

Primary cold agglutinin disease (CAD) is a rare autoimmune hemolytic anemia caused by a distinct type of B-cell lymphoproliferative disease of the bone marrow.1,2 We have recently evaluated the use of bendamustine-rituximab therapy for CAD and found that B cell–directed therapy is highly efficient and safe and may be considered first line therapy for relatively fit patients with CAD.3 Hemolysis is mediated by binding of monoclonal immunoglobulin M antibodies (cold agglutinins) to the erythrocyte surface I antigen at temperatures below central body temperature, followed by agglutination and complement classical pathway activation.1,4 Immunoglobulins are almost exclusively encoded by heavy chain variable gene IGHV4-34 and mostly by κ light chain variable gene IGKV3-20 or similar IGHV3-15.5 We recently revealed recurrent KMT2D and CARD11 gene mutations in CAD-associated B-cell lymphoproliferative disease.6
Chromosome instability is one of the hallmarks of cancer, and it has implications in disease diagnostics, prognostics, and response to therapy. Copy number variations (CNVs), a gain or loss of copies of DNA segments larger than 1 kb in length, are associated with chromosome instability. Chromosome instability has not been studied in great detail in CAD. However, some studies published more than 20 years ago have indicated that CNV is a feature of CAD or CAD-associated malignant lymphoproliferative disorders.7-9 In this study, we have analyzed 15 cases of well-defined primary CAD for CNV using new high-throughput methods to further characterize the genetic background of the disease.
We have analyzed 13 CAD samples from the CAD5 study3 using cytogenetic microarrays (OncoScan CNV Assay; Thermo Fisher Scientific) and exome sequencing to detect CNVs. In addition, we present data from 2 samples with only exome sequencing.6 The study was approved by the Regional Committee for Medical and Health Research Ethics of Southeast Norway (REK-SØ 2012/131). B cells were isolated from the bone marrow using fluorescence-activated cell sorting before analysis, as previously described.5 Exome sequencing reads were aligned to the hg38 reference genome with BWA software.10 Postprocessing involved Picard (https://broadinstitute.github.io/picard/) and GATK11-13 tools and consisted of quality score recalibration, realignment around indels, and marking of duplicates. The exome sequencing data were analyzed for CNV using GATK411-13 and Control-FREEC14 software to confirm our findings. Major findings were confirmed by both methods (detailed material and methods are available in the supplemental Data).
Complete or partial gain of chromosome 3 (+3 or +3q) was detected in all samples, except for one (14/15) (Table 1; Figure 1; supplemental Figure 1). This case without gain of chromosome 3 is an outlier with regard to other molecular characteristics (unpublished data). Further, most cases showed either gain of chromosome 12 or 18 (11/15); 5/15 showed gain of chromosome 12 and 6/15 showed gain of chromosome 18 (Table 1; Figure 1; supplemental Figure 1). Additional small regions of recurrent gains or losses were also detected in other chromosomes. The recurrent CNVs detected in at least 4 samples are: +1p36.31-p36.13, −8p21.3-p21.1, +9q34.2-q34.3, +11q13.1-q13.3, +17q25.1-q25.3, +21q22.2-q22.3, and +22q13.31-q13.33 (supplemental Table 1). Gains and losses of large parts of chromosomes are revealed by both cytogenetic microarrays and exome sequencing CNV analysis (supplemental Figure 1). However, some of the smaller CNVs detected by cytogenetic microarrays could not consistently be confirmed by exome sequencing CNV analysis (Table 1; supplemental Table 1). This is probably due to the very limited material available, inherent to CAD-associated B-cell lymphoproliferative disease, to perform exome sequencing. The major CNVs have a copy number around 3, whereas most of the small CNVs have a copy number around 2.5, indicating that these small CNVs are present only in a subset of cells.
CNVs in CAD patient samples detected by both cytogenetic microarray assay and exome sequencing CNV analysis
| Major recurrent CNVs | Other CNVs* | Response to therapy (CAD5) | |||
|---|---|---|---|---|---|
| Samples | chr3 | chr12 | chr18 | ||
| CAD-1.06 | +3 | +12 | — | N/A | Partial remission |
| CAD-1.07 | +3 | — | +18 | N/A | No response |
| CAD-1.22 | +3 | — | +18 | — | Partial remission |
| CAD-1.23 | +3 | — | — | −7q31.2-q32.3 | Complete remission |
| CAD-1.24 | +3 | +12 | — | +21† | Partial remission |
| CAD-1.25 | +3† | — | — | +1q23.1-q31.3; +1q42.13-q44; −3p21.31-p21.2; −6q23.3-q24.1; −8p23.1-p21.1‡; −10q23.31‡ | Partial remission |
| CAD-1.26 | +3 | — | — | — | Complete remission |
| CAD-1.30 | +3 | — | +18 | −6q16.1‡ −8p21.3-p21.1‡ | Complete remission |
| CAD-1.31 | +3 | — | +18 | −2q37.3‡; +10q24.32‡ | No response |
| CAD-1.32 | — | — | — | — | Complete remission |
| CAD-1.34 | +3 | — | +18q | +14q12‡ | No response |
| CAD-1.37 | +3q | +12 | — | −8p23.3-p11.23‡ | Partial remission |
| CAD-2.02 | +3q | +12† | — | −8p23.3-p12; −8q11.1q12.1; -X | Complete remission |
| CAD-5§ | +3q | +12 | — | N/A | N/A |
| CAD-7§ | +3 | — | +18 | N/A | N/A |
| Major recurrent CNVs | Other CNVs* | Response to therapy (CAD5) | |||
|---|---|---|---|---|---|
| Samples | chr3 | chr12 | chr18 | ||
| CAD-1.06 | +3 | +12 | — | N/A | Partial remission |
| CAD-1.07 | +3 | — | +18 | N/A | No response |
| CAD-1.22 | +3 | — | +18 | — | Partial remission |
| CAD-1.23 | +3 | — | — | −7q31.2-q32.3 | Complete remission |
| CAD-1.24 | +3 | +12 | — | +21† | Partial remission |
| CAD-1.25 | +3† | — | — | +1q23.1-q31.3; +1q42.13-q44; −3p21.31-p21.2; −6q23.3-q24.1; −8p23.1-p21.1‡; −10q23.31‡ | Partial remission |
| CAD-1.26 | +3 | — | — | — | Complete remission |
| CAD-1.30 | +3 | — | +18 | −6q16.1‡ −8p21.3-p21.1‡ | Complete remission |
| CAD-1.31 | +3 | — | +18 | −2q37.3‡; +10q24.32‡ | No response |
| CAD-1.32 | — | — | — | — | Complete remission |
| CAD-1.34 | +3 | — | +18q | +14q12‡ | No response |
| CAD-1.37 | +3q | +12 | — | −8p23.3-p11.23‡ | Partial remission |
| CAD-2.02 | +3q | +12† | — | −8p23.3-p12; −8q11.1q12.1; -X | Complete remission |
| CAD-5§ | +3q | +12 | — | N/A | N/A |
| CAD-7§ | +3 | — | +18 | N/A | N/A |
N/A, not available.
CNVs detected by both cytogenetic microarrays and exome sequencing CNV analysis.
Almost entire chromosome.
CNVs <3 Mb detected by both cytogenetic microarray assay and exome sequencing CNV analysis.
Analyzed by exome sequencing CNV analysis only.
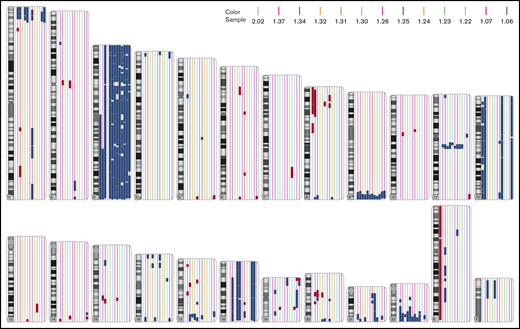
Chromosomal gains and losses in each chromosome detected by cytogenetic microarrays. Chromosomal gains (blue) and losses (red) for each sample. Each sample is indicated by a different color. Only changes >3 Mb are shown.
Chromosomal gains and losses in each chromosome detected by cytogenetic microarrays. Chromosomal gains (blue) and losses (red) for each sample. Each sample is indicated by a different color. Only changes >3 Mb are shown.
Gain of chromosome 3 has previously been reported in 9 of 26 patients with CAD, but is not a specific feature of CAD.7,8 Gain of this chromosome is also frequently found in marginal zone lymphoma (MZL) and diffuse large B-cell lymphoma of activated B-cell type.15,16 Moreover, gain of chromosomes 12 and 18, demonstrated in our study, is also a feature of MZL.15 Of note, gain of chromosomes 12 and 18 was mutually exclusive. Furthermore, the high frequency of KMT2D mutation we previously reported in CAD6 is also found in nodal MZL.15 These findings, together with the immunophenotype of CAD-associated lymphoproliferative disease, suggest that the CAD-associated lymphoproliferative disease, although exclusively present in the bone marrow, might be related to MZL.
We also explored whether the presence or absence of trisomy 12 and 18 was associated with response to therapy in 13 patients. Previous studies have indicated that gain of chromosome 18 may be associated with an adverse prognosis in MZL.17,18 Although our series is small, we found a trend toward poorer response in patients with trisomy 18 compared with patients with trisomy 12. Of note, the 3 patients with no response to therapy had trisomy 18 or +18q. On the contrary, 3 patients without either trisomy 12 or 18 had the best responses (Table 1). Despite the limited number of cases, the Pearson correlation coefficient was statistically significant (P = .009). However, more samples need to be analyzed to confirm these results. A correlation between response to therapy and presence of trisomy 12 or 18 has previously been demonstrated for other B-cell lymphomas. Trisomy 12 is frequent in patients with small lymphocytic lymphoma (28%-36%) and chronic lymphocytic leukemia (10%-25%), and in the latter +12 is associated with intermediate prognosis.19,20 Trisomy 3 and 18 are found to be correlated with advanced-stage extranodal MZL.17 There is evidence from previous studies that trisomy 18 may be associated with upregulation of BCL2, an antiapoptotic gene located on chromosome 18. Chromosome 18q21 amplification, leading to high BCL2 protein levels, is observed in some patients with mantle cell lymphoma.21 Iqbal et al22 have shown that diffuse large B-cell lymphoma of activated B-cell type have a higher frequency of chromosome 18q21 amplification, higher BCL2 expression, and worse survival. In addition, Galteland et al23 showed increased BCL2 expression in samples with gain of BCL2 in B-cell non-Hodgkin lymphoma. Whether BCL2 is upregulated in CAD with trisomy 18, and whether it might contribute to therapy resistance, needs to be further investigated. If so, venetoclax, a BCL2 inhibitor, might be considered for therapy of patients not responding to current therapy, as suggested by Berentsen.24 Therapy including venetoclax has recently been tested for relapsed/refractory non-Hodgkin lymphoma.25
We conclude that gain of chromosome 3 is a highly recurrent finding in CAD-associated lymphoproliferative disease. Further gain of chromosomes 12 and 18 might be predictors of therapy outcome. These genetic findings are similar to what has previously been demonstrated in nodal and extranodal MZL, suggesting that CAD-associated lymphoproliferative disease, a disease of the bone marrow, might be related to MZL.
In accordance with Norwegian legislation and the ethics approval of the study, all sensitive data are stored in protective systems at Oslo University Hospital. On request, the data will be made available for other institutions. For the original data, please contact gto@ous-hf.no.
Acknowledgments:
The authors are grateful for the sequencing services provided by the Helse Sør-Øst Genomics Core Facility at Oslo University Hospital.
This study was funded by South-Eastern Norway Regional Health Authority (Helse Sør-Øst) grant and by Fondsstiftelsen, Oslo University Hospital.
Contribution: A.M., J.D., A.T., S.B., G.E.T., and G.T. designed the study; A.M., I.Ø., and G.T. performed the analyses; J.D., A.T., S.B., G.E.T., and G.T. supervised the study; J.D., A.T., U.R., S.B., and G.E.T. reviewed the diagnostic patient samples and collected the clinical data; A.M., J.D., and G.T. prepared the manuscript; and all authors have critically read the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Gunhild Trøen, Department of Pathology, Oslo University Hospital, Montebello, 0379 Oslo, Norway; e-mail: gto@ous-hf.no.

