both complement and interferon pathways in patients with TA-TMA and offer potential new therapeutic targets for TA-TMA and other disorders presenting with thrombotic microangiopathies, especially in patients with lack of response to eculizumab therapy.
Methods
Study subjects and clinical data
Cincinnati Children’s Hospital Medical Center has an institutional review board-approved HSCT specimen repository and clinical database. Patients are consented before transplant, prospectively enrolled, and blood samples are collected and stored starting before transplant and continuing weekly until day 100 after HSCT. For the current study, we selected patients with neuroblastoma who developed TA-TMA after autologous HSCT and had received the terminal complement blocker eculizumab. Controls were autologous HSCT recipients with neuroblastoma of similar age, who received identical high-dose chemotherapy and did not develop TA-TMA. Controls were evaluated at equivalent timepoints after HSCT as subjects with TA-TMA. Cases and time-matched controls had available peripheral blood mononuclear cells (PBMCs) samples stored within 1 week before transplant conditioning chemotherapy (baseline), at the time of TA-TMA diagnosis, and when eculizumab was discontinued (or equivalent timepoints in controls) (supplemental Figure 1). Patient demographics and disease characteristics were captured from the clinical database. We also selected PBMCs of 3 allogenic HSCT recipients, all of whom developed TA-TMA and had a complete response to eculizumab. PBMSs were collected at TA-TMA diagnosis and TA-TMA resolution (after documenting full engraftment with donor cells). A baseline sample was not used because it represents recipient PBMCs. All patients at our institution are prospectively monitored for TA-TMA, with daily blood counts for hemoglobin, platelets and schistocytes, renal panels, twice weekly lactate dehydrogenase, weekly haptoglobin, weekly urinalyses with random urine protein/creatinine ratio and weekly Cystatin C estimated glomerular filtration rate (GFR; CystC GFR), calculated by the Larsson formula. Soluble terminal complement complex activity (sC5b-9) was measured twice per week when TA-TMA was suspected (normal sC5b-9 < 244 ng/mL). TA-TMA diagnosis was made using published criteria (supplemental Table 1). Patients with TA-TMA and high-risk features (elevated blood sC5b-9 and nephrotic range proteinuria) were offered eculizumab as first-line therapy for TA-TMA. Eculizumab was dosed using personalized pharmacokinetics/pharmacodynamics monitoring as previously published, and was discontinued in all subjects after clinical resolution of TA-TMA.9,15
Gene expression analyses
RNAseq analyses were performed by the Genomics, Epigenomics, and Sequencing Core at the University of Cincinnati.
Automatic RNAseq library preparation.
Using PrepX mRNA Library kits (WaferGen) and the Apollo 324 next-generation sequencing (NGS) automatic library prep system, depleted or isolated RNA was RNase III fragmented, adaptor-ligated and Superscript III reverse transcriptase (Lifetech, Grand Island, NY) converted into complement DNA, followed by automatic purification using Agencourt AMPure XP beads (Beckman Coulter, Indianapolis, IN). Next, using the universal (SR) and index-specific primer with limited polymerase chain reaction (PCR) cycle number (∼13), sample-specific index was added to each ligated complement DNA sample and the amplified library was enriched by AMPure XP bead purification with the final elution volume of 16 µL. To check the quality and yield of the purified library, 1 µL library was analyzed by a Bioanalyzer (Agilent, Santa Clara, CA) using its DNA high-sensitivity chip. To accurately quantify the library concentration for the clustering, the library was 1:104 diluted in dilution buffer (10 mM Tris-HCl, pH 8.0 with 0.05% Tween 20), and quantitative PCR measured by the Kapa Library Quantification kit (Kapabiosystem, Woburn, MA) using ABI's 9700HT real-time PCR system (Lifetech).
Cluster generation and HiSeq sequencing.
Individually indexed libraries were proportionally pooled (20-50 million reads per sample) for clustering in the cBot system (Illumina, San Diego, CA). Libraries at the final concentration of 12.0 pM were clustered onto a flow cell using Illumina’s TruSeq SR Cluster kit v3 and sequenced for 50 cycles using TruSeq SBS kits on the Illumina HiSeq system.
Bioinformatics analysis.
Sequence reads were aligned to the human reference genome (hg19) using the TopHat2 aligner16 and reads aligning to each known transcript were counted using Bioconductor packages for NGS data analysis.17 The differential expression analysis between different samples was performed using the negative binomial statistical model of read counts as implemented in the edgeR Bioconductor package.18 To account for patient heterogeneity, the matched analysis was performed by including the “patient” covariate in the edgeR generalized linear model. Statistical significance of differential expressions was established for false discovery rate (FDR)–adjusted P values.19 The differences between log2-reads per kilobase per million mapped reads20 for each patient were used for visualizing data in heatmaps. The gene list enrichment analysis was performed in 2 steps. First, the Fisher's exact test was used to identify KEGG pathways,21 transcription factor target gene lists from the TFacts database22 and from the TREG signatures of ENCODE transcription factor binding ChIP-seq datasets,23 and MSigDB Hallmark genes sets24 enriched by the genes upregulated at the TA-TMA diagnosis timepoint. A stringent Bonferroni adjustment was used for P value adjustment in this analysis. Second, the Random Set25 enrichment analysis, as implemented in the CLEAN package,26 of upregulated genes against all MSigDB gene sets27 was performed to identify additional complement and interferon-related categories. Standard FDR P value adjustment was also used in this analysis.
Results
Clinical data
We selected 7 autologous HSCT recipients (range, 2-5 years old) with neuroblastoma who all received identical high-dose chemotherapy conditioning with carboplatin, etoposide, and melphalan, a regimen reported to be associated with a high-risk of TA-TMA and had stored samples available for this study.28,29 Four patients (cases) had a diagnosis of TA-TMA and 3 patients (controls) did not have any evidence of TA-TMA. In cases, TA-TMA was diagnosed 1 to 49 days after transplant. All patients with TA-TMA had hematologic TMA features, severe hypertension requiring more than 2 antihypertensive medications or continuous antihypertensive medication infusion, nephrotic range proteinuria (random urine protein/creatinine ratio ≥2 mg/mg), more than 70% reduction in Cystatin C GFR from their pretransplant baseline, evidence of terminal complement activation at TA-TMA diagnosis with elevated blood sC5b-9 concentrations and end-organ injury, in addition to the expected bone marrow aplasia and mucositis from high dose chemotherapy. All 4 cases were treated with eculizumab and received 7 to 19 doses, after which they all had resolution of TA-TMA and normalization of their blood sC5b-9 concentration and recovery of organ function. Eculizumab was successfully discontinued in all treated cases without any evidence of TA-TMA recurrence. All 4 cases were alive and well at 1 year after transplant. The remaining 3 included patients who did not develop TA-TMA (controls) were evaluated at equivalent timepoints after HSCT. Demographics and disease characteristics are summarized in supplemental Table 2.
Three allogeneic transplant recipients were diagnosed with TA-TMA within first 100 days after HSCT and responded to eculizumab therapy using the same monitoring criteria as described for autologous HSCT recipients. Demographics and relevant disease characteristic are listed in supplemental Table 3.
Gene expression in autologous HSCT cases with TA-TMA
All 4 autologous HSCT cases with TA-TMA had a complete clinical response to treatment with the terminal complement inhibitor eculizumab, leading us to expect that patients would have elevated expression of complement genes at TA-TMA diagnosis, and that expression would return to baseline values after resolution of TA-TMA. To test our hypothesis, we compared gene expression at the time of TA-TMA diagnosis (but before starting eculizumab) to the pretransplant baseline and found elevated expression of 960 genes in the samples collected at TA-TMA diagnosis (FDR < 0.1). An enrichment analysis of KEGG pathways,21 transcription factor targets,22,23 and MSigDB hallmark gene sets24 identified significant activation of complement, complement-related genes, and interferon-responsive genes (Table 1). The involvement of differentially expressed genes in upregulated complement and interferon-related pathways, including signal transducer and activator of transcription 1 and 2 (STAT1 and STAT2) targets, is illustrated in supplemental Figure 2. The comprehensive analysis of MSigDB identified additional complement and interferon-related gene sets enriched for upregulated genes (Table 2).
Upregulated pathways and transcription factor targets at the time of TA-TMA diagnosis as compared with pretransplant baseline
| Upregulated pathways and TF targets | Bonferroni adjusted P* |
|---|---|
| KEGG pathways | |
| Staphylococcus aureus infection | 2.73e-10 |
| Osteoclast differentiation | 1.09e-06 |
| Complement and coagulation cascades | 1.84e-05 |
| TF targets (TFactS database) | |
| STAT1 | .00041 |
| SP1 | .00052 |
| SPI1 | .00112 |
| TF targets (ENCODE) | |
| STAT1 targets K562 interferon-α 30 min | 2.73e-07 |
| STAT1 targets K562 interferon-α 6 h | 6.08e-07 |
| STAT2 targets K562 interferon-α 6 h | 1.31e-06 |
| STAT2 targets K562 interferon-α 30 min | .00027 |
| MSigDb hallmark gene sets | |
| Hallmark interferon-γ response | 2.73e-07 |
| Hallmark inflammatory response | 6.08e-07 |
| Hallmark interferon-α response | 1.31e-06 |
| Hallmark G2M checkpoint | .00027 |
| Upregulated pathways and TF targets | Bonferroni adjusted P* |
|---|---|
| KEGG pathways | |
| Staphylococcus aureus infection | 2.73e-10 |
| Osteoclast differentiation | 1.09e-06 |
| Complement and coagulation cascades | 1.84e-05 |
| TF targets (TFactS database) | |
| STAT1 | .00041 |
| SP1 | .00052 |
| SPI1 | .00112 |
| TF targets (ENCODE) | |
| STAT1 targets K562 interferon-α 30 min | 2.73e-07 |
| STAT1 targets K562 interferon-α 6 h | 6.08e-07 |
| STAT2 targets K562 interferon-α 6 h | 1.31e-06 |
| STAT2 targets K562 interferon-α 30 min | .00027 |
| MSigDb hallmark gene sets | |
| Hallmark interferon-γ response | 2.73e-07 |
| Hallmark inflammatory response | 6.08e-07 |
| Hallmark interferon-α response | 1.31e-06 |
| Hallmark G2M checkpoint | .00027 |
TF, transcription factor.
Bonferroni adjusted P < .01.
Interferon- and complement-related MSigDB gene sets upregulated at the time of TA-TMA diagnosis in autologous HSCT recipients as compared with pretransplant baseline
| Pathway | Direction of change and FDR |
|---|---|
| Interferon pathways | |
| Browne interferon responsive genes | ↑ 3.59e-37 |
| Hallmark interferon-α response | ↑ 1.38e-38 |
| Bosco interferon induced antiviral module | ↑ 5.16e-27 |
| Zhang interferon response | ↑ 3.75e-18 |
| Control vs interferon-treated macrophages | ↑ 1.98e-17 |
| Moserle interferon response | ↑ 6.36e-17 |
| Chiang liver cancer subclass interferon up | ↑ 4.17e-16 |
| Reactome interferon α β signaling | ↑ 1.30e-14 |
| Reactome interferon signaling | ↑ 6.00e-12 |
| Interferon-β response up | ↑ 1.19e-11 |
| Urosevic response to imiquimod | ↑ 2.68e-11 |
| Interferon-α response up | ↑ 2.69e-10 |
| Interferon-γ response up | ↑ 6.23e-07 |
| Natsume response to interferon β | ↑ 8.02e-06 |
| Farmer breast cancer cluster | ↑ 1.94e-05 |
| Reactome γ interferon signaling | ↑ 3.87e-05 |
| Geiss response to DSRNA up | ↑ 4.13e-04 |
| VSIRF Q6 | ↑ 0.001 |
| VSICSBP Q6 | ↑ 0.004 |
| Interferon-γ biosynthetic process | ↑ 0.005 |
| VsIRF7 01 | ↑ 0.006 |
| Regulation of interferon-γ biosynthetic process | ↑ 0.01 |
| Interferon-γ production | ↑ 0.01 |
| Complement pathways | |
| Initial triggering of complement | ↑ 2.7e-105 |
| Classical complement pathway | ↑ 1.6e-102 |
| Complement pathway | ↑ 6.35e-85 |
| Complement cascade | ↑ 6.4e-59 |
| Complement and coagulation cascades | ↑ 2.2e-44 |
| Complement | ↑ 2.1e-30 |
| Lectin-induced complement pathway | ↑ 0.0008 |
| Regulation of complement cascade | ↑ 0.001 |
| Pathway | Direction of change and FDR |
|---|---|
| Interferon pathways | |
| Browne interferon responsive genes | ↑ 3.59e-37 |
| Hallmark interferon-α response | ↑ 1.38e-38 |
| Bosco interferon induced antiviral module | ↑ 5.16e-27 |
| Zhang interferon response | ↑ 3.75e-18 |
| Control vs interferon-treated macrophages | ↑ 1.98e-17 |
| Moserle interferon response | ↑ 6.36e-17 |
| Chiang liver cancer subclass interferon up | ↑ 4.17e-16 |
| Reactome interferon α β signaling | ↑ 1.30e-14 |
| Reactome interferon signaling | ↑ 6.00e-12 |
| Interferon-β response up | ↑ 1.19e-11 |
| Urosevic response to imiquimod | ↑ 2.68e-11 |
| Interferon-α response up | ↑ 2.69e-10 |
| Interferon-γ response up | ↑ 6.23e-07 |
| Natsume response to interferon β | ↑ 8.02e-06 |
| Farmer breast cancer cluster | ↑ 1.94e-05 |
| Reactome γ interferon signaling | ↑ 3.87e-05 |
| Geiss response to DSRNA up | ↑ 4.13e-04 |
| VSIRF Q6 | ↑ 0.001 |
| VSICSBP Q6 | ↑ 0.004 |
| Interferon-γ biosynthetic process | ↑ 0.005 |
| VsIRF7 01 | ↑ 0.006 |
| Regulation of interferon-γ biosynthetic process | ↑ 0.01 |
| Interferon-γ production | ↑ 0.01 |
| Complement pathways | |
| Initial triggering of complement | ↑ 2.7e-105 |
| Classical complement pathway | ↑ 1.6e-102 |
| Complement pathway | ↑ 6.35e-85 |
| Complement cascade | ↑ 6.4e-59 |
| Complement and coagulation cascades | ↑ 2.2e-44 |
| Complement | ↑ 2.1e-30 |
| Lectin-induced complement pathway | ↑ 0.0008 |
| Regulation of complement cascade | ↑ 0.001 |
Focused analysis of gene expression levels of complement and interferon-related genes showed significant changes in expression of 76 complement and interferon-related genes (Figure 1). A majority (63 of 76, 83%) of these genes upregulated during active TA-TMA showed a statistically significant decrease after clinical TA-TMA resolution. Specifically, all 9 genes in the complement and coagulation cascade KEGG pathway that were upregulated during active TA-TMA returned to their pretransplant baseline when TA-TMA resolved after eculizumab therapy. The genes that were still upregulated after resolution of TA-TMA were related to proliferation, and increased expression is likely appropriate during stem cell engraftment and blood count recovery.

Gene expression changes between clinical time points of active TA-TMA and TA-TMA resolution in 4 autologous HSCT recipients with TA-TMA. The figure shows a section of the heatmap that illustrates the change in complement and interferon gene expression profiles in patients with TA-TMA. “TMA diagnosis” indicates gene expression changes from pretransplant baseline compared with when TA-TMA was diagnosed posttransplant, but before initiating complement-blocking therapy with eculizumab. “TMA resolution” indicates gene expression changes from when TA-TMA was diagnosed compared with clinical resolution of TA-TMA, after completing eculizumab therapy. Genes shown are upregulated at the time of TA-TMA diagnosis (FDR < 0.1) and belong to at least of 1 of the enriched pathway or gene lists shown in Table 1. Green bars on the right side indicate statistically significant changes at the time of TA-TMA resolution (FDR < 0.1). Data indicate that complement pathways are highly upregulated at TA-TMA diagnosis in subjects with TA-TMA and normalize after resolution of TA-TMA.
Gene expression changes between clinical time points of active TA-TMA and TA-TMA resolution in 4 autologous HSCT recipients with TA-TMA. The figure shows a section of the heatmap that illustrates the change in complement and interferon gene expression profiles in patients with TA-TMA. “TMA diagnosis” indicates gene expression changes from pretransplant baseline compared with when TA-TMA was diagnosed posttransplant, but before initiating complement-blocking therapy with eculizumab. “TMA resolution” indicates gene expression changes from when TA-TMA was diagnosed compared with clinical resolution of TA-TMA, after completing eculizumab therapy. Genes shown are upregulated at the time of TA-TMA diagnosis (FDR < 0.1) and belong to at least of 1 of the enriched pathway or gene lists shown in Table 1. Green bars on the right side indicate statistically significant changes at the time of TA-TMA resolution (FDR < 0.1). Data indicate that complement pathways are highly upregulated at TA-TMA diagnosis in subjects with TA-TMA and normalize after resolution of TA-TMA.
Pathway analyses confirmed significant upregulation of multiple complement pathways, including upregulation of the alternative, classical, and lectin pathways at TA-TMA diagnosis as compared with pretransplant baseline. No complement pathways were downregulated. Specifically, the data showed a marked upregulation of expression of the complement component C1Q C chain (22.8-fold, P = 2.2e-13), B chain (15-fold, P = 1.6e-12), and A chain (7.9-fold, P = 3.7e-8) initiating steps in the classical pathway of complement, at time of TA-TMA, compared with pretransplant baseline. Upregulation of C2 (elevated 2.9-fold, P = .0002), in addition to C1Q supports a central role for the classical pathway in initiation of TA-TMA. Factor D was also upregulated (1.8-fold, P = .05), indicating upregulation of the rapid loop of complement activation, perhaps contributing to the clinical severity of TA-TMA in some cases. Additional changes in expression of key genes in the alternative, classical, and lectin pathways of complement activation are displayed in Figure 2.

Complement gene expression in 4 patients with TA-TMA at time of clinical diagnosis. This figure illustrates gene expression in alternative, classical, and lectin complement pathways among cases with TA-TMA at the time of clinical diagnosis but before initiation of complement-blocking therapy with eculizumab. Red, orange, and yellow colors indicate upregulated pathways and the degree of upregulation (red > orange > yellow). Blue colors indicate downregulated and the degree of downregulation (dark blue > light blue). Red letters and red outlines indicate statistically significant changes in gene expression from pretransplant baseline to the time of TA-TMA clinical diagnosis.
Complement gene expression in 4 patients with TA-TMA at time of clinical diagnosis. This figure illustrates gene expression in alternative, classical, and lectin complement pathways among cases with TA-TMA at the time of clinical diagnosis but before initiation of complement-blocking therapy with eculizumab. Red, orange, and yellow colors indicate upregulated pathways and the degree of upregulation (red > orange > yellow). Blue colors indicate downregulated and the degree of downregulation (dark blue > light blue). Red letters and red outlines indicate statistically significant changes in gene expression from pretransplant baseline to the time of TA-TMA clinical diagnosis.
Expression of interferon-regulated genes in autologous HSCT cases and controls
Our data identified a strong downstream interferon signature in autologous HSCT cases with TA-TMA. Because of our interest in identifying new targetable endothelial injury pathways, we investigated if interferon activation was associated with TA-TMA, or just with the transplantation process itself, by comparing gene expression profiles at TA-TMA diagnosis and resolution in cases with TA-TMA and at equivalent timepoints in controls without TA-TMA. The data displayed in Figure 3 show broad upregulation of expression of interferon-regulated genes which are members of interferon-related gene sets (Hallmark interferon-γ response and hallmark interferon-α response) and transcription factor target gene lists (STAT1 and STAT2 targets K562 IFN-α 30 minutes, STAT1 and STAT2 targets K562 IFN-α 6 hours) at TA-TMA diagnosis. Expression of these genes decreased in conjunction with clinical resolution of TA-TMA. Most of the genes in interferon related pathways were not significantly upregulated in controls without TA-TMA at equivalent timepoints posttransplant.
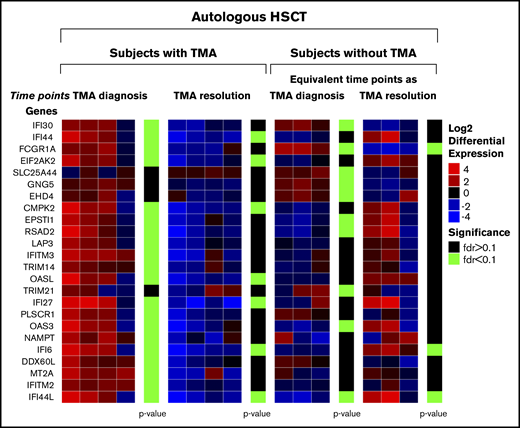
Changes in interferon gene expression between clinical time points in 4 cases with TA-TMA and 3 controls without TA-TMA after autologous HSCT. The figure illustrates changes in interferon gene expression profiles in autologous HSCT recipients with TA-TMA and without TA-TMA. In cases with TA-TMA, “TMA diagnosis” indicates gene expression changes at time of clinical diagnosis of TA-TMA compared with pretransplant baseline, but before the start of complement-blocking therapy with eculizumab. “TMA resolution” illustrates gene expression change at the time of TA-TMA clinical diagnosis compared with TA-TMA resolution, after completing eculizumab therapy. In controls without TA-TMA, this evaluation was performed at matched timepoints after transplant. Genes shown were upregulated in the “TMA diagnosis” comparison for patients with or without TA-TMA and are members of interferon-related gene sets (Hallmark interferon-γ response and hallmark interferon-α response) and transcription factor target gene lists (STAT1 and STAT2 targets K562 IFN-α 30 minutes, STAT1 and STAT2 targets K562 IFN-α 6 hours). Green bars indicate statistically significant differences between time points. Data indicate that interferon pathways are highly upregulated at clinical diagnosis of TA-TMA in cases with TA-TMA and normalize after therapy with eculizumab. Interferon pathways are not upregulated in controls without TA-TMA at matched time points after transplant.
Changes in interferon gene expression between clinical time points in 4 cases with TA-TMA and 3 controls without TA-TMA after autologous HSCT. The figure illustrates changes in interferon gene expression profiles in autologous HSCT recipients with TA-TMA and without TA-TMA. In cases with TA-TMA, “TMA diagnosis” indicates gene expression changes at time of clinical diagnosis of TA-TMA compared with pretransplant baseline, but before the start of complement-blocking therapy with eculizumab. “TMA resolution” illustrates gene expression change at the time of TA-TMA clinical diagnosis compared with TA-TMA resolution, after completing eculizumab therapy. In controls without TA-TMA, this evaluation was performed at matched timepoints after transplant. Genes shown were upregulated in the “TMA diagnosis” comparison for patients with or without TA-TMA and are members of interferon-related gene sets (Hallmark interferon-γ response and hallmark interferon-α response) and transcription factor target gene lists (STAT1 and STAT2 targets K562 IFN-α 30 minutes, STAT1 and STAT2 targets K562 IFN-α 6 hours). Green bars indicate statistically significant differences between time points. Data indicate that interferon pathways are highly upregulated at clinical diagnosis of TA-TMA in cases with TA-TMA and normalize after therapy with eculizumab. Interferon pathways are not upregulated in controls without TA-TMA at matched time points after transplant.
Expression of complement-regulated genes in autologous HSCT cases and controls
Table 3 summarizes changes in complement-associated gene expression between pretransplant baseline and time of diagnosis of TA-TMA in cases with TA-TMA and controls without TA-TMA. Expression of some critical components of the complement system were increased in cases with TA-TMA but unaltered in controls without TA-TMA. C1INH, an interferon-inducible gene and a negative regulator of the complement classical pathway, was elevated fivefold in cases with TA-TMA. Expression of C2, a key component of the classical pathway, was significantly elevated in cases with TA-TMA, as was CFD, a promoter of rapid activation of complement via the alternative pathway. Expression of both C2 and CFD were unchanged from baseline in controls without TA-TMA. Similarly, expression of key receptors for activated C5, C3, and C4 were elevated only in cases with TA-TMA. Expression of the central complement component C3 was significantly reduced in controls without TA-TMA (reduced fivefold, P = 9.6 × 10−9), whereas expression was nonsignificantly reduced (less than twofold; P = .51) in cases with TA-TMA, perhaps indicating an inappropriately high level of expression in the cases with TA-TMA. CFI, a negative regulator of complement, was significantly upregulated (more than eightfold) in controls without TA-TMA and was nonsignificantly upregulated (3.2-fold) in cases with TA-TMA, also perhaps indicating inappropriately low expression in the setting of intense complement activation. Expression of C1QA, B, and C was upregulated in both cases with TA-TMA and controls, but the magnitude of the upregulation was notably greater in the cases with TA-TMA (eg, C1QA was elevated 22.8-fold in children with TA-TMA vs 7.6-fold in children without TA-TMA). Expression of C3AR1, a key receptor for activated C3, was upregulated to a similar extent in cases with TA-TMA and controls, whereas expression of the receptor for C3d (CR2) was downregulated more in controls without TA-TMA than in case with TA-TMA, potentially exposing cells from children with TA-TMA to relatively greater complement activation.
Comparison of complement gene expression profiles between cases with TA-TMA and controls after autologous HSCT
| Gene | Children with TA-TMA after autologous HSCT | Children without TA-TMA after autologous HSCT | Complement protein function and regulation by interferons (data from Interferon Web site v. 2.01, accessed 24 March 2019) | ||
|---|---|---|---|---|---|
| Fold change in expression | Adjusted P | Fold change in expression | Adjusted P | ||
| Gene expression significantly changed only in children with TA-TMA | |||||
| SERPING1 (C1INH) | 5.0 | .0029 | 0.63 | .28 | Inhibits activated C1r and C1S: negative regulator of classical pathway. Upregulated by types I and II interferons. |
| C2 | 2.88 | .00023 | 0.88 | .77 | Necessary component of classical pathway of complement. Upregulated by types I and II interferons. |
| CFD | 1.81 | .05 | 1.12 | .7 | Majority of production is extrahepatic; This protease catalyzes the cleavage of factor B, the rate-limiting step of the alternative pathway of complement activation; a net promoter of complement activation. Upregulated by types I and II interferons in majority of datasets. |
| C5AR1 | 2.3 | .005 | 1.29 | .47 | Binds C5a: Receptor activation stimulates chemotaxis, granule enzyme release, intracellular calcium release and superoxide anion production. Downregulated by types I and II interferons. |
| C5AR2 | 2.08 | .03 | 1.3 | .39 | Receptor for the chemotactic and inflammatory C3a, C4a, and C5a anaphylatoxin peptide. Data mixed on interferon regulation upregulated in 1 and downregulated in 2 datasets. |
| PLAUR | 1.91 | .04 | 1.08 | .88 | Acts as a receptor for urokinase plasminogen activator. Plays a role in localizing and promoting plasmin formation. Downregulated by types I and II interferons. |
| Gene expression significantly changed only in children without TA-TMA | |||||
| C3 | 0.56 | .51 | 0.18 | 9.6 × 10−9 | C3 plays a central role in the activation of the complement system. Processing of C3 by C3 convertase is the central reaction in both classical and alternative complement pathways. Downregulated by types I and II interferons. |
| CFI | 3.2 | .39 | 8.5 | .0049 | Responsible for cleaving the α-chains of C4b and C3b in the presence of the cofactors C4-binding protein and factor H respectively. Negative regulator of complement. Not reported to be regulated by interferons. |
| GENE expression significantly changed in children with and without TA-TMA | |||||
| C1QC | 22.8 | 2.2 × 10−13 | 7.6 | 8.7 × 10−16 | C1QC, B, and A combine to form C1Q, which is the recognition molecule for activation of complement via the classical pathway; majority of production is extrahepatic. Upregulated by interferon-γ. |
| C1QB | 15 | 1.7 × 10−12 | 3.7 | 1 × 10−9 | |
| C1QA | 7.87 | 3.7 × 10−8 | 2.95 | 4 × 10−7 | |
| C3AR1 | 2.66 | .0021 | 2.74 | 5.1 × 10−7 | Receptor for activated C3: binding of C3a by the encoded receptor activates chemotaxis, granule enzyme release, superoxide anion production, and bacterial opsonization. Data mixed on interferon regulation; majority of datasets report upregulation. |
| CR2 | 0.06 | 5.32 × 10−5 | 0.03 | 3.23 × 10−11 | Receptor for C3d, and EBV, participates in B-cell activation. Data mixed on interferon regulation; 1 dataset reports upregulation and 1 downregulation. |
| Gene | Children with TA-TMA after autologous HSCT | Children without TA-TMA after autologous HSCT | Complement protein function and regulation by interferons (data from Interferon Web site v. 2.01, accessed 24 March 2019) | ||
|---|---|---|---|---|---|
| Fold change in expression | Adjusted P | Fold change in expression | Adjusted P | ||
| Gene expression significantly changed only in children with TA-TMA | |||||
| SERPING1 (C1INH) | 5.0 | .0029 | 0.63 | .28 | Inhibits activated C1r and C1S: negative regulator of classical pathway. Upregulated by types I and II interferons. |
| C2 | 2.88 | .00023 | 0.88 | .77 | Necessary component of classical pathway of complement. Upregulated by types I and II interferons. |
| CFD | 1.81 | .05 | 1.12 | .7 | Majority of production is extrahepatic; This protease catalyzes the cleavage of factor B, the rate-limiting step of the alternative pathway of complement activation; a net promoter of complement activation. Upregulated by types I and II interferons in majority of datasets. |
| C5AR1 | 2.3 | .005 | 1.29 | .47 | Binds C5a: Receptor activation stimulates chemotaxis, granule enzyme release, intracellular calcium release and superoxide anion production. Downregulated by types I and II interferons. |
| C5AR2 | 2.08 | .03 | 1.3 | .39 | Receptor for the chemotactic and inflammatory C3a, C4a, and C5a anaphylatoxin peptide. Data mixed on interferon regulation upregulated in 1 and downregulated in 2 datasets. |
| PLAUR | 1.91 | .04 | 1.08 | .88 | Acts as a receptor for urokinase plasminogen activator. Plays a role in localizing and promoting plasmin formation. Downregulated by types I and II interferons. |
| Gene expression significantly changed only in children without TA-TMA | |||||
| C3 | 0.56 | .51 | 0.18 | 9.6 × 10−9 | C3 plays a central role in the activation of the complement system. Processing of C3 by C3 convertase is the central reaction in both classical and alternative complement pathways. Downregulated by types I and II interferons. |
| CFI | 3.2 | .39 | 8.5 | .0049 | Responsible for cleaving the α-chains of C4b and C3b in the presence of the cofactors C4-binding protein and factor H respectively. Negative regulator of complement. Not reported to be regulated by interferons. |
| GENE expression significantly changed in children with and without TA-TMA | |||||
| C1QC | 22.8 | 2.2 × 10−13 | 7.6 | 8.7 × 10−16 | C1QC, B, and A combine to form C1Q, which is the recognition molecule for activation of complement via the classical pathway; majority of production is extrahepatic. Upregulated by interferon-γ. |
| C1QB | 15 | 1.7 × 10−12 | 3.7 | 1 × 10−9 | |
| C1QA | 7.87 | 3.7 × 10−8 | 2.95 | 4 × 10−7 | |
| C3AR1 | 2.66 | .0021 | 2.74 | 5.1 × 10−7 | Receptor for activated C3: binding of C3a by the encoded receptor activates chemotaxis, granule enzyme release, superoxide anion production, and bacterial opsonization. Data mixed on interferon regulation; majority of datasets report upregulation. |
| CR2 | 0.06 | 5.32 × 10−5 | 0.03 | 3.23 × 10−11 | Receptor for C3d, and EBV, participates in B-cell activation. Data mixed on interferon regulation; 1 dataset reports upregulation and 1 downregulation. |
Expression of complement and interferon genes in allogeneic HSCT recipients with TA-TMA
The allogeneic HSCT recipients had elevated expression of interferon and complement gene sets at TA-TMA diagnosis, similar to our findings in autologous HSCT recipients with neuroblastoma. Supplemental Table 4 displays GSEA complement and interferon-related gene sets, enriched for the genes that were upregulated at the TA-TMA diagnosis time point compared with the TA-TMA resolution time point. We examined only TA-TMA diagnosis and TA-TMA resolution timepoints and not the pretransplant baseline sample in these subjects because in the allogeneic HSCT setting the host genome is present before transplant and the donor genome at the latter 2 timepoints.
Discussion
TA-TMA is an important cause of organ injury and mortality after HSCT. HSCT is a planned procedure and TA-TMA is frequent in our patient population so biological samples can be collected before, during, and after TA-TMA, allowing mechanistic studies that are impossible with less predictable forms of thrombotic microangiopathies, like atypical hemolytic uremic syndrome (aHUS). TA-TMA can be caused by or amplified by graft-versus-host disease (GVHD), infections, or calcineurin inhibitors after allogeneic HSCT, all of which may complicate biological findings.30-33 We opted to study a more homogeneous group of patients developing TA-TMA after autologous HSCT for neuroblastoma, in whom TA-TMA is uncomplicated by GVHD and calcineurin inhibitors and viral infections are markedly less frequent. The cause of endothelial injury in patients with neuroblastoma and TA-TMA is likely related to high-dose chemotherapy, and we were able to study children receiving exactly the same chemotherapy regimen to eliminate another potential source of heterogeneity.28,29
We considered at length the suitability of PBMC as a source of RNA for this study, rather than liver, traditionally thought to be the major source of complement components. PBMC have the important advantage of being accessible for serial collection, a strategy that would not be possible with liver tissue from human patients. The complement components C1q, C7, properdin, and CFD are predominantly produced by myeloid cells, particularly monocytes and dendritic cells.34 It is also apparent that under inflammatory conditions both hepatic and extrahepatic production of complement is enhanced, particularly for larger proteins such as C1q, or those that are rapidly cleaved such as C3.35 Of note, C3 from bone marrow-derived cells can restore normal lymphoid organ-dependent antibody responses in mice lacking C3.36 A recent review of production of complement proteins by immune cells supports the importance of PBMC as a key source of proteins in local complement activation and under inflammatory conditions.37 Taken together, we believe these data support our strategy of studying PBMC to allow serial study of complement activation and expression of other key genes in a human setting of acute illness with extensive inflammation.
All subjects undergoing HSCT likely have some degree of endothelial injury from high-dose chemotherapy, medications, and infectious agents. Although we saw marked elevation of expression in complement and interferon genes in cases we diagnosed with TA-TMA, there was more modest elevation in controls, suggesting perhaps that it is the magnitude of the complement/interferon response that determines whether clinical TA-TMA is manifest.
In previous work, we have shown that both complement gene polymorphisms and non-Caucasian race are associated with increased risk of TA-TMA.38 The frequency of complement gene polymorphisms is higher in persons of non-Caucasian race, providing 1 potential explanation for increased risk. Our new observation of the role of interferon expression in TA-TMA offers an additional potential mechanism for the association with non-Caucasian race. A recent publication from Cole et al analyzed variation in the expression of genes involved in inflammation and type I interferon using peripheral blood transcriptome profiles. This cohort study of normal young adults demonstrated increased upregulation of interferon gene expression in Blacks and Asians relative to non-Hispanic whites.39
We observed upregulation of interferon-responsive complement genes at onset of TA-TMA that returned to baseline after resolution of TA-TMA. These novel findings are in agreement with the highly inflammatory clinical phenotype of children presenting with severe TA-TMA, and may explain, at least in part, incomplete response to complement blocking therapy in some patients, especially after allogeneic transplant, despite achievement of therapeutic drug concentration in the blood.
Our study goal was to examine the transcriptome in patients during TA-TMA and after TA-TMA resolution with eculizumab. Because we purposely selected subjects that were eculizumab therapy responders, we do not directly demonstrate that anti-interferon agents are beneficial. We are hoping to document the need or benefit of interferon blockade in poor responders in the future, by testing augmentation of therapy with the targeted anti-interferon agent emapalumab in children with an inadequate or delayed response to complement blockade.
Interferons and complement are both key components of innate immunity, and interferons induce expression of many complement genes. In particular, the key initiators of complement activation by the classical pathway, C1QC, B, and A, are upregulated by interferons, in addition to the amplifier of cascade activation, CFD, and the classical pathway component C2. Our current data demonstrate activation of the classical, lectin, and alternative complement pathways in TA-TMA. These data contrast with findings in aHUS where complement activation is thought to occur largely via the alternative pathway.40 This observation opens opportunities to study novel druggable targets in patients with incomplete response to complement blockers. Our previously reported clinical experience indicates that complement blockade alone allows resolution of TA-TMA in many instances, but not all, despite achievement of therapeutic drug concentration and full complement blockade, measured by a very low complement hemolytic activity (CH50) in the blood of treated patients.9 It is possible that elevated levels of interferons perpetuate endothelial injury in those patients despite effective terminal complement blockade. Moreover, the benefits of complement blockade may not manifest for more than 7 days after start of therapy, and additional organ injury or even death can occur in that time. Immediate and brief interferon blockade in severe cases might interrupt endothelial injury loop and reduce inflammation and organ injury until complement blockade can extinguish the underlying process.
Our data show that gene expression returns to baseline levels with no residual elevation of complement-related pathways after eculizumab therapy in responding patients and in general TA-TMA reactivation has not occurred after stopping therapy. Data in a small group of allogeneic transplant recipients supported a similar mechanism. These data further support our usual strategy of discontinuing eculizumab after resolution of TA-TMA.9 Prediagnosis samples are not generally available in other thrombotic microangiopathies, like aHUS, which occurs unpredictably, but samples at diagnosis and resolution could be studied. A similar strategy may identify patients with other thrombotic microangiopathies in whom short course of complement blockers or combined complement and interferon blocking therapy may also be indicated.
In this report, we describe a close interaction between elevation of interferons and complement in initiating and maintaining inflammation and organ injury in TA-TMA for the first time. Based on our data in TA-TMA and published reports of a reciprocal relationship between interferon genes and complement genes in other clinical and biological settings, we propose a model of an “interferon-complement loop” that can perpetuate endothelial injury and thrombotic microangiopathy (Figure 4). Interferons promote expression of complement genes, such as C1Q, which initiates the classical complement pathway and ultimately leads to formation of the lytic membrane attack complex (MAC or C5b-9) and endothelial injury presenting as TMA. Intracellular complement C5a receptor production can maintain interferon γ secretion and sustain endothelial damage. Injured endothelial cells release interleukin-8 (IL-8), causing neutrophil activation and formation of neutrophil extracellular traps (NETs). NETs become activated during rapid neutrophil engraftment after HSCT, a period associated with a high incidence of TA-TMA. NET formation promotes complement system activation via factor P. NET production can also be further stimulated by interferon-γ.41 Interferon-α and interferon-β increase differentiation of B cells into plasma cells that can produce anti-factor H antibodies, preventing inhibition of the alternative complement pathway via CFH. NETs can activate plasmacytoid dendritic cells to produce high levels of interferon α, as has been shown in systemic lupus erythematosus.42 Interferon-α, like in Dego disease, directly activates complement via C5b-9, resulting in vascular endothelial injury. High levels of interferons administered exogenously as a therapeutic agent can be directly damaging to the endothelium and are known to result in TMA.43-47 BK virus has been shown to trigger high interferon-γ production in renal transplant recipients and also to be associated with an increased risk of complement mediated TA-TMA in HSCT recipients.48 In addition, BK virus can directly injure endothelial cells and promote release of interferon-γ.49 Inflammatory cytokines like IL-6, IL-8, and interferon-γ are also released from circulating activated T cells, NK cells, monocytes, and tissue macrophages as a result of BK virus infection, GVHD and in disorders like hemophagocytic lymphohistiocytosis, with high interferon production resulting in thrombotic microangiopathy.49,50
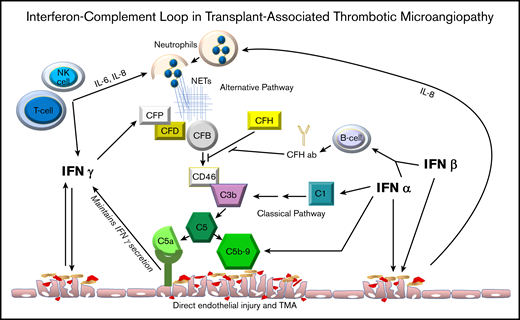
Interferon-complement loop. This figure displays the relationship between interferon and complement activation that perpetuates vascular endothelial injury in clinical conditions presenting with thrombotic microangiopathy where both interferon and complement pathways are activated. CFP, CFD, CFB, CFH: complement factors P, D, B and H.
Interferon-complement loop. This figure displays the relationship between interferon and complement activation that perpetuates vascular endothelial injury in clinical conditions presenting with thrombotic microangiopathy where both interferon and complement pathways are activated. CFP, CFD, CFB, CFH: complement factors P, D, B and H.
Our study has strengths and limitations. Our study benefited from a careful, prospective TA-TMA phenotyping in HSCT recipients using established diagnostic and risk criteria, and our experience using eculizumab in these complex patients. TA-TMA offers a unique opportunity for serial study and comparison of baseline state with disease state, a strategy that is more challenging in other microangiopathies, such as aHUS, in which there is rarely opportunity to collect samples immediately before disease presentation. We studied a modest number of autologous HSCT cases and controls, and demonstrated similar findings in small group of allogeneic HSCT recipients, but the strength of the associations we report suggest that larger patient numbers will not importantly alter the conclusions of the study. Moreover, neuroblastoma patients enrolled were homogeneous, with the same diagnosis and similar age, and they had been treated with the same chemotherapy regimen, an important strength of the analysis. We do recognize that although examining complement gene expression in PBMC in this clinical circumstance is valuable, there may be important changes in hepatic production of complement components that we were unable to study.
In summary, we observed that interferon gene expression in addition to the activation of all 3 complement pathways may represent new targetable mechanism of endothelial injury in thrombotic microangiopathies. It is possible that brief, combined anti-complement and anti-interferon treatment may facilitate rapid disease control in TA-TMA and other interferon- and complement-driven diseases. Steroids can likely modify interferon production to some degree and might be considered as an adjunct to complement blocking therapy in some patients with TA-TMA like who are not receiving steroids for other clinical indications. We speculate that response to steroids would depend on the magnitude of interferon system activation because clinical responses to the interferon blocker emapalumab are observed in patients with hemophagocytic lymphohistiocytosis, an interferon-driven disease, after failing to achieve disease remission with high-dose dexamethasone.
This is a novel but testable hypothesis as both a complement-blocking agent (eculizumab) and an interferon-γ-blocking agent (emapalumab) are now available for clinical use. Also, anifrolumab is a monoclonal antibody against the type I interferon receptor that inhibits the activity of all type I interferons and is being tested in patients with systemic lupus erythematosus.51 Our data suggest studying these therapies in patients with TA-TMA may benefit other thrombotic microangiopathies, especially in those with refractory disease.
The generalizability of these data to more complex clinical settings such as allogeneic stem cell transplant needs to be established, and such RNAseq studies are under way. We are also interested in additional variables such as age, presence of concomitant GVHD and infections, race, and use of specific preparative regimens; these will also be tested in specific targeted studies of interferon in the future.
RNAseq data have been deposited in the Gene Expression Omnibus database (accession number GSE134979).
Acknowledgments
Samples used in this study were collected and provided by the staff of the Cincinnati Children’s Hospital Bone Marrow Tissue Repository, whom we thank for outstanding ongoing technical assistance.
Part of the research reported in this publication was supported by the National Institutes of Health, Eunice Kennedy Shriver National Institute of Child Health and Human Development (R01HD093773) (S.J. and S.M.D.) and National Institute of Diabetes and Digestive and Kidney Diseases (K23 DK101600) (B.L.L.).
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Authorship
Contribution: S.J., S.M.D., and B.L.L. planned and performed this study and wrote the manuscript; N.L., M.M., and J.C. performed RNAseq studies and analyses and prepared figures; C.E.D. collected and analyzed study subject data; and all authors approved the final version of the manuscript.
Conflict-of-interest disclosure: S.J. and S.M.D. have National Institutes of Health–funded research for TA-TMA and received free drug for study subjects from Alexion, unrelated to this work. S.J. received travel support and honoraria for presentation at the scientific meeting from Omeros and consultancy fees from Arcus Medica and Magnolia Innovations, unrelated to this work. M.D. received consultancy fees from Novartis and research funding and travel support from Prolacta, all unrelated to this work. B.L.L. has received consulting fees from Jazz Pharmaceuticals, unrelated to this work. The remaining authors have no disclosures to report.
Correspondence: Sonata Jodele, Division of Bone Marrow Transplantation and Immune Deficiency, Cincinnati Children’s Hospital Medical Center, 3333 Burnet Ave, MLC 11027, Cincinnati, OH 45229; e-mail sonata.jodele@cchmc.org.

