

Key Points
Expression of several thrombotic, inflammatory, and HIF-regulated genes is increased in granulocytes and platelets of PV and ET patients.
Functional studies investigating the role of tissue factor, P-selectin, interleukin-10, and VEGFA in thrombosis may lead to targeted therapies.

Abstract
Thrombosis is a major cause of morbidity and mortality in polycythemia vera (PV) and essential thrombocythemia (ET). The pathophysiology of thrombosis in these disorders remains unclear, and we hypothesized that upregulation of thrombotic, inflammatory, and hypoxia-inducible factor (HIF)–regulated genes may play a role in it. We performed unbiased RNA sequencing in granulocytes and platelets of PV patients and found differential expression of several thrombotic, inflammatory, and HIF-regulated genes. The expression of many of these genes positively correlated with JAK2 expression and JAK2V617F allelic burden. We then validated these findings by quantitative polymerase chain reaction analyses of selected gene transcripts in a larger number of PV and ET granulocytes and platelets (58 patients) and in 28 controls, and we compared these findings in patients with and without thrombosis. The study included 29 females and 29 males; of these, 28 had a history of thrombosis. We found that transcripts of several selected genes were upregulated in patients with PV or ET compared with controls. In granulocytes, the expression levels of F3, SELP, VEGFA, and SLC2A1 were significantly higher in patients with a history of thrombosis compared with those who did not have thrombosis. Patients with a history of thrombosis have significantly higher expression of IL1RAP (P < .05) in platelets compared with those without thrombosis. Our study confirms the presence of a thrombo-inflammatory state and augmented HIF activity in PV and ET and its role in thrombosis. These data may provide the background for targeted therapies in PV and ET.
Introduction
Philadelphia chromosome–negative myeloproliferative neoplasms (MPNs) include polycythemia vera (PV) and essential thrombocythemia (ET), which are characterized by increased risk of thrombosis and occur in about 20% of patients at diagnosis.1,2 Thrombosis is a major cause of morbidity and mortality in these patients, and a major goal of treatment is to prevent thrombotic complications.3 In MPNs, thrombosis can occur at unusual anatomic sites, such as splanchnic veins or cerebral venous sinuses. MPNs are also the most common cause of noncirrhotic and nonmalignant extrahepatic portal vein obstruction and Budd-Chiari syndrome.4 Age >60 years, history of thrombosis and other cardiovascular risk factors, high leukocyte count, and JAK2V617F allelic burden are known risk factors for thrombosis in MPN patients.5-10 Qualitative abnormalities in blood cells and increased adhesion of leukocytes and platelets have been postulated to contribute to the increased risk of thrombosis in PV and ET, but the exact mechanism contributing to this risk is unclear.11
Although increased blood viscosity from high hematocrit in PV has been suggested to contribute to thrombosis, this notion has been challenged because it does not explain increased thrombotic risk in other MPN phenotypes such as ET and primary myelofibrosis.2,12,13 This is further supported by the occurrence of thrombotic events in MPN patients with normal blood counts. Additional mechanisms beyond increased hematocrit and blood counts have been elucidated in thrombotic risk. Chronic inflammatory state as a result of paracrine secretion of inflammatory cytokines by a neoplastic clone is thought to be responsible for constitutional symptoms in MPN patients.14 This may also contribute to premature atherosclerosis and risk of arterial and venous thrombosis. Furthermore, the presence of a JAK2V617F mutation in endothelial cells of patients with MPN has also been reported and suggests a role for dysfunctional endothelium.15,16 Chuvash polycythemia (CP) is the first and best studied of the several disorders characterized by congenital augmentation of hypoxia-inducible factor (HIF) levels, and the rate of arterial and venous thromboses in CP is higher than in PV.17,18 Therefore, in this study, we further interrogated augmented HIF signaling that we and others have previously reported in PV,19,20 and sought to study the role of the HIF-signaling pathway in MPN thrombogenesis.
Leukocytes have recently come to the forefront in the thrombogenesis of MPN,21 and platelets play a major role in hemostasis and inflammation. Hence, we performed unbiased RNA sequencing (RNA-seq) in both granulocytes and platelets of PV patients. We then used a targeted approach by determining the expression of known thrombo-inflammatory and HIF-regulated genes in granulocytes and platelets from PV and ET patient samples.
Methods
Patients
The patients were recruited from the MPN clinic at the Huntsman Cancer Center and Veterans Affairs Hospital in Salt Lake City, Utah. The study was approved by the Institutional Review Board of the University of Utah, and all the participants provided informed consent.
Unbiased RNA sequencing.
Whole blood was collected from PV patients and controls, and granulocytes and platelets were separated from whole blood samples by Ficoll-Paque/density gradient centrifugation, using the methodology described previously.22 Briefly, whole blood was collected in acid citrate dextrose tubes and centrifuged at 99g for 10 minutes at room temperature. The upper layer of plasma that contained the platelets was transferred to new tubes and centrifuged at 400g for 10 minutes at room temperature and the plasma was removed. Peripheral blood granulocytes were obtained from the bottom layer. Red blood cell lysis buffer was added to the bottom layer and incubated for 10 minutes. The sample was again centrifuged at 400g for 10 minutes at room temperature, and the hemolytic supernatant containing lysed reticulocytes was removed. This step was repeated until the pellet was no longer visibly red. The pellet was resuspended with 2 mL of Tri reagent, and 1 mL was transferred to each 1.5-mL tube and stored at –80°C. We have previously shown that granulocytes obtained by this method were 97.5% pure morphologically.22
RNA was isolated by using the RNeasy Mini Kit (Qiagen). Cytoplasmic and mitochondrial ribosomal RNA were removed by using Ribo-Zero Gold (Illumina Inc.). Stranded RNA sequencing libraries were prepared with the Illumina TruSeq Stranded Total RNA Kit with Ribo-Zero Gold (RS-122-2301 and RS-122-2302). The quality of the libraries was tested on an Agilent Technologies 2200 TapeStation using a D1000 ScreenTape assay (Catalog No. 5067-5582 and No. 5067-5583). The molarity of adapter-modified molecules was defined by quantitative polymerase chain reaction (PCR) using the Kapa Library Quant Kit (Kapa Biosystems; Catalog No. KK4824). For Illumina sequence analysis, 10 nM of each library was prepared.
RNA-seq used 25 pM of each library. First, the libraries were chemically denatured. They were applied to an Illumina HiSeq v4 paired-end flow cell by using Illumina cBot. Using an Illumina HiSeq PE Cluster Kit v4-cBot (PE-401-4001), hybridized DNA was amplified and annealed to sequencing primers. Then, the flow cell was transferred to an Illumina HiSeq 2500 instrument (HCS v2.2.38 and RTA v1.18.61). By using HiSeq SBS Kit v4 sequencing reagents (FC-401-4003), a 125-cycle paired-end sequence was run. Eight samples were sequenced together per lane on the instrument. Library preparation and sequencing were performed by High Throughput Genomics Core at the University of Utah.
Whole transcriptome data were analyzed using Useq package (useq.sourceforge.net). The reads were mapped to a reference genome (Hg19) using Novoalign. Aligned files (sequence alignment map [SAM] files) were converted into binary alignment map (BAM) files by using the Sam Transcriptome Parser. The Ensembl Biomart database was used for annotation of genes. Differential gene expression was determined by DEseq2. Pathway analysis was performed using the Reactome pathway database via Panther version 14.1 (http://www.pantherdb.org).23
Quantitative analysis of thrombotic, inflammatory, and HIF-regulated gene transcripts.
In a separate analysis, whole blood was collected from PV and ET patients and controls; granulocytes and platelets were isolated using the method described above. RNA was extracted from these cells using TRI reagent according to the manufacturer’s protocol (Molecular Research Center, Cincinnati, OH). RNA was then reverse-transcribed to make complementary DNA using the SuperScript VILO complementary DNA Synthesis Kit (Invitrogen). Expression levels of selected genes involved in thrombo-inflammatory and HIF pathways were determined in granulocytes and platelets by quantitative reverse transcription PCR (qRT-PCR) using TaqMan Expression Assays (Applied Biosystems, Foster City, CA). Expression levels were normalized against RPL13A (calculated by ΔΔCt method) and were expressed as fold change, which was described previously.24 We tested several housekeeping genes, including HPRT1, GAPDH, ACTB, and RPL13A. Among these genes, expression levels of RPL13A in MPN patients were similar to those of controls and were not affected by therapy.
Genes involved in thrombo-inflammatory and HIF pathways that were selected for the study include tissue factor (F3), P-selectin (SELP), serpin peptidase inhibitor clade E member 1 (SERPINE1) which encodes plasminogen activator inhibitor (PAI-1), thrombospondin 1 (THBS1), interleukin-1 receptor-associated kinase 1 (IRAK1), interleukin-1 receptor accessory protein (IL1RAP), and two HIF-regulated genes (vascular endothelial growth factor A [VEGFA] and solute carrier family 2 [SLC2A1]), which is a facilitated glucose transporter. These genes were selected to validate the findings from RNA-seq, and their selection was based on previous literature that suggested their role in the pathogenesis of MPN and/or thrombosis and on our previous work indicating that HIF activity is upregulated in PV granulocytes, as demonstrated by increased glucose transport and augmented transcription of HIF-regulated genes.17,20,25-30
Measurement of JAK2V617F allelic burden.
The JAK2V617F allelic burden was measured with purified neutrophils by the method previously described.31 Unlike previously reported correlations of the influence of JAK2V617F allelic burden performed in whole blood, total leukocytes, or bone marrow cells, our analysis was more representative because we used purified neutrophils. The neutrophils represent an MPN clone unlike entire leukocytes or marrow cells which consist of a mixture of MPN clonal cells, polyclonal T cells, and non-MPN polyclonal cells. Correlation of dysregulated genes with JAK2V617F allelic burden was determined by Prism (GraphPad).
Results
Patient characteristics
This study included 58 patients, 39 with PV (38 JAK2V617F-mutation positive and 1 JAK2 exon 12–mutation positive) and 19 with ET (12 JAK2V617F-mutation positive, 5 calreticulin-mutation positive, and 2 with unmutated JAK2V617F, calreticulin, and MPL). The study group contained 29 females and 29 males. Of these, 28 patients had a history of thrombosis (16 females and 12 males). The characteristics of the patients with and without thrombosis are shown in Table 1. The mean age of study participants with thrombosis was 55.9 years (standard deviation, ± 18.5 years) and those without thrombosis was 65.6 years (standard deviation ± 12.1 years). There were 22 patients with venous thrombosis and 6 with arterial thrombotic events. Thrombotic events included abdominal vein thrombosis, stroke, myocardial infarction, deep vein thrombosis in lower extremities, pulmonary embolism, cerebral venous sinus thrombosis, or arterial clots in extremities. There was no significant difference in complete blood count for participants with or without thrombosis. However, patients with thrombosis had a higher JAK2V617F allelic burden compared with those without thrombosis (39.8 ± 37.3 vs 19.8 ± 20.9; P = .022) (Table 1).
Characteristics of patients with and without thrombosis
| Variable | N | With thrombosis (n = 28) | Without thrombosis (n = 30) | P | ||||
|---|---|---|---|---|---|---|---|---|
| n | Mean | SD | n | Mean | SD | |||
| Age, y | 55.9 | 18.5 | 65.6 | 12.1 | ||||
| Sex | ||||||||
| Male | 29 | 12 | 17 | |||||
| Female | 29 | 16 | 13 | |||||
| Diagnosis | ||||||||
| PV | 39 | 19 | 20 | |||||
| ET | 19 | 9 | 10 | |||||
| Laboratory tests | ||||||||
| White blood cell count, × 109/L | 10.4 | 9.3 | 6.9 | 3.3 | .076 | |||
| Hemoglobin, g/dL | 15.1 | 6.3 | 14.7 | 2.5 | .764 | |||
| Platelets, × 109/L | 648.0 | 597.8 | 470.4 | 330.6 | .173 | |||
| Patients with JAK2V617F mutation | 27 | 23 | ||||||
| JAK2V617F allelic burden | 50 | 39.8 | 37.3 | 19.8 | 20.9 | .022 | ||
| Thrombosis type | NA | |||||||
| Venous | 22 | |||||||
| Arterial | 6 | |||||||
| Medications* | ||||||||
| Aspirin | 3 | 19 | ||||||
| Warfarin | 14 | — | ||||||
| Rivaroxaban | 2 | — | ||||||
| Aspirin plus clopidogrel | 2 | — | ||||||
| Statins | 4 | 9 | ||||||
| NSAIDs | — | 2 | ||||||
| Hydroxyurea | 11 | 13 | ||||||
| Pegylated IFN-α | 9 | 7 | ||||||
| Ruxolitinib | 2 | 1 | ||||||
| Busulfan | 1 | — | ||||||
| Variable | N | With thrombosis (n = 28) | Without thrombosis (n = 30) | P | ||||
|---|---|---|---|---|---|---|---|---|
| n | Mean | SD | n | Mean | SD | |||
| Age, y | 55.9 | 18.5 | 65.6 | 12.1 | ||||
| Sex | ||||||||
| Male | 29 | 12 | 17 | |||||
| Female | 29 | 16 | 13 | |||||
| Diagnosis | ||||||||
| PV | 39 | 19 | 20 | |||||
| ET | 19 | 9 | 10 | |||||
| Laboratory tests | ||||||||
| White blood cell count, × 109/L | 10.4 | 9.3 | 6.9 | 3.3 | .076 | |||
| Hemoglobin, g/dL | 15.1 | 6.3 | 14.7 | 2.5 | .764 | |||
| Platelets, × 109/L | 648.0 | 597.8 | 470.4 | 330.6 | .173 | |||
| Patients with JAK2V617F mutation | 27 | 23 | ||||||
| JAK2V617F allelic burden | 50 | 39.8 | 37.3 | 19.8 | 20.9 | .022 | ||
| Thrombosis type | NA | |||||||
| Venous | 22 | |||||||
| Arterial | 6 | |||||||
| Medications* | ||||||||
| Aspirin | 3 | 19 | ||||||
| Warfarin | 14 | — | ||||||
| Rivaroxaban | 2 | — | ||||||
| Aspirin plus clopidogrel | 2 | — | ||||||
| Statins | 4 | 9 | ||||||
| NSAIDs | — | 2 | ||||||
| Hydroxyurea | 11 | 13 | ||||||
| Pegylated IFN-α | 9 | 7 | ||||||
| Ruxolitinib | 2 | 1 | ||||||
| Busulfan | 1 | — | ||||||
Common comorbidities for patients with thrombosis included portal hypertension, gastroesophageal reflux disease, and hypothyroidism; for patients without thrombosis, they included hypertension, gastroesophageal reflux disease, and diabetes.
NA, not applicable; NSAIDs, nonsteroidal anti-inflammatory drugs; SD, standard deviation.
Comorbidities and medications are reported at the time of sample collection.
All the study samples were collected during routine clinic visits, and none of the patients had any concurrent active inflammatory condition or infection or were receiving corticosteroids at the time of sample collection. Because our center is a tertiary referral center, some of the patients had documented thrombosis several years before our initial evaluation and some of them were receiving anticoagulation therapy at the time of sample collection. The median time from thrombosis to sample collection was about 2 years (interquartile range, 0.5-8.5 years). For those without thrombosis, the median time from diagnosis of MPN to sample collection was about 6 months (interquartile range, 0-3 years). At the time of sample collection, 24 patients were being treated with hydroxyurea, 16 with pegylated interferon α (IFN-α), 3 with ruxolitinib, and 1 with busulfan; 14 patients were not receiving any cytoreductive therapy (a majority of the patients were newly diagnosed). Twenty-two patients were taking aspirin, 14 were taking warfarin, 2 were receiving dual antiplatelet therapy with aspirin and clopidogrel, 2 were taking rivaroxaban, 13 were taking statins, and 2 were taking nonsteroidal anti-inflammatory drugs at the time of sample collection. Twenty-eight controls (19 females and 9 males) without a history of cancer or thrombosis were also tested in parallel.
Unbiased RNA-seq of PV granulocytes and platelets
Granulocyte analysis.
We performed unbiased RNA-seq in 22 PV patients and compared those patients with 10 controls. We confirmed high expression of granulocyte markers (FCGR3B, CEACAM4, EMR3, LRG1, and CEACAM8). A total of 23 500 genes were analyzed. In granulocytes, 1408 genes were upregulated and 425 genes were downregulated in PV patients (adjusted P < .05; log2 fold change >1) compared with controls. The heatmap showed that there were 2 groups in PV patients (Figure 1A). Those in group 1 had less JAK2V617F positivity (average, 26.0%) in granulocytes compared with those in group 2 (average, 58.3%) (P = .0069), except for 1 PV patient (PV13) with low JAK2V617F allelic burden (8.3%) who was grouped with normal controls. Differentially expressed genes were involved in coagulation such as formation of fibrin clot and platelet activation. They are also associated with immune response such as cytokine signaling and the innate immune system (Figure 2A).
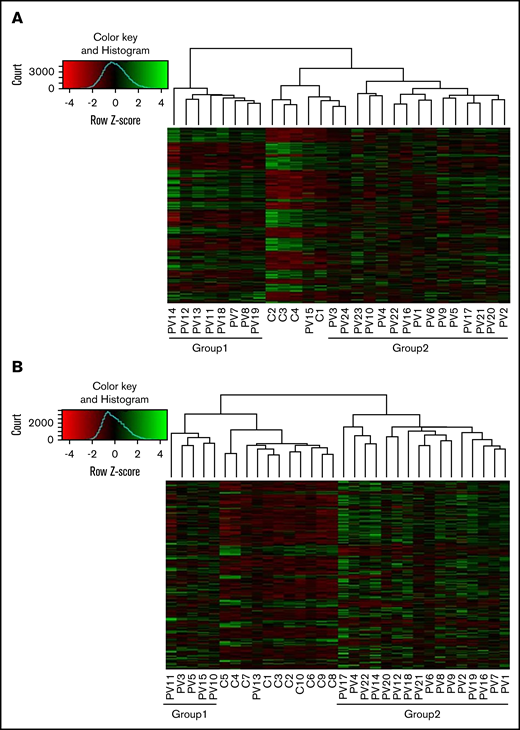
Differential expression of genes in granulocytes and platelets of PV patients compared with controls. Differentially expressed genes (adjusted P < .05; log2 fold change >1) in granulocytes (A) and platelets (B) of PV patients compared with controls were analyzed using the DEseq2 package. There were 2 groups in PV patients (group 1 and group 2). In granulocytes, patients in group 1 had less JAK2V617F positivity than those in group 2; whereas in platelets, there was no difference in JAK2V617F positivity, white blood cell counts, platelet counts, or type of treatment between group 1 and group 2.
Differential expression of genes in granulocytes and platelets of PV patients compared with controls. Differentially expressed genes (adjusted P < .05; log2 fold change >1) in granulocytes (A) and platelets (B) of PV patients compared with controls were analyzed using the DEseq2 package. There were 2 groups in PV patients (group 1 and group 2). In granulocytes, patients in group 1 had less JAK2V617F positivity than those in group 2; whereas in platelets, there was no difference in JAK2V617F positivity, white blood cell counts, platelet counts, or type of treatment between group 1 and group 2.

Pathway analysis of dysregulated genes in granulocytes and platelets of PV patients. Pathway enrichment analysis of dysregulated genes in granulocytes (A) and platelets (B) was performed by using the Reactome database. FCERI, Fc epsilon receptor; FDR, false discovery rate; LAB, linker for activation of B cells; LAT1, L-type amino acid transporter 1; MAPK, mitogen-activated protein kinase; MHC, major histocompatibility complex; NTAL, non–T cell activation linker.
Pathway analysis of dysregulated genes in granulocytes and platelets of PV patients. Pathway enrichment analysis of dysregulated genes in granulocytes (A) and platelets (B) was performed by using the Reactome database. FCERI, Fc epsilon receptor; FDR, false discovery rate; LAB, linker for activation of B cells; LAT1, L-type amino acid transporter 1; MAPK, mitogen-activated protein kinase; MHC, major histocompatibility complex; NTAL, non–T cell activation linker.
Expression of genes involved in thrombosis was analyzed, and among the 364 coagulation-related genes expressed in granulocytes, 49 were upregulated and 2 were downregulated in PV patients compared with controls (adjusted P < .05; log2 fold change >1) (Figure 3A). Among 49 upregulated genes, 13 were positively correlated with JAK2V617F allelic burden and total JAK2 expression levels in granulocytes (GNA15, THBD, NFE2, GNG5, KIF15, F8, SLC7A11, F5, MAPK14, CDK2, FCER1G, F3, MERTK), and 9 genes were inversely correlated (ANGPT1, PTK2, EGF, CLU, ITGB3, SPARC, GUCY1A3, ITGA2B, SERPINB2) with JAK2V617F allelic burden and total JAK2 expression levels (Table 2). Among the 2 downregulated genes, ANGPT1 inversely correlated with them (Table 2).

Dysregulated genes involved in thrombosis, HIF target genes, and inflammation in PV patients compared with controls by RNA-seq. Dysregulated thrombosis genes in PV granulocytes (A) and platelets (D). HIF-regulated genes in PV granulocytes (B) and platelets (E). Inflammatory genes in PV granulocytes (C) and platelets (F). Black dots represent genes that do not have significantly different expression levels compared with controls. Green dots are genes with log2 fold change >1 but not adjusted P (Adjp) < .05. Blue dots are genes with adjusted P < .05 but not log2 fold change >1. Genes labeled with red dots are differentially expressed genes with adjusted P < .05 and log2 fold change >1. F3, SELP, SERPINE1, VEGFA, and SLC2A1 were selected to validate expression levels by qRT-PCR (see Figure 5).
Dysregulated genes involved in thrombosis, HIF target genes, and inflammation in PV patients compared with controls by RNA-seq. Dysregulated thrombosis genes in PV granulocytes (A) and platelets (D). HIF-regulated genes in PV granulocytes (B) and platelets (E). Inflammatory genes in PV granulocytes (C) and platelets (F). Black dots represent genes that do not have significantly different expression levels compared with controls. Green dots are genes with log2 fold change >1 but not adjusted P (Adjp) < .05. Blue dots are genes with adjusted P < .05 but not log2 fold change >1. Genes labeled with red dots are differentially expressed genes with adjusted P < .05 and log2 fold change >1. F3, SELP, SERPINE1, VEGFA, and SLC2A1 were selected to validate expression levels by qRT-PCR (see Figure 5).
Correlation of JAK2V617F and JAK2 transcript expression levels with HIF target, inflammatory, and coagulation genes in granulocytes
| Gene | Correlation with: | |||
|---|---|---|---|---|
| JAK2V617F | JAK2 transcript levels | |||
| r | P | r | P | |
| HIF target genes | ||||
| CDKN1A | 0.4677 | .0282 | 0.5067 | .0161 |
| CXCL2 | 0.5705 | .0056 | 0.5348 | .0103 |
| EGLN3 | −0.4550 | .0382 | ||
| FHL1 | −0.6081 | .0027 | −0.5111 | .0151 |
| LDHA | 0.5357 | .0102 | 0.6786 | .0005 |
| NBN | 0.4272 | .0474 | ||
| NDRG1 | 0.6020 | .0030 | 0.5056 | .0164 |
| PKM | 0.5017 | .0174 | ||
| SLC2A1 | 0.6950 | .0003 | 0.6509 | .0010 |
| TNFSF15 | 0.5105 | .0181 | ||
| VEGFA | 0.4947 | .0192 | ||
| Inflammatory genes | ||||
| CXCL3 | 0.5861 | .0042 | 0.6316 | .0016 |
| F3 | 0.5081 | .0158 | 0.5640 | .0063 |
| HAMP | 0.5660 | .0060 | ||
| IGHG1 | −0.4737 | .0301 | −0.5388 | .0117 |
| IL10 | 0.5618 | .0065 | 0.6825 | .0005 |
| MMP9 | 0.6270 | .0023 | ||
| PTX3 | 0.4760 | .0339 | ||
| TNFRSF4 | −0.7270 | .0001 | −0.6069 | .0027 |
| Coagulation genes | ||||
| ANGPT1 | −0.5688 | .0057 | −0.4978 | .0216 |
| APP | 0.4263 | .0479 | ||
| CDK2 | 0.5665 | .0060 | 0.5898 | .0039 |
| CLU | −0.4887 | .0210 | −0.4739 | .0259 |
| EGF | −0.5395 | .0096 | −0.4684 | .0279 |
| F13A1 | −0.4607 | .0310 | ||
| F3 | 0.5081 | .0158 | 0.5640 | .0063 |
| F5 | 0.6487 | .0011 | 0.6751 | .0006 |
| F8 | 0.4598 | .0313 | 0.5052 | .0165 |
| FCER1G | 0.7437 | < .0001 | 0.7241 | .0001 |
| GATA2 | −0.5093 | .0184 | ||
| GNA15 | 0.6367 | .0014 | 0.5071 | .0160 |
| GNG5 | 0.5788 | .0048 | 0.7451 | < .0001 |
| GP1BA | −0.5750 | .0051 | ||
| GUCY1A3 | −0.5177 | .0136 | −0.4815 | .0233 |
| ITGA2B | −0.4914 | .0202 | −0.4977 | .0184 |
| ITGB3 | −0.5069 | .0161 | −0.4782 | .0244 |
| KIF15 | 0.6476 | .0011 | 0.7440 | < .0001 |
| MAPK14 | 0.6371 | .0014 | 0.5782 | .0048 |
| MERTK | 0.7959 | < .0001 | 0.8446 | < .0001 |
| MGLL | −0.5061 | .0192 | ||
| MMRN1 | −0.4321 | .0446 | ||
| NFE2 | 0.7565 | < .0001 | 0.8320 | < .0001 |
| PDE1B | 0.5020 | .0173 | ||
| PTK2 | −0.5562 | .0072 | −0.4388 | .0410 |
| SERPINB2 | −0.4330 | .0441 | −0.4723 | .0265 |
| SLC7A11 | 0.7042 | .0003 | 0.5832 | .0044 |
| SPARC | −0.6651 | .0007 | −0.5110 | .0151 |
| TFPI | −0.5057 | .0164 | ||
| THBD | 0.6760 | .0006 | 0.6175 | .0022 |
| VEGFA | 0.4947 | .0192 | ||
| Gene | Correlation with: | |||
|---|---|---|---|---|
| JAK2V617F | JAK2 transcript levels | |||
| r | P | r | P | |
| HIF target genes | ||||
| CDKN1A | 0.4677 | .0282 | 0.5067 | .0161 |
| CXCL2 | 0.5705 | .0056 | 0.5348 | .0103 |
| EGLN3 | −0.4550 | .0382 | ||
| FHL1 | −0.6081 | .0027 | −0.5111 | .0151 |
| LDHA | 0.5357 | .0102 | 0.6786 | .0005 |
| NBN | 0.4272 | .0474 | ||
| NDRG1 | 0.6020 | .0030 | 0.5056 | .0164 |
| PKM | 0.5017 | .0174 | ||
| SLC2A1 | 0.6950 | .0003 | 0.6509 | .0010 |
| TNFSF15 | 0.5105 | .0181 | ||
| VEGFA | 0.4947 | .0192 | ||
| Inflammatory genes | ||||
| CXCL3 | 0.5861 | .0042 | 0.6316 | .0016 |
| F3 | 0.5081 | .0158 | 0.5640 | .0063 |
| HAMP | 0.5660 | .0060 | ||
| IGHG1 | −0.4737 | .0301 | −0.5388 | .0117 |
| IL10 | 0.5618 | .0065 | 0.6825 | .0005 |
| MMP9 | 0.6270 | .0023 | ||
| PTX3 | 0.4760 | .0339 | ||
| TNFRSF4 | −0.7270 | .0001 | −0.6069 | .0027 |
| Coagulation genes | ||||
| ANGPT1 | −0.5688 | .0057 | −0.4978 | .0216 |
| APP | 0.4263 | .0479 | ||
| CDK2 | 0.5665 | .0060 | 0.5898 | .0039 |
| CLU | −0.4887 | .0210 | −0.4739 | .0259 |
| EGF | −0.5395 | .0096 | −0.4684 | .0279 |
| F13A1 | −0.4607 | .0310 | ||
| F3 | 0.5081 | .0158 | 0.5640 | .0063 |
| F5 | 0.6487 | .0011 | 0.6751 | .0006 |
| F8 | 0.4598 | .0313 | 0.5052 | .0165 |
| FCER1G | 0.7437 | < .0001 | 0.7241 | .0001 |
| GATA2 | −0.5093 | .0184 | ||
| GNA15 | 0.6367 | .0014 | 0.5071 | .0160 |
| GNG5 | 0.5788 | .0048 | 0.7451 | < .0001 |
| GP1BA | −0.5750 | .0051 | ||
| GUCY1A3 | −0.5177 | .0136 | −0.4815 | .0233 |
| ITGA2B | −0.4914 | .0202 | −0.4977 | .0184 |
| ITGB3 | −0.5069 | .0161 | −0.4782 | .0244 |
| KIF15 | 0.6476 | .0011 | 0.7440 | < .0001 |
| MAPK14 | 0.6371 | .0014 | 0.5782 | .0048 |
| MERTK | 0.7959 | < .0001 | 0.8446 | < .0001 |
| MGLL | −0.5061 | .0192 | ||
| MMRN1 | −0.4321 | .0446 | ||
| NFE2 | 0.7565 | < .0001 | 0.8320 | < .0001 |
| PDE1B | 0.5020 | .0173 | ||
| PTK2 | −0.5562 | .0072 | −0.4388 | .0410 |
| SERPINB2 | −0.4330 | .0441 | −0.4723 | .0265 |
| SLC7A11 | 0.7042 | .0003 | 0.5832 | .0044 |
| SPARC | −0.6651 | .0007 | −0.5110 | .0151 |
| TFPI | −0.5057 | .0164 | ||
| THBD | 0.6760 | .0006 | 0.6175 | .0022 |
| VEGFA | 0.4947 | .0192 | ||
We then analyzed the expression of 248 known HIF target genes and 90 inflammatory pathway genes. Among 248 HIF target genes, 16 (CDKN1A, CXCL2, LDHA, NDRG1, EDN1, PKM, SLC2A1, NBN, EGLN3, RET, SERPINE1, TNFSF15, VEGFA, TMEM45A, SLC7A5, and FHL1) were upregulated and 3 (CKB, SLC29A1, and FLT1) were downregulated in PV patients compared with controls (Figure 3B). Of the 16 upregulated genes, 8 (CDKN1A, CXCL2, LDHA, NDRG1, PKM, SLC2A1, TNFSF15, and VEGFA) were positively correlated with JAK2V617F allelic burden, and 2 (ELGN3 and FHL1) were inversely correlated with JAK2V617F allelic burden (Table 2). Among upregulated genes, 5 (CDKN1A, CXCL2, LDHA, NDRG1, SLC2A1) were also positively correlated with total JAK2 expression levels, and 1 (FHL1) was negatively correlated (Table 2). Among 90 inflammation-related genes, 10 (F3, IL10, CXCL3, IL2RA, IL15, PTX3, C4BP4, MMP9, CD80, SELP, and HAMP) were upregulated and 2 (TNFRSF4 and IGHG1) were downregulated (Figure 3C). Six (F3, IL10, CXCL3, PTX3, MMP9, and HAMP) of the 10 upregulated genes were positively correlated with JAK2V617F allelic burden, and 2 downregulated genes (TNFRSF4 and IGHG1) were inversely correlated (Table 2). Among the 6 genes that positively correlated with JAK2V617F allelic burden, 3 (IL10, F3, and CXCL3) were also positively correlated with total JAK2 transcript expression. Two downregulated genes were also inversely correlated with total JAK2 transcript expression (Table 2).
Among the 22 PV patients, 8 had a history of thrombosis. Seventy-two genes were upregulated and 25 genes were downregulated in granulocytes of PV patients with thrombosis (adjusted P < .05; log2 fold change >1) compared with those without thrombosis. Among these 72 upregulated genes, 48 were upregulated in patients with PV compared with controls. Only 4 of the 25 downregulated genes showed lower expression in controls compared with PV patients. Among 16 upregulated HIF target genes, 4 (CDKN1A, LDHA, NDRG1, and SLC2A1) were upregulated in those with thrombosis (Figure 4A). Thrombosis and inflammation-related genes (F3, IL10, and IL15) were upregulated in those with a history of thrombosis compared with those without thrombosis and controls (Figure 4B).

Differential expression of HIF-regulated genes and thrombo-inflammatory genes among PV patients with or without thrombosis by RNA-seq. Expression levels of 4 HIF target genes (A) and 3 inflammatory genes (B) in granulocytes from PV patients with or without thrombosis. (C) Expression levels of 2 inflammatory genes in platelets of PV patients with or without thrombosis. *P ≤ .05, **P ≤ .01, ***P ≤ .001.
Differential expression of HIF-regulated genes and thrombo-inflammatory genes among PV patients with or without thrombosis by RNA-seq. Expression levels of 4 HIF target genes (A) and 3 inflammatory genes (B) in granulocytes from PV patients with or without thrombosis. (C) Expression levels of 2 inflammatory genes in platelets of PV patients with or without thrombosis. *P ≤ .05, **P ≤ .01, ***P ≤ .001.
Platelet analysis.
Whole platelet transcriptomes from 24 PV patients and 4 controls were analyzed, and they showed upregulation of 1601 genes and downregulation of 823 genes in PV patients compared with controls (adjusted P < .05; log2 fold change >1). In an independent analysis, we confirmed high expression of platelet markers (ITGB3, PDGFRA, MYLP, PF4V1, and PF4) in whole transcriptome data. Two groups of PV patients are shown on the heat map in Figure 1B. However, there was no significant difference in JAK2V617F positivity between the groups, nor was there a difference in white blood cell count, platelet count, or type of treatment. Differentially expressed genes in PV were associated with hemostasis, the innate immune system, cell surface interactions at the vascular wall, and antibody-mediated complement activation (Figure 2B).
In platelets, 63 coagulation-related genes were upregulated in PV patients compared with controls (Figure 3D). Among those 63 genes, 14 were positively correlated with JAK2V617F allelic burden. Forty-three of the 63 upregulated genes were positively correlated with total JAK2 expression levels in platelets, and only 3 genes (ATP1B1, PIK3R3, SERPING1) were positively correlated with both JAK2V617F allelic burden and total JAK2 transcript levels (Table 3)
Correlation of JAK2V617F and JAK2 transcript expression levels to HIF target, inflammatory, and coagulation genes in platelets
| Gene | Correlation with: | |||
|---|---|---|---|---|
| JAK2V617F | JAK2 transcript levels | |||
| r | P | r | P | |
| HIF target genes | ||||
| ADM | 0.5488 | .0082 | ||
| BHLHE40 | –0.5425 | .0075 | ||
| CDKN1A | 0.6067 | .0021 | ||
| CKB | 0.4378 | .0471 | 0.5833 | .0035 |
| CXCL2 | 0.6447 | .0012 | ||
| DDIT4 | –0.5729 | .0043 | ||
| ENO1 | 0.6625 | .0006 | ||
| GAPDH | 0.6091 | .0020 | ||
| KDR | 0.6586 | .0006 | ||
| NFIL3 | 0.6042 | .0029 | ||
| PLAUR | 0.4884 | .0211 | ||
| TEK | 0.6942 | .0002 | ||
| TIMP1 | 0.8378 | <.0001 | ||
| Inflammatory gene | ||||
| BCL2 | –0.4535 | .0297 | ||
| CCL28 | −0.4923 | .0170 | ||
| CHI3L1 | –0.5119 | .0106 | ||
| CXCL1 | 0.4426 | .0344 | ||
| IGKC | −0.5322 | .0108 | ||
| MMP9 | 0.4337 | .0437 | –0.5553 | .0049 |
| PGLYRP1 | 0.4499 | .0312 | –0.5586 | .0046 |
| PLAU | 0.4893 | .0178 | ||
| TNF | –0.6267 | .0011 | ||
| Coagulation genes | ||||
| ALDOA | 0.6753 | .0003 | ||
| ATP1B1 | 0.5347 | .0104 | 0.6105 | .0020 |
| ATP2A3 | 0.6187 | .0013 | ||
| BSG | 0.6532 | .0005 | ||
| CAV1 | 0.7983 | <.0001 | ||
| CD63 | 0.6133 | .0014 | ||
| CFL1 | 0.7678 | <.0001 | ||
| DOCK4 | 0.4455 | .0331 | ||
| DOCK5 | 0.4794 | .0178 | ||
| EHD2 | 0.7901 | <.0001 | ||
| F13B | 0.6143 | .0014 | ||
| FCER1G | 0.8390 | <.0001 | ||
| GATA1 | 0.7489 | <.0001 | ||
| GNA15 | 0.4505 | .0272 | ||
| GNAI2 | 0.4357 | .0333 | ||
| GNG5 | 0.5925 | .0029 | ||
| GP1BB | 0.7395 | <.0001 | ||
| GP9 | 0.7901 | <.0001 | ||
| HBE1 | 0.6971 | .0002 | ||
| HBG2 | 0.4964 | .0136 | ||
| ITGA2B | 0.7519 | <.0001 | ||
| ITGAX | 0.5199 | .0110 | ||
| ITGB3 | 0.7678 | <.0001 | ||
| KCNMB1 | 0.7445 | <.0001 | ||
| KIF3C | 0.7617 | <.0001 | ||
| KIF4A | 0.6389 | .0008 | ||
| KIFAP3 | 0.9091 | <.0001 | ||
| LCP2 | 0.6407 | .0010 | ||
| MMP1 | 0.7059 | .0001 | ||
| MRVI1 | 0.8298 | <.0001 | ||
| P2RX1 | 0.7452 | <.0001 | ||
| PDE2A | 0.8370 | <.0001 | ||
| PIK3R2 | 0.5299 | .0112 | ||
| PIK3R3 | 0.8281 | <.0001 | ||
| PLAU | 0.4893 | .0178 | ||
| PLAUR | 0.4884 | .0211 | ||
| PRKAR1B | 0.6883 | .0002 | ||
| PRKCD | 0.5719 | .0035 | ||
| PRKG2 | 0.5898 | .0031 | 0.5284 | .0079 |
| PSAP | 0.4932 | .0143 | ||
| PTGIR | 0.7563 | <.0001 | ||
| SERPINA1 | 0.5603 | .0067 | ||
| SERPING1 | 0.3986 | .0596 | 0.4405 | .0312 |
| SHC1 | 0.6179 | .0013 | ||
| SIRPA | 0.5049 | .0140 | ||
| SLC16A3 | 0.4563 | .0328 | ||
| SLC7A11 | 0.6874 | .0002 | ||
| SOS1 | 0.8916 | <.0001 | ||
| TEK | 0.6790 | .0003 | ||
| TGFB1 | 0.5578 | .0046 | ||
| TIMP1 | 0.8465 | <.0001 | ||
| TREM1 | 0.5070 | .0160 | ||
| VEGFC | 0.8644 | <.0001 | ||
| ZFPM2 | 0.8947 | <.0001 | ||
| Gene | Correlation with: | |||
|---|---|---|---|---|
| JAK2V617F | JAK2 transcript levels | |||
| r | P | r | P | |
| HIF target genes | ||||
| ADM | 0.5488 | .0082 | ||
| BHLHE40 | –0.5425 | .0075 | ||
| CDKN1A | 0.6067 | .0021 | ||
| CKB | 0.4378 | .0471 | 0.5833 | .0035 |
| CXCL2 | 0.6447 | .0012 | ||
| DDIT4 | –0.5729 | .0043 | ||
| ENO1 | 0.6625 | .0006 | ||
| GAPDH | 0.6091 | .0020 | ||
| KDR | 0.6586 | .0006 | ||
| NFIL3 | 0.6042 | .0029 | ||
| PLAUR | 0.4884 | .0211 | ||
| TEK | 0.6942 | .0002 | ||
| TIMP1 | 0.8378 | <.0001 | ||
| Inflammatory gene | ||||
| BCL2 | –0.4535 | .0297 | ||
| CCL28 | −0.4923 | .0170 | ||
| CHI3L1 | –0.5119 | .0106 | ||
| CXCL1 | 0.4426 | .0344 | ||
| IGKC | −0.5322 | .0108 | ||
| MMP9 | 0.4337 | .0437 | –0.5553 | .0049 |
| PGLYRP1 | 0.4499 | .0312 | –0.5586 | .0046 |
| PLAU | 0.4893 | .0178 | ||
| TNF | –0.6267 | .0011 | ||
| Coagulation genes | ||||
| ALDOA | 0.6753 | .0003 | ||
| ATP1B1 | 0.5347 | .0104 | 0.6105 | .0020 |
| ATP2A3 | 0.6187 | .0013 | ||
| BSG | 0.6532 | .0005 | ||
| CAV1 | 0.7983 | <.0001 | ||
| CD63 | 0.6133 | .0014 | ||
| CFL1 | 0.7678 | <.0001 | ||
| DOCK4 | 0.4455 | .0331 | ||
| DOCK5 | 0.4794 | .0178 | ||
| EHD2 | 0.7901 | <.0001 | ||
| F13B | 0.6143 | .0014 | ||
| FCER1G | 0.8390 | <.0001 | ||
| GATA1 | 0.7489 | <.0001 | ||
| GNA15 | 0.4505 | .0272 | ||
| GNAI2 | 0.4357 | .0333 | ||
| GNG5 | 0.5925 | .0029 | ||
| GP1BB | 0.7395 | <.0001 | ||
| GP9 | 0.7901 | <.0001 | ||
| HBE1 | 0.6971 | .0002 | ||
| HBG2 | 0.4964 | .0136 | ||
| ITGA2B | 0.7519 | <.0001 | ||
| ITGAX | 0.5199 | .0110 | ||
| ITGB3 | 0.7678 | <.0001 | ||
| KCNMB1 | 0.7445 | <.0001 | ||
| KIF3C | 0.7617 | <.0001 | ||
| KIF4A | 0.6389 | .0008 | ||
| KIFAP3 | 0.9091 | <.0001 | ||
| LCP2 | 0.6407 | .0010 | ||
| MMP1 | 0.7059 | .0001 | ||
| MRVI1 | 0.8298 | <.0001 | ||
| P2RX1 | 0.7452 | <.0001 | ||
| PDE2A | 0.8370 | <.0001 | ||
| PIK3R2 | 0.5299 | .0112 | ||
| PIK3R3 | 0.8281 | <.0001 | ||
| PLAU | 0.4893 | .0178 | ||
| PLAUR | 0.4884 | .0211 | ||
| PRKAR1B | 0.6883 | .0002 | ||
| PRKCD | 0.5719 | .0035 | ||
| PRKG2 | 0.5898 | .0031 | 0.5284 | .0079 |
| PSAP | 0.4932 | .0143 | ||
| PTGIR | 0.7563 | <.0001 | ||
| SERPINA1 | 0.5603 | .0067 | ||
| SERPING1 | 0.3986 | .0596 | 0.4405 | .0312 |
| SHC1 | 0.6179 | .0013 | ||
| SIRPA | 0.5049 | .0140 | ||
| SLC16A3 | 0.4563 | .0328 | ||
| SLC7A11 | 0.6874 | .0002 | ||
| SOS1 | 0.8916 | <.0001 | ||
| TEK | 0.6790 | .0003 | ||
| TGFB1 | 0.5578 | .0046 | ||
| TIMP1 | 0.8465 | <.0001 | ||
| TREM1 | 0.5070 | .0160 | ||
| VEGFC | 0.8644 | <.0001 | ||
| ZFPM2 | 0.8947 | <.0001 | ||
Fourteen HIF-regulated genes (KDR, TEK, CKB, ADM, TIMP1, SLC29A1, CXCL2, PLAUR, NFIL3, CDKN1A, GAPDH, ENO1, BHLHE40, and DDIT4) were highly expressed in platelets of PV patients compared with controls (adjusted P < .05; log2 fold-change >1) (Figure 3E). Five of the 14 genes (CKB, ADM, CXCL2, PLAUR, and NFIL3) were positively correlated with JAK2V617F allelic burden, and CKB was positively correlated with total JAK2 expression as well. (Table 3). Eight inflammation-related genes (CCL2, PLAU, IL1RN, CXCL1, CHI3L1, PGLYRP1, MMP9, and TNF) were also upregulated in PV (Figure 3F). Of these, 4 genes (PGLYRP1, PLAU, CXCL1, and MMP9) were positively correlated with JAK2V617F allelic burden, and 2 (PGLYRP1 and MMP9) were inversely correlated with total JAK2 transcript levels in platelets (Table 3). Four inflammation-related genes (IGHG1, CCL28, BCL2, and IGKC) were downregulated in PV (Figure 3F), and CCL28 and IGKC levels were inversely correlated with JAK2V617F allelic burden (Table 3).
We also compared differential gene expression between patients with thrombosis (n = 7) and those without (n = 17). Only 3 genes, including DTNA (dystrobrevin), PCSK1 (proprotein convertase subtilisin/kexin type 1), and LAMA2 (laminin α2) were upregulated in platelets of PV patients with thrombosis compared with platelets in patients without thrombosis. Among these 3 genes, only LAMA2 had higher expression levels in PV patients compared with controls. Expression of inflammation genes CHI3L1 and CCL28 was downregulated in platelets of PV patients with thrombosis compared with those without (Figure 4C). Expression of SELP, THBS1, and VEGFA (P < .05) was increased in PV patients with thrombosis compared with controls.
Quantitative analysis of thrombotic, inflammatory, and HIF-regulated genes in patients with PV or ET
To verify the quantitative changes of expression and to extend the findings from RNA-seq to a larger number of patients, we recruited additional patients (58 total; 39 with PV and 19 with ET) and 28 healthy volunteers. Of these, 28 patients had a history of thrombosis. Only 9 PV patients analyzed by RNA-seq were included in the qRT-PCR analysis, and the remainder were newly recruited. We analyzed selected genes in thrombo-inflammatory and HIF pathways (F3, SELP, SERPINE1, THBS1, IRAK1, IL1RAP, VEGFA, and SLC2A1) by using qRT-PCR. F3 and SLC2A1 were selected because RNA-seq showed increased expression in granulocytes of PV patients compared with those of controls; this expression was higher in those with thrombosis compared with those without (F3, P < .01; SLC2A1, P < .05). In addition, the expression of SELP (P < .05), SERPINE1 (P < .05), and VEGFA (P < .01) was higher in PV patients with thrombosis compared with controls. Tissue factor and VEGFA were previously shown to have a role in angiogenesis and thrombosis in gastrointestinal and hematologic cancers.32,33 THBS1, IRAK1, and IL1RAP were selected because previous literature suggested their role in MPN pathogenesis.34-36 Targeted therapies were also under development for some of these proteins.
Granulocyte analysis.
In granulocytes, the relative expression levels of all the selected genes listed above was higher in patients with PV or ET compared with normal controls (Figure 5A). In patients with thrombosis, the expression of F3 and VEGFA was increased ∼13-fold, and the expression of SERPINE1 was increased ∼11-fold over that in controls. The expression of SELP, THBS1, IRAK1, IL1RAP, and SLC2A1 were significantly increased three- to fivefold. The expression levels of F3, SELP, VEGFA, and SLC2A1 were significantly higher in patients with a history of thrombosis compared with those without thrombosis (Figure 5A). Compared with JAK2-negative patients (n = 7), SELP, IRAK1, IL1RAP, VEGFA, SLC2A1, THBS1, and SERPINE1 were significantly upregulated in JAK2-positive patients (n = 51) (Figure 6A).
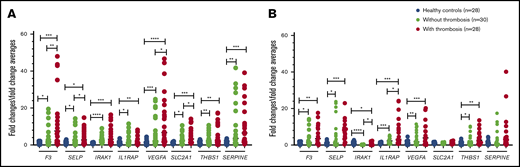
Relative expression levels of the tested genes in granulocytes and platelets of patients with PV or ET by qRT-PCR. Targeted genes expression levels were measured in granulocytes (A) and platelets (B). *P < .05; **P < .01; ***P < .001; ****P < .0001. F3, tissue factor; IRAK1, interleukin-1 receptor-associated kinase 1; IL1RAP, interleukin-1 receptor accessory protein; SELP, P-selectin; SERPINE1, serpin peptidase inhibitor clade E member 1; SLC2A1, solute carrier family 2; THBS1, thrombospondin 1; VEGFA, vascular endothelial growth factor A.
Relative expression levels of the tested genes in granulocytes and platelets of patients with PV or ET by qRT-PCR. Targeted genes expression levels were measured in granulocytes (A) and platelets (B). *P < .05; **P < .01; ***P < .001; ****P < .0001. F3, tissue factor; IRAK1, interleukin-1 receptor-associated kinase 1; IL1RAP, interleukin-1 receptor accessory protein; SELP, P-selectin; SERPINE1, serpin peptidase inhibitor clade E member 1; SLC2A1, solute carrier family 2; THBS1, thrombospondin 1; VEGFA, vascular endothelial growth factor A.

Relative expression levels of the tested genes in JAK2-mutated and JAK2-nonmutated patients by qRT-PCR. Relative gene expression levels were measured in granulocytes (A) and platelets (B) of patients with and without JAK2 mutation. *P ≤ .05, **P ≤ .01, ***P ≤ .001. Cont, healthy controls (n = 28); Neg, patients without JAK2 mutation (n = 7); Pos, patients with JAK2 mutation (n = 51; 1 patient with JAK2 exon 12 mutation).
Relative expression levels of the tested genes in JAK2-mutated and JAK2-nonmutated patients by qRT-PCR. Relative gene expression levels were measured in granulocytes (A) and platelets (B) of patients with and without JAK2 mutation. *P ≤ .05, **P ≤ .01, ***P ≤ .001. Cont, healthy controls (n = 28); Neg, patients without JAK2 mutation (n = 7); Pos, patients with JAK2 mutation (n = 51; 1 patient with JAK2 exon 12 mutation).
Platelet analysis.
In platelets, expression levels of F3, SELP, THBS1, IL1RAP, and VEGFA were higher in patients with PV or ET, with or without thrombosis compared with controls (P < .05 to P < .001). IRAK1 expression was lower in patients with PV or ET compared with controls (P < .05 in those with thrombosis, and P < .0001 in those without thrombosis); however, patients with thrombosis had higher expression compared with those without (P < .05). IL1RAP expression was significantly higher in patients with a history of thrombosis compared with those without (P < .05) (Figure 5B). In the JAK2-positive group, F3 and SELP were expressed more in comparison with the JAK2-negative group (Figure 6B).
To further validate the RNA sequencing findings, we measured the expression levels of 2 upregulated inflammatory genes (IL10 and IL15) and HIF target genes (EDN1 and LDHA) by qRT-PCR in granulocytes from 58 patients with PV or ET and 28 controls. The results showed that PV patients had higher expression of inflammation-associated genes and HIF-target genes compared with controls. We also validated the increased expression levels of 2 upregulated inflammatory genes (PLAU and TNF) and HIF target genes (SLC29A1 and CKB) by qRT-PCR in platelets from 46 patients with PV or ET and 21 controls (data not shown).
Expression levels of thrombotic, inflammatory, and HIF-regulated genes on the basis of type of treatment.
We compared the expression levels of the above selected genes by qRT-PCR in granulocytes and platelets from newly diagnosed untreated patients and in patients who were treated with hydroxyurea and IFN-α. There was no significant difference in the expression of these genes between the hydroxyurea-treated group and the IFN-α–treated group except for IRAK1, which has lower expression in the IFN-α–treated group compared with hydroxyurea-treated group in granulocytes (Figure 7). Ruxolitinib is known to decrease inflammation, but the small number of ruxolitinib-treated patients (n = 3) precluded any meaningful analysis.
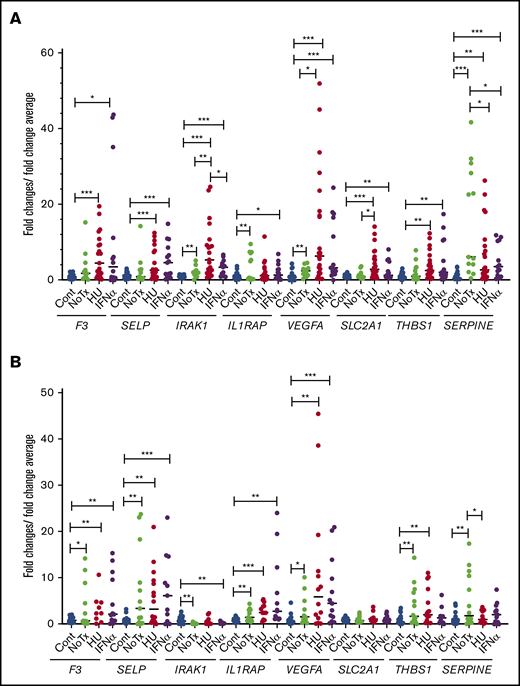
Relative expression levels of the tested genes in patients treated with hydroxyurea and pegylated IFN-α. Relative gene expression levels were determined by qRT-PCR in granulocytes (A) and platelets (B) of controls and patients. *P < .05; **P < .01; ***P < .001. Cont, controls (n = 28); HU, treated with hydroxyurea (n = 24); IFN-α, treated with pegylated interferon α (n = 16); NoTx, no treatment (n = 14).
Relative expression levels of the tested genes in patients treated with hydroxyurea and pegylated IFN-α. Relative gene expression levels were determined by qRT-PCR in granulocytes (A) and platelets (B) of controls and patients. *P < .05; **P < .01; ***P < .001. Cont, controls (n = 28); HU, treated with hydroxyurea (n = 24); IFN-α, treated with pegylated interferon α (n = 16); NoTx, no treatment (n = 14).
Discussion
We found increased expression levels of several thrombo-inflammatory and HIF-regulated genes in granulocytes and platelets of patients with PV or ET, especially in those with a history of thrombosis. The transcript levels of these genes differed in granulocytes and platelets, reflecting the fact that their transcriptional regulation is cell specific. RNA-seq findings were validated by increased transcript levels of selected genes by qRT-PCR. Our data further underline the presence of a thrombo-inflammatory state in MPNs, with several target genes identified by both RNA-seq and qRT-PCR: F3, SELP, SERPINE1, THBS1, VEGFA, SLC2A1, IL10, IL15, EDN1, LDHA, PLAU, and TNF. The expression of F3, IL10, VEGFA, LDHA, and SLC2A1 in granulocytes positively correlated with JAK2V617F allelic burden. In addition, some of the genes identified by RNA-seq correlated with total JAK2 transcript levels in granulocytes and platelets. Expression of F3, SELP, VEGFA, IL10, LDHA, and SLC2A1 was higher in patients with thrombosis compared with those without. Patients with JAK2 mutation had higher expression levels of several of these genes compared with JAK2 mutation–negative patients.
Tissue factor is a principal initiator of coagulation, and we found increased expression in granulocytes of PV patients by RNA-seq. High levels of tissue factor transcripts were seen in granulocytes and platelets of patients with PV or ET by qRT-PCR. In granulocytes, this expression was significantly higher in patients with a history of thrombosis compared with those with no history of thrombosis. SELP encodes P-selectin, which is an adhesion molecule on the surface of activated endothelial cells and platelets. It is a marker of platelet activation and facilitates adhesion of leukocytes to endothelium and platelets. Increased P-selectin levels are implicated in cardiovascular diseases.25 We found that the transcripts of SELP are increased in granulocytes and platelets of patients with PV or ET and are significantly higher in granulocytes of patients with thrombosis. SELP is known to be expressed in platelets, and we also showed SELP expression in granulocytes by both RNA-seq and qRT-PCR. MPN patients with thrombosis have a higher concentration of platelet-granulocyte and platelet-granulocyte-monocyte aggregates.37 Hence, to exclude contamination in granulocytes, we confirmed that granulocytes isolated by density gradient had little contamination (<1% of platelets), as verified by their morphologic evaluation. The amount of RNA in granulocytes is ∼293 times more than the amount in platelets.38 Thus, the observation of SELP expression in granulocytes suggests the presence of a low transcript of this gene in granulocytes, and our data indicate that in patients with PV, this low expression is about fourfold greater than in controls. A previous study also reported SELP expression in neutrophils on RNA-seq.39 Vascular endothelial expression of JAK2V617F in MPN patients promotes a prothrombotic state because of increased expression of P-selectin.15 Furthermore, P-selectin blockade and treatment with hydroxyurea reduced the increased propensity toward thrombosis. Guadall et al40 showed that JAK2V617F-positive endothelial cells were characterized by overexpression of both von Willebrand factor and P-selectin and increased expression of genes associated with inflammation and cell adhesion. Previous studies have shown that monocytes are the principal source of tissue factor in healthy participants.41 However, platelet surface tissue factor and P-selectin were found to be higher in patients with PV or ET compared with controls and increased with JAK2V617F allelic burden.42 We showed increased tissue factor expression in granulocytes from PV patients by RNA-seq and in granulocytes and platelets from patients with PV or ET by qRT-PCR. It is possible that granulocytes may transcribe tissue factor in patients with PV or ET, unlike normal controls.
SERPINE1 encodes PAI-1, increased levels of which are associated with prothrombotic conditions such as diabetes, hypertension, obesity, and cerebrovascular disorders.30 Thrombospondin-1, which is encoded by THBS1, mediates cell-to-cell and cell-to-matrix interactions. It also plays a role in platelet aggregation and arterial thrombosis and contributes to vaso-occlusive complications, pulmonary vascular remodeling, and vasoconstriction.43 HIF-2α is implicated in pulmonary regulation of THBS1,44 and THBS1 is upregulated in peripheral blood mononuclear cells and granulocytes in patients with CP, particularly those with thrombosis.43 SERPINE1 and THBS1 dysregulation have been reported in PV patients by using oligonucleotide microarray technology.36 We found that SERPINE1 was differentially expressed in PV granulocytes on RNA-seq. There were increased transcripts of SERPINE1 and THBS1 in granulocytes, and THBS1 in platelets of patients with PV or ET when compared with controls.
Leukocytosis is an independent risk factor for thrombosis in MPN patients, and 1 aim of cytoreduction in MPN patients is to keep the white cell count within normal range. Inflammation has been clearly linked to atherosclerosis and venous thromboembolism,45 and anti-inflammatory therapy targeting the interleukin-1β innate immunity pathway leads to a lower rate of recurrent cardiovascular events.46 Inflammation and activation of leukocytes have been shown to play a major role in constitutional symptoms of MPN patients and may link this association to atherosclerosis and thrombotic risk.47-49 Indeed, the rate of major thromboses was higher in PV and ET patients with elevated levels of high sensitivity C-reactive protein, and it correlated with JAK2V617F allelic burden.50 Whole transcriptome analysis of granulocytes and platelets showed differentially expressed genes in PV patients compared with controls. These genes were involved in inflammatory response pathways such as cytokine signaling, innate immune system, signaling by interleukins, coagulation, and platelet activation, which further support the presence of a thrombo-inflammatory state in MPNs. A previous study showed a distinct gene expression signature with upregulation of JAK-STAT targeted genes in granulocytes of MPN patients,51 and our analysis focused on thrombosis, inflammation, and HIF pathways.
The NF-κB pathway, which has a key role in inflammation and promoting tumor development, cross-talks with HIFs and results in selective upregulation of proinflammatory and prothrombotic mediators of atherosclerosis, as well as in mutual upregulation with HIF-1.28,52 NF-κB pathway activation is implicated in the release of cytokines in primary myelofibrosis,53 and we hypothesized that it may provide the link between chronic inflammation and atherosclerosis, as well as with hypoxia in MPN patients. We studied 2 genes in this pathway. The first, IRAK1, is a key mediator of the toll-like receptor signaling pathway and activates the PI3K/AKT pathway, which plays a major role in platelet activation, thrombosis, and atherosclerosis.54,55 The second, IL1RAP, is a subunit of the IL-1 type 1 receptor that contributes to the affinity of the receptor for IL-1 and is required for IL-1 signal transduction. It is overexpressed in hematopoietic stem cells and granulocyte-monocyte progenitors of patients with myeloid neoplasms such as acute myeloid leukemia, myelodysplastic syndrome, and chronic myeloid leukemia.34,35 We found that both IRAK1 and IL1RAP transcripts are increased in granulocytes of patients with PV or ET. IRAK1 transcripts are lower in platelets, whereas IL1RAP is higher compared with controls. Patients with thrombosis have higher IL1RAP expression in platelets compared those without thrombosis. IL10 and IL15 were upregulated by RNA-seq and qRT-PCR, and their expression is higher in patients with thrombosis compared with those without. Studies confirming inflammation in thrombosis in patients with PV or ET may play a role in designing prospective studies that evaluate the potential role of statins and anti-inflammatory agents specifically directed at interleukins, IRAK1 and IL1RAP. C-reactive protein and fibrinogen play a major role in thrombosis. RNA-seq data showed that C-reactive protein and FGB and FBA (which encode fibrinogen) were not expressed in granulocytes and platelets of either controls or MPN patients. These clinical parameters could not be obtained in our patients because these tests were not performed in routine clinical practice, and plasma samples were not available for this type of testing.
Hypoxia in the cells within a venous thrombosis results in HIF-mediated increase in the production of angiogenic targets such as VEGF. This enhances neovascularization within the thrombus, vein recanalization, and thrombus resolution.56 Our group has previously shown that living at a high altitude and the associated hypoxia increase the risk of thrombotic complications in PV patients, likely mediated by the hypoxia-driven upregulation of HIF.57 Our previous work indicated that HIF activity is upregulated in PV granulocytes as demonstrated by increased glucose transport and augmented transcription of HIF-regulated genes.20 Furthermore, in CP, which is characterized by congenital upregulation of hypoxia sensing, the rate of arterial and venous thromboses is higher than in PV.17 A commonly encountered situation of intermittent hypoxia is seen with obstructive sleep apnea, which is associated with a prothrombotic state. This is related to hypoxia secondary to changes in airflow during sleep.58 Resolution of platelet function abnormalities after therapy for obstructive sleep apnea underscores the role of hypoxia-driven HIF pathways in the mediation of coagulation abnormalities.59 High HIF activity in CP induces VEGF and PAI-1 levels which are also associated with high rates of thrombosis.17,60 Whole transcriptome analysis of platelets and granulocytes showed that several HIF target genes were upregulated in PV patients. Specifically, we found increased transcripts of HIF regulated genes VEGFA, SLC2A1, and SERPINE1 in granulocytes of patients with PV or ET by RNA-seq and qRT-PCR, which were significantly higher in those with thrombosis. VEGFA expression was also higher in platelets in both these analyses. Hypoxia reduces tissue factor pathway inhibitor messenger RNA and protein levels and increases tissue factor messenger RNA in a dose-dependent manner in endothelial cells, which demonstrates that tissue factor expression is also HIF regulated.61 In breast cancer cells, HIF-2α/endothelial PAS domain-containing protein 1 (EPAS1) is involved in the regulation of tissue factor pathway inhibitor gene expression. Activation of coagulation and the thrombotic risk observed in breast cancer patients may correlate with local hypoxic regulation of coagulation factors and their inhibitors.62
We evaluated the effect of treatment with hydroxyurea and IFN-α on gene expression levels. There was no significant difference between these 2 treatments with the exception of IRAK1 level, which decreased after treatment with IFN-α in granulocytes. We previously showed that treatment with IFN-α reduces tumor necrosis factor α expression levels,63 which is 1 of the target genes of NF-κB,64 and IRAK1 regulates the NF-κB signaling pathway.65 These data are supported by the multicenter phase 3 trials66,67 and by our previous study,63 which failed to confirm the expected superiority of IFN-α over hydroxyurea. Anti-inflammatory agents, aspirin, and statin treatment may change these gene transcript levels. However, medication history was obtained at the time of sample collection, and thrombosis might have occurred before starting these medications in some of our study patients. Ruxolitinib decreases inflammation, but the small number of ruxolitinib-treated patients precluded meaningful analysis.68
We acknowledge some limitations of our study. The length of time between the initial diagnosis, thrombotic episode, and sample collection varied greatly among the patients, which may affect the expression profile of certain genes. Some patients included in our study were referred to our center several years after MPN diagnosis. It is possible that the levels of several markers included in our study fluctuated with clinical course of their disease, and serial measurement of these markers may provide additional information. However, increased expression of inflammatory and HIF-regulated genes in patients with thrombosis, despite the different length of time to sample collection, lends weight to the fact that pro-inflammatory and pro-coagulant states in patients with thrombosis may be long-term. Although age is a known risk factor for thrombosis, the average age of our patients with thrombosis is lower than those without, reflecting the fact that younger patients with MPN and thrombosis were more likely to be referred to our specialized MPN clinic by other hematologists. MPN may also increase the risk of developing thrombosis at younger age.
In conclusion, the expression of several thrombo-inflammatory and HIF-regulated genes is increased in granulocytes and platelets in patients with PV or ET, which may play a role in increased thrombotic risk. Whether the increased thrombosis in MPNs is primarily from augmented HIF signaling, increased inflammatory milieu, or the direct contribution of augmented JAK2-STAT signaling was not resolved by our study. It is possible that several of these pathways may interact and contribute to the risk of thrombosis in MPNs. Of particular interest is the high expression of F3, SELP, and VEGFA because of the growing evidence of their role in thrombosis and the availability of targeted agents. However, the findings of increased transcripts of several putative prothrombotic genes need to be extended to the protein levels, and even more importantly, to the functional analyses of these genes, which may lead to the identification of possible therapeutic targets in the future.
For information regarding original data, please contact Jihyun Song (jihyun.song@utah.edu) and Tsewang Tashi (tsewang.tashi@utah.edu).
Acknowledgments
This work was supported by a grant from the National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, under Ruth L. Kirschstein National Research Service Awards (5T32DK007115) (R.G. and J.T.P.), from the National Heart, Lung, and Blood Institute (2T32HL007576-31) (J.S.), by a Joseph M. Quagliana and Paula Quagliana fellow research award (R.G.), and by the VA Merit Review Award (J.T.P.).
Authorship
Contribution: R.G., P.T., J.S., and J.T.P. contributed to the study design; R.G. and S.J.K. performed qRT-PCR and analyzed data; R.G., J.T.P., and T.T. collected and critically analyzed clinical data; S.J.K. and J.S. performed RNA sequencing; J.S. analyzed RNA sequencing data; R.G., J.S., and J.T.P. wrote the manuscript; and B.N.R., P.T., K.M.S., R.G., J.S., S.J.K., T.T., and J.T.P. critically revised the manuscript for important intellectual content and approved the final manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Radhika Gangaraju, Department of Medicine, University of Alabama at Birmingham, 1600 7th Ave South, Lowder 500, Birmingham, AL 35233; e-mail: rgangaraju@uabmc.edu; and Josef T. Prchal, Hematology, SOM 5C310, University of Utah School of Medicine, Salt Lake City, UT 84132-2408; e-mail: josef.prchal@hsc.utah.edu.

