

Key Points
Loncastuximab tesirine demonstrated an acceptable safety and tolerability profile in adults with R/R B-cell ALL.
Loncastuximab tesirine showed modest efficacy, with 3 complete responses in a heavily pretreated R/R B-cell ALL population.

Abstract
Relapsed or refractory (R/R) B-cell acute lymphoblastic leukemia (B-ALL) remains a therapeutic challenge. Loncastuximab tesirine is an antibody-drug conjugate against CD19, an antigen expressed in many B-cell malignancies. This open-label, single-arm, dose-escalation, dose-expansion study assessed the safety, tolerability, pharmacokinetics (PKs), immunogenicity, and preliminary clinical activity of loncastuximab tesirine in adults with R/R B-ALL. A total of 35 patients were enrolled, with a median age of 55 years (range, 20-80) and a median of 3 prior therapies (range, 1-15). All patients received at least 1 IV infusion of loncastuximab tesirine at 15 to 150 μg/kg once every 3 weeks (Q3W; n = 30) or 50 μg/kg IV weekly (n = 5). Common treatment-emergent adverse events (TEAEs) were nausea (42.9%), febrile neutropenia (37.1%), and reversible liver test abnormalities. Grade ≥3 TEAEs were reported in 85.7% patients, most commonly febrile neutropenia and other hematologic abnormalities and reversible liver test abnormalities. There were no treatment-related deaths. Four patients (11.4%) had grade 2 infusion-related reactions, and 1 patient (150 μg/kg Q3W) had a dose-limiting toxicity of hyperbilirubinemia that resolved within 6 days without further action. The maximum tolerated dose was not reached. Three patients achieved complete responses, 1 each at 30, 120, and 150 μg/kg Q3W. PK studies showed marked interpatient variability, with target-mediated drug disposition seeming to contribute to time- and dose-dependent disposition. No clinically relevant anti–drug-antibody formation occurred. The trial was terminated in the dose-escalation phase because of slow accrual. This trial was registered at www.clinicaltrials.gov as NCT02669264.
Introduction
Advances in firstline therapy for acute lymphoblastic leukemia (ALL) have led to ∼80% to 90% complete response (CR) rates in adult patients with ALL with initial induction.1 Despite high initial response rates, a majority of patients eventually relapse. Outcomes for relapsed or refractory (R/R) ALL remain dismal, with survival times typically <6 months and a CR rate of <40% with conventional chemotherapy salvage treatments.2 Novel therapies are of increasing interest to improve treatment outcomes for patients with R/R ALL,1 and several novel agents have recently been approved for the treatment of R/R ALL of B-cell lineage (B-ALL), including blinatumomab3 (an anti-CD19 bispecific T cell–engaging antibody), inotuzumab ozogamicin4 (an anti-CD22 antibody-drug conjugate [ADC]), and tisagenlecleucel5 (an anti-CD19 chimeric antigen receptor T-cell [CAR-T] treatment).
ADCs permit targeting of potent cytotoxic agents to cancer cells that express specific antigens, with the potential to maximize efficacy while minimizing systemic toxicities.6 The human CD19 antigen is a transmembrane glycoprotein that is expressed during B-cell development and in B-cell lineage malignancies, including B-ALL.7 CD19 plays a vital role in the regulation of B-cell receptor signaling and is efficiently internalized upon antigen binding, making it an attractive target for antibody-based therapeutics to treat B-cell malignancies. CD19 has been clinically validated as a therapeutic target with several approaches, including naked and bispecific antibodies, ADCs, and CAR-T cells.8
Loncastuximab tesirine (also known as ADCT-402) is an ADC comprising a humanized anti-CD19 antibody, stochastically conjugated through a cathepsin-cleavable valine-alanine linker to SG3199, a pyrrolobenzodiazepine (PBD) dimer toxin. The mechanism of SG3199 for DNA crosslinking contributes to persistence in cells,9 and SG3199 has had picomolar antitumor activity against panels of human hematologic tumor cell lines in in vitro studies.10
In preclinical studies, loncastuximab tesirine has demonstrated potent dose-dependent antitumor activity against CD19-expressing B-cell malignancies in both in vitro and in vivo preclinical models.11 Moreover, loncastuximab tesirine has shown an acceptable safety and pharmacokinetic (PK) profile, with excellent stability and tolerability in preclinical studies, supporting further investigation in clinical trials.11 A first-in-human study of loncastuximab tesirine has been completed in non-Hodgkin lymphoma (NHL), and a phase 2 trial has been initiated based on the high response rate in the R/R diffuse large B-cell lymphoma (DLBCL) cohort.12
This phase 1 trial was designed to evaluate the safety, maximum tolerated dose (MTD), PKs, immunogenicity, and preliminary clinical activity of loncastuximab tesirine monotherapy in patients with R/R B-ALL in 2 parts: dose escalation (part 1) and dose expansion (part 2). The study was terminated during dose escalation because of slow accrual. Here, we present data on all 35 patients enrolled in this clinical trial.
Methods
This was an open-label, single-arm, phase 1 study conducted at 11 US centers in adult patients with R/R B-ALL.13 The study was performed in accordance with the International Council for Harmonisation good clinical practice guidelines and the ethical principles of the Declaration of Helsinki and was approved by the institutional review board of each study center. All patients provided written informed consent. The primary objectives of the study were to evaluate the safety and tolerability of loncastuximab tesirine and determine the MTD and recommended dose for expansion in patients with R/R B-ALL. Secondary objectives were to evaluate the preliminary clinical activity and PK profile of loncastuximab tesirine in R/R B-ALL and evaluate induction of anti-drug antibodies (ADAs) to loncastuximab tesirine.
Study population
Patients aged ≥12 years with R/R B-ALL for whom standard therapies had failed, who were intolerant to standard therapies, or for whom no other treatment options were available, in the opinion of the investigator, were eligible for the study. Other key inclusion criteria included Eastern Cooperative Oncology Group (ECOG) performance status of 0 to 2 and white blood cell count <15 × 109/L before day 1. Key exclusion criteria were known active central nervous system leukemia, Burkitt leukemia/lymphoma, active autoimmune disease, allogeneic stem cell transplantation within 60 days, active graft-versus-host disease, and major surgery or anticancer treatment within 14 days before day-1 treatment (complete inclusion/exclusion criteria are listed in the study protocol included in the data supplement).
Study design and treatment
Loncastuximab tesirine was administered IV over 60 minutes once every 3 weeks (Q3W; day 1 of each 21-day cycle) or once weekly (QW; days 1, 8, and 15 of each 21-day cycle). If the first infusion was well tolerated, the infusion duration of subsequent doses could be shortened to 30 minutes. Patients were assigned to treatment according to a 3 + 3 dose-escalation design and overseen by a dose-escalation steering committee (DESC).
A hematologic dose-limiting toxicity (DLT) was defined as a grade ≥3 event of neutropenia or thrombocytopenia or grade 4 anemia, with hypocellular bone marrow lasting for ≥6 weeks, in the absence of residual leukemia (ie, with <5% blasts in the bone marrow). In the case of normocellular bone marrow with <5% blasts, grade ≥3 pancytopenia lasting 8 weeks was considered a DLT. A nonhematologic DLT was defined as grade 4 tumor lysis syndrome, grade ≥3 adverse event (AE; including nausea, vomiting, diarrhea, and electrolyte imbalances lasting >48 hours despite optimal therapy; excluding all grades of alopecia), grade ≥3 hypersensitivity reaction, or grade ≥3 skin ulceration. The protocol-defined DLT observation period for dose escalation was the duration of cycle 1. No intrapatient dose escalation was permitted. The MTD was defined as the highest dose level at which none of the first 3 treated patients or ≤1 of the first 6 treated patients experienced a DLT. The DESC determined if a Q3W or QW schedule was appropriate during dose escalation based on available safety and tolerability data and/or the PK profile of loncastuximab tesirine in patients. In QW dosing, patients were administered a cumulative dose each cycle comparable to the highest dose tested in Q3W without a DLT.
The trial was continuously monitored for safety, efficacy, and PK profile. Additional patients could be added to a dose level, provided there were ≥1 patients who had achieved a partial response (PR) or better.
Patients were treated until disease progression, initiation of new anticancer treatment, or significant toxicity. Patients who discontinued treatment for any reason other than disease progression were followed until disease progression or start of new anticancer treatment. All patients were followed by telephone or medical record review for up to 12 months after documentation of disease progression or start of new anticancer treatment.
Study end points
Safety end points included AEs graded according to the National Cancer Institute Common Terminology for Criteria for Adverse Events (version 4.0), serious AEs, and DLTs, as assessed by periodic 12-lead electrocardiogram recordings, physical examinations, vital sign measurements, ECOG performance status, and laboratory abnormalities.
According to National Comprehensive Cancer Network guidelines, preliminary assessments of clinical activity were based on investigator assessment of response to treatment using bone marrow samples and included CR, CR with incomplete blood count recovery, PR, no response, or progressive disease.14
PK concentrations of total antibody, PBD-conjugated antibody, and free SG3199 in serum were measured using validated bioanalytic methods, with lower limits of quantification (LLOQs) of 20 ng/mL, 5.06 ng/mL, and 25 pg/mL, respectively. Serum concentrations above the LLOQs were used for the PK analysis. PK parameters (ie, maximum concentration [Cmax] and area under the concentration-time curve [AUC]) were determined by noncompartmental analysis. For the Q3W schedule, PK samples were collected during cycles 1 and 2 on days 1 (0 hours, end of infusion, 3 hours, and 6 hours), 2, 3, 5, 8, and 15; during cycle 3 on day 1 (0 hours and end of infusion); at end of treatment; and during the follow-up period. For the QW schedule, samples were collected in cycle 1 during week 1, days 1 (0 hours, end of infusion, and 5 hours), 2, 3, and 5; week 2, days 8 (0 hours, end of infusion, and 5 hours), 9, 10, and 12; and week 3, day 15 (0 hours and end of infusion) and in cycle 2 and subsequent cycles on days 1 (0 hours and end of infusion), 8 (0 hours and end of infusion), and 15 (0 hours and end of infusion); at end of treatment; and during the follow-up period. ADA levels in serum before, during, and after treatment were measured using validated bioanalytic methods.
Statistical analysis
Descriptive statistics and listings were used to report safety, efficacy, and PK parameters. Patients were evaluable for efficacy and toxicity if they had received ≥1 dose of loncastuximab tesirine. The population for PK analysis comprised all treated patients with sufficient concentration data available for PK analysis.
All authors had full access to the data in this study and were involved in data analysis and interpretation. Additional details of the study design are available in the study protocol included in the data supplement.
Results
Patient characteristics
Thirty-five patients were accrued and received treatment in the dose-escalation phase from April 2016 through May 2018. The study was terminated by the study sponsor during the dose-escalation phase. After study termination, patients were to be followed for 30 days after the last dose of study medication or until initiation of new treatment. Baseline demographics and clinical characteristics of the study population are reported in Table 1. The median age was 55 years (range, 20-80). Philadelphia chromosome (4 [11%] of 35) and hypodiploid chromosomes (3 [9%] of 35) were the most common cytogenetic abnormalities. Patients had received a median of 3 prior systemic therapies (range, 1-15). All patients had received prior chemotherapy. Overall, 21 patients (60%) had received ≥1 prior B-cell–directed therapies (Table 1); approximately half had received prior blinatumomab (16 [46%] of 35).
Patient baseline characteristics
| Characteristic | All patients (N = 35) |
|---|---|
| Age, y | |
| Median (range) | 55 (20-80) |
| Sex, n (%) | |
| Male | 19 (54) |
| Female | 16 (46) |
| ECOG score, median (range) | 1 (0-3) |
| ALL cytogenetic characteristics, n (%) | |
| Philadelphia chromosome | 4 (11) |
| MLL rearrangement | 0 (0) |
| E2A-PBX1 | 1 (3) |
| TEL-AML1 | 0 (0) |
| IL3-IGH | 1 (3) |
| Hyperdiploid >50 | 1 (3) |
| Hypodiploid | 3 (9) |
| Other (mixed/unknown) | 25 (71) |
| Disease status at enrollment, n (%) | |
| Primary refractory | 6 (17) |
| Relapsed | 29 (83) |
| Extramedullary involvement, n (%) | |
| Yes | 2 (6) |
| No | 33 (94) |
| Number of prior systemic therapies,*median (range) | 3 (1-15) |
| 1, n (%) | 4 (11) |
| 2, n (%) | 8 (23) |
| 3, n (%) | 8 (23) |
| ≥4, n (%) | 15 (43) |
| Prior B cell–directed therapies, n (%) | |
| Blinatumomab | 16 (46) |
| Inotuzumab ozogamicin | 6 (17) |
| Rituximab | 6 (17) |
| CAR-T | 2 (6) |
| Ofatumumab | 1 (3) |
| Prior HSCT, n (%) | |
| Allogeneic | 11 (31) |
| Autologous | 2 (6) |
| Characteristic | All patients (N = 35) |
|---|---|
| Age, y | |
| Median (range) | 55 (20-80) |
| Sex, n (%) | |
| Male | 19 (54) |
| Female | 16 (46) |
| ECOG score, median (range) | 1 (0-3) |
| ALL cytogenetic characteristics, n (%) | |
| Philadelphia chromosome | 4 (11) |
| MLL rearrangement | 0 (0) |
| E2A-PBX1 | 1 (3) |
| TEL-AML1 | 0 (0) |
| IL3-IGH | 1 (3) |
| Hyperdiploid >50 | 1 (3) |
| Hypodiploid | 3 (9) |
| Other (mixed/unknown) | 25 (71) |
| Disease status at enrollment, n (%) | |
| Primary refractory | 6 (17) |
| Relapsed | 29 (83) |
| Extramedullary involvement, n (%) | |
| Yes | 2 (6) |
| No | 33 (94) |
| Number of prior systemic therapies,*median (range) | 3 (1-15) |
| 1, n (%) | 4 (11) |
| 2, n (%) | 8 (23) |
| 3, n (%) | 8 (23) |
| ≥4, n (%) | 15 (43) |
| Prior B cell–directed therapies, n (%) | |
| Blinatumomab | 16 (46) |
| Inotuzumab ozogamicin | 6 (17) |
| Rituximab | 6 (17) |
| CAR-T | 2 (6) |
| Ofatumumab | 1 (3) |
| Prior HSCT, n (%) | |
| Allogeneic | 11 (31) |
| Autologous | 2 (6) |
HSCT, hematopoietic stem cell transplantation; MLL, mixed lineage leukemia.
Prior stem cell transplantation is included. For patients who underwent autologous transplantation, the mobilization regimen was considered a line of therapy if it was chemotherapy based and distinct from the other previous lines of treatment.
All patients received ≥1 infusions of loncastuximab tesirine. A majority of patients (30 of 35) received loncastuximab tesirine Q3W at doses ranging from 15 to 150 μg/kg; 5 patients received 50 μg/kg of loncastuximab tesirine QW. Patients received a median of 2 infusions (range, 1-10) of loncastuximab tesirine, with a median overall cumulative dose of 120.2 μg/kg (range, 15.0-741.2). The median treatment duration was 3.1 weeks (range, 0.1-35.4).
Safety
All patients experienced ≥1 treatment-emergent AEs (TEAEs). TEAEs reported in ≥20% of patients are summarized in Table 2, and the most common included nausea, febrile neutropenia, and liver test abnormalities (AST increased, GGT increased, alanine aminotransferase increased, or blood alkaline phosphatase increased). Five patients (15.3%) had peripheral edema during the study. Three patients (8.6%) had other effusion-related TEAEs: 1 patient had ascites, pericardial effusion, and pleural effusion, all grade 2 and considered possibly related to study drug; 1 patient had grade 2 fluid overload, considered unrelated to study drug; and 1 patient had grade 2 ascites considered unrelated to study drug and 2 events of grade 3 fluid overload, the second of which was considered related to study drug. Four patients (11.4%) experienced infusion-related reactions, all of which were grade 2 and resolved on the same day or the next day, and all affected patients continued to receive the study drug.
Any grade TEAEs reported by ≥20% of patients (safety analysis set)
| TEAE, n (%) | Dose, μg/kg | |||||||
|---|---|---|---|---|---|---|---|---|
| Q3W | QW | Total (N = 35) | ||||||
| 15 (n = 5) | 30 (n = 7) | 60 (n = 3) | 90 (n = 4) | 120 (n = 5) | 150 (n = 6) | 50 (n = 5) | ||
| Any | 5 (100) | 7 (100) | 3 (100) | 4 (100) | 5 (100) | 6 (100) | 5 (100) | 35 (100) |
| Nausea | 1 (20.0) | 2 (28.6) | 1 (33.3) | 2 (50.0) | 3 (60.0) | 5 (83.3) | 1 (20.0) | 15 (42.9) |
| Febrile neutropenia | 0 (0) | 1 (14.3) | 1 (33.3) | 2 (50.0) | 3 (60.0) | 4 (66.7) | 2 (40.0) | 13 (37.1) |
| AST increased | 0 (0) | 0 (0) | 2 (66.7) | 2 (50.0) | 2 (40.0) | 4 (66.7) | 1 (20.0) | 11 (31.4) |
| GGT increased | 1 (20.0) | 0 (0) | 0 (0) | 2 (50.0) | 3 (60.0) | 3 (50.0) | 1 (20.0) | 10 (28.6) |
| Alanine aminotransferase increased | 0 (0) | 0 (0) | 1 (33.3) | 2 (50.0) | 2 (40.0) | 2 (33.3) | 2 (40.0) | 9 (25.7) |
| Blood alkaline phosphatase increased | 0 (0) | 0 (0) | 2 (66.7) | 1 (25.0) | 1 (20.0) | 3 (50.0) | 2 (40.0) | 9 (25.7) |
| Fatigue | 0 (0) | 3 (42.9) | 1 (33.3) | 3 (75.0) | 2 (40.0) | 0 (0) | 0 (0) | 9 (25.7) |
| Headache | 1 (20.0) | 2 (28.6) | 1 (33.3) | 0 (0) | 2 (40.0) | 2 (33.3) | 1 (20.0) | 9 (25.7) |
| Rash maculopapular | 0 (0) | 1 (14.3) | 1 (33.3) | 1 (25.0) | 2 (40.0) | 2 (33.3) | 2 (40.0) | 9 (25.7) |
| Vomiting | 0 (0) | 1 (14.3) | 0 (0) | 1 (25.0) | 3 (60.0) | 3 (50.0) | 1 (20.0) | 9 (25.7) |
| Abdominal pain | 0 (0) | 1 (14.3) | 1 (33.3) | 1 (25.0) | 0 (0) | 3 (50.0) | 2 (40.0) | 8 (22.9) |
| Dizziness | 0 (0) | 0 (0) | 1 (33.3) | 1 (25.0) | 2 (40.0) | 2 (33.3) | 2 (40.0) | 8 (22.9) |
| Diarrhea | 1 (20.0) | 1 (14.3) | 1 (33.3) | 2 (50.0) | 0 (0) | 1 (16.7) | 1 (20.0) | 7 (20.0) |
| Dyspnea | 0 (0) | 1 (14.3) | 1 (33.3) | 3 (75.0) | 0 (0) | 1 (16.7) | 1 (20.0) | 7 (20.0) |
| TEAE, n (%) | Dose, μg/kg | |||||||
|---|---|---|---|---|---|---|---|---|
| Q3W | QW | Total (N = 35) | ||||||
| 15 (n = 5) | 30 (n = 7) | 60 (n = 3) | 90 (n = 4) | 120 (n = 5) | 150 (n = 6) | 50 (n = 5) | ||
| Any | 5 (100) | 7 (100) | 3 (100) | 4 (100) | 5 (100) | 6 (100) | 5 (100) | 35 (100) |
| Nausea | 1 (20.0) | 2 (28.6) | 1 (33.3) | 2 (50.0) | 3 (60.0) | 5 (83.3) | 1 (20.0) | 15 (42.9) |
| Febrile neutropenia | 0 (0) | 1 (14.3) | 1 (33.3) | 2 (50.0) | 3 (60.0) | 4 (66.7) | 2 (40.0) | 13 (37.1) |
| AST increased | 0 (0) | 0 (0) | 2 (66.7) | 2 (50.0) | 2 (40.0) | 4 (66.7) | 1 (20.0) | 11 (31.4) |
| GGT increased | 1 (20.0) | 0 (0) | 0 (0) | 2 (50.0) | 3 (60.0) | 3 (50.0) | 1 (20.0) | 10 (28.6) |
| Alanine aminotransferase increased | 0 (0) | 0 (0) | 1 (33.3) | 2 (50.0) | 2 (40.0) | 2 (33.3) | 2 (40.0) | 9 (25.7) |
| Blood alkaline phosphatase increased | 0 (0) | 0 (0) | 2 (66.7) | 1 (25.0) | 1 (20.0) | 3 (50.0) | 2 (40.0) | 9 (25.7) |
| Fatigue | 0 (0) | 3 (42.9) | 1 (33.3) | 3 (75.0) | 2 (40.0) | 0 (0) | 0 (0) | 9 (25.7) |
| Headache | 1 (20.0) | 2 (28.6) | 1 (33.3) | 0 (0) | 2 (40.0) | 2 (33.3) | 1 (20.0) | 9 (25.7) |
| Rash maculopapular | 0 (0) | 1 (14.3) | 1 (33.3) | 1 (25.0) | 2 (40.0) | 2 (33.3) | 2 (40.0) | 9 (25.7) |
| Vomiting | 0 (0) | 1 (14.3) | 0 (0) | 1 (25.0) | 3 (60.0) | 3 (50.0) | 1 (20.0) | 9 (25.7) |
| Abdominal pain | 0 (0) | 1 (14.3) | 1 (33.3) | 1 (25.0) | 0 (0) | 3 (50.0) | 2 (40.0) | 8 (22.9) |
| Dizziness | 0 (0) | 0 (0) | 1 (33.3) | 1 (25.0) | 2 (40.0) | 2 (33.3) | 2 (40.0) | 8 (22.9) |
| Diarrhea | 1 (20.0) | 1 (14.3) | 1 (33.3) | 2 (50.0) | 0 (0) | 1 (16.7) | 1 (20.0) | 7 (20.0) |
| Dyspnea | 0 (0) | 1 (14.3) | 1 (33.3) | 3 (75.0) | 0 (0) | 1 (16.7) | 1 (20.0) | 7 (20.0) |
AST, aspartate aminotransferase; GGT, γ-glutamyl transferase.
Overall, 85.7% (30 of 35) of patients experienced a grade ≥3 TEAE. Grade ≥3 TEAEs reported in ≥10% of patients are presented in Table 3 and most commonly included febrile neutropenia and other hematologic abnormalities and reversible liver test abnormalities (AST increased, blood bilirubin increased, and GGT increased). Many patients had hematologic abnormalities at baseline because of the nature of the disease (supplemental Table 2). There was a trend toward more grade ≥3 TEAEs with increasing dose.
Grade ≥3 TEAEs reported by ≥10% of patients (safety analysis set)
| TEAE, n (%) | Dose, μg/kg | |||||||
|---|---|---|---|---|---|---|---|---|
| Q3W | QW | Total (N = 35) | ||||||
| 15 (n = 5) | 30 (n = 7) | 60 (n = 3) | 90 (n = 4) | 120 (n = 5) | 150 (n = 6) | 50 (n = 5) | ||
| Any | 5 (100) | 4 (57.1) | 3 (100) | 3 (75.0) | 5 (100) | 6 (100) | 4 (80.0) | 30 (85.7) |
| Febrile neutropenia | 0 (0) | 1 (14.3) | 1 (33.3) | 2 (50.0) | 2 (40.0) | 3 (50.0) | 1 (20.0) | 10 (28.6) |
| AST increased | 0 (0) | 0 (0) | 0 (0) | 2 (50.0) | 1 (20.0) | 2 (33.3) | 1 (20.0) | 6 (17.1) |
| Blood bilirubin increased | 0 (0) | 0 (0) | 1 (33.3) | 0 (0) | 1 (20.0) | 2 (33.3) | 1 (20.0) | 5 (14.3) |
| GGT increased | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 3 (60.0) | 1 (16.7) | 1 (20.0) | 5 (14.3) |
| Abdominal pain | 0 (0) | 1 (14.3) | 0 (0) | 1 (25.0) | 0 (0) | 1 (16.7) | 1 (20.0) | 4 (11.4) |
| Anemia | 0 (0) | 1 (14.3) | 1 (33.3) | 1 (25.0) | 0 (0) | 0 (0) | 1 (20.0) | 4 (11.4) |
| Bacteremia | 0 (0) | 0 (0) | 0 (0) | 2 (50.0) | 0 (0) | 1 (16.7) | 1 (20.0) | 4 (11.4) |
| Neutrophil count decreased | 0 (0) | 2 (28.6) | 0 (0) | 0 (0) | 1 (20.0) | 1 (16.7) | 0 (0) | 4 (11.4) |
| Sepsis | 0 (0) | 2 (28.6) | 0 (0) | 0 (0) | 1 (20.0) | 0 (0) | 1 (20.0) | 4 (11.4) |
| TEAE, n (%) | Dose, μg/kg | |||||||
|---|---|---|---|---|---|---|---|---|
| Q3W | QW | Total (N = 35) | ||||||
| 15 (n = 5) | 30 (n = 7) | 60 (n = 3) | 90 (n = 4) | 120 (n = 5) | 150 (n = 6) | 50 (n = 5) | ||
| Any | 5 (100) | 4 (57.1) | 3 (100) | 3 (75.0) | 5 (100) | 6 (100) | 4 (80.0) | 30 (85.7) |
| Febrile neutropenia | 0 (0) | 1 (14.3) | 1 (33.3) | 2 (50.0) | 2 (40.0) | 3 (50.0) | 1 (20.0) | 10 (28.6) |
| AST increased | 0 (0) | 0 (0) | 0 (0) | 2 (50.0) | 1 (20.0) | 2 (33.3) | 1 (20.0) | 6 (17.1) |
| Blood bilirubin increased | 0 (0) | 0 (0) | 1 (33.3) | 0 (0) | 1 (20.0) | 2 (33.3) | 1 (20.0) | 5 (14.3) |
| GGT increased | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 3 (60.0) | 1 (16.7) | 1 (20.0) | 5 (14.3) |
| Abdominal pain | 0 (0) | 1 (14.3) | 0 (0) | 1 (25.0) | 0 (0) | 1 (16.7) | 1 (20.0) | 4 (11.4) |
| Anemia | 0 (0) | 1 (14.3) | 1 (33.3) | 1 (25.0) | 0 (0) | 0 (0) | 1 (20.0) | 4 (11.4) |
| Bacteremia | 0 (0) | 0 (0) | 0 (0) | 2 (50.0) | 0 (0) | 1 (16.7) | 1 (20.0) | 4 (11.4) |
| Neutrophil count decreased | 0 (0) | 2 (28.6) | 0 (0) | 0 (0) | 1 (20.0) | 1 (16.7) | 0 (0) | 4 (11.4) |
| Sepsis | 0 (0) | 2 (28.6) | 0 (0) | 0 (0) | 1 (20.0) | 0 (0) | 1 (20.0) | 4 (11.4) |
A total of 80% (28 of 35) of patients experienced a serious TEAE of any grade, and 20% (7 of 35) of patients had a TEAE with a fatal outcome; a fatal event of worsening ALL was also reported as a TEAE but was considered to be disease progression. TEAEs resulting in death were infections (6 [21.4%] of 28; sepsis, n = 3; lung infection, n = 2; and bacteremia, n = 1) and subarachnoid hemorrhage (1 [3.5%] of 28). Disease progression was the most common reason for death (18 [64.3%] of 28). No deaths were considered related to the study drug by investigators.
Approximately half of the patients (19 [54.3%] of 35) experienced TEAEs considered by the investigator to be at least possibly related to the study treatment. The most common were gastrointestinal disorders (12 [34.3%] of 35) and laboratory investigations (9 [25.7%] of 35), including liver test and hematologic abnormalities. There was a trend toward more study treatment–related AEs at higher dose levels, with 11 (68.7%) of 16 patients in the higher-dose groups (120 and 150 μg/kg Q3W and 50 μg/kg QW) and 8 (42.1%) of 19 patients in the lower-dose groups (≤90 μg/kg) experiencing AEs related to study treatment. TEAEs that led to dose delay or treatment interruption occurred in 7 (20%) of 35 patients; 6 had dose delays and 1 had a dose interruption because of an infusion-related reaction.
One patient experienced a DLT. This patient was in the 150 μg/kg Q3W group and had worsening grade 3 blood bilirubin increased, considered by the investigator to be possibly related to study treatment. The event resolved after 6 days without requiring further action. The patient had concurrent AST increased in laboratory tests. One patient in the 50 μg/kg QW dose group had a TEAE of GGT increased that led to treatment discontinuation, but it was considered unrelated to study treatment, because the patient had disease progression with liver involvement. The MTD was not reached.
PKs, pharmacodynamics, and immunogenicity
PK data of cycle 1 were available for analysis for PBD-conjugated antibody, total antibody, and SG3199 in 28, 26, and 13 patients, respectively, on the Q3W dosing schedule. For cycle 2, PK data were available for analysis for PBD-conjugated antibody, total antibody, and SG3199 in 18, 18, and 8 patients, respectively, on the Q3W dosing schedule. Because of the low number of patients, limited PK data were available for the QW dosing schedule. Patients with insufficient data or levels of SG3199 below the LLOQ were excluded from the PK analysis.
Serum concentrations of PBD-conjugated antibody and total antibody seemed to increase with dose in general and displayed significant degrees of variability (Figure 1). Similar observations were made for the associated PK metrics of Cmax and AUC from time 0 to the last measurable time point in the respective cycle (Table 4; supplemental Table 1). For SG3199, PK profiles for a majority of patients were found to be largely below the LLOQ immediately after dosing (LLOQ, 25 pg/mL).10
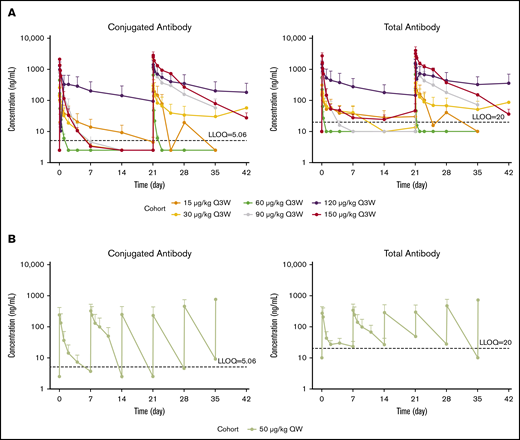
Serum concentrations of PBD-conjugated antibody and total antibody by dose over time. Semilog plot of mean (+ standard error) concentration of PBD-conjugated antibody and total antibody in serum vs time by dose for patients on the Q3W dosing regimen (A) and patients on the QW dosing regimen (B). Concentrations below the LLOQ are inputted as 1/2 LLOQ.
Serum concentrations of PBD-conjugated antibody and total antibody by dose over time. Semilog plot of mean (+ standard error) concentration of PBD-conjugated antibody and total antibody in serum vs time by dose for patients on the Q3W dosing regimen (A) and patients on the QW dosing regimen (B). Concentrations below the LLOQ are inputted as 1/2 LLOQ.
Summary of PK exposure in serum of patients on the Q3W dosing regimen
| Cohort, µg/kg | Arithmetic mean (CV%); n | |||
|---|---|---|---|---|
| Cycle 1 | Cycle 2 | |||
| Cmax, µg/L* | AUClast, d × µg/L | Cmax, µg/L* | AUClast, d × µg/L | |
| Conjugated Ab | ||||
| 15 | 178 (72.0); 4 | 337 (193); 4 | 2150 (-); 1 | 1596 (—); 1 |
| 30 | 258 (62.9); 7 | 127 (180); 7 | 339 (81.5); 4 | 735 (190); 4 |
| 60 | 449 (62.7); 3 | 43.0 (86.9); 3 | 661 (62.3); 2 | 50.6 (18.0); 2 |
| 90 | 1058 (84.4); 3 | 671 (162); 3 | 1813 (91.0); 2 | 3370 (135); 2 |
| 120 | 1291 (79.0); 5 | 3907 (209); 5 | 1515 (66.0); 5 | 8744 (203); 5 |
| 150 | 2145 (49.1); 6 | 688 (82.9); 6 | 3033 (48.1); 4 | 7553 (9.62); 4 |
| Total Ab | ||||
| 15 | 205 (78.8); 4 | 610 (195); 4 | 1251 (138); 2 | 1012 (129); 2 |
| 30 | 317 (61.1); 7 | 408 (153); 7 | 366 (68.5); 4 | 1140 (184); 4 |
| 60 | 717 (59.8); 2 | 59.9 (72.6); 2 | 689 (55.4); 2 | 40.2 (0.0608); 2 |
| 90 | 1049 (85.1); 3 | 558 (161); 3 | 1845 (90.8); 2 | 3671 (138); 2 |
| 120 | 1722 (45.6); 4 | 5718 (169); 4 | 2125 (44.4); 4 | 13 519 (169); 4 |
| 150 | 2761 (62.2); 6 | 1091 (61.6); 6 | 4338 (56.7); 4 | 10 493 (19.3); 4 |
| SG3199 | ||||
| 15 | — | — | — | — |
| 30 | 0.0428 (0.165); 2 | 0.00355 (41.5); 2 | 0.0459 (—); 1 | 0.00522 (—); 1 |
| 60 | — | — | 0.0697 (—); 1 | 0.0129 (—); 1 |
| 90 | 0.0617 (38.7); 2 | 0.00930 (72.6); 2 | 0.0551 (—); 1 | 0.00830 (—); 1 |
| 120 | 0.0810 (35.6); 3 | 0.0194 (119); 3 | 0.108 (22.1); 3 | 0.0433 (73.0); 3 |
| 150 | 0.0907 (55.2); 6 | 0.0338 (44.2); 6 | 0.0550 (50.4); 2 | 0.00927 (95.1); 2 |
| Cohort, µg/kg | Arithmetic mean (CV%); n | |||
|---|---|---|---|---|
| Cycle 1 | Cycle 2 | |||
| Cmax, µg/L* | AUClast, d × µg/L | Cmax, µg/L* | AUClast, d × µg/L | |
| Conjugated Ab | ||||
| 15 | 178 (72.0); 4 | 337 (193); 4 | 2150 (-); 1 | 1596 (—); 1 |
| 30 | 258 (62.9); 7 | 127 (180); 7 | 339 (81.5); 4 | 735 (190); 4 |
| 60 | 449 (62.7); 3 | 43.0 (86.9); 3 | 661 (62.3); 2 | 50.6 (18.0); 2 |
| 90 | 1058 (84.4); 3 | 671 (162); 3 | 1813 (91.0); 2 | 3370 (135); 2 |
| 120 | 1291 (79.0); 5 | 3907 (209); 5 | 1515 (66.0); 5 | 8744 (203); 5 |
| 150 | 2145 (49.1); 6 | 688 (82.9); 6 | 3033 (48.1); 4 | 7553 (9.62); 4 |
| Total Ab | ||||
| 15 | 205 (78.8); 4 | 610 (195); 4 | 1251 (138); 2 | 1012 (129); 2 |
| 30 | 317 (61.1); 7 | 408 (153); 7 | 366 (68.5); 4 | 1140 (184); 4 |
| 60 | 717 (59.8); 2 | 59.9 (72.6); 2 | 689 (55.4); 2 | 40.2 (0.0608); 2 |
| 90 | 1049 (85.1); 3 | 558 (161); 3 | 1845 (90.8); 2 | 3671 (138); 2 |
| 120 | 1722 (45.6); 4 | 5718 (169); 4 | 2125 (44.4); 4 | 13 519 (169); 4 |
| 150 | 2761 (62.2); 6 | 1091 (61.6); 6 | 4338 (56.7); 4 | 10 493 (19.3); 4 |
| SG3199 | ||||
| 15 | — | — | — | — |
| 30 | 0.0428 (0.165); 2 | 0.00355 (41.5); 2 | 0.0459 (—); 1 | 0.00522 (—); 1 |
| 60 | — | — | 0.0697 (—); 1 | 0.0129 (—); 1 |
| 90 | 0.0617 (38.7); 2 | 0.00930 (72.6); 2 | 0.0551 (—); 1 | 0.00830 (—); 1 |
| 120 | 0.0810 (35.6); 3 | 0.0194 (119); 3 | 0.108 (22.1); 3 | 0.0433 (73.0); 3 |
| 150 | 0.0907 (55.2); 6 | 0.0338 (44.2); 6 | 0.0550 (50.4); 2 | 0.00927 (95.1); 2 |
—, not available; Ab, antibody; AUClast, AUC from time 0 to last measurable time point in respective cycle; CV, coefficient of variation.
Approximates the concentration at the end of infusion for conjugated and total Ab (0.480-4.56 hours) and after the end of infusion for SG3199 (0.960-47.5 hours).
Despite limited data and highly variable apparent PK parameters of clearance and volume of distribution at terminal phase estimations, the apparent half-life of total antibody and conjugated antibody at higher doses (≥90 µg/kg, Q3W) seemed similar, indicating good stability of loncastuximab tesirine in the circulation (Table 5). However, the apparent half-life of loncastuximab tesirine conjugated and total antibody (<1 day) at the first dose cycle in B-ALL patients was much shorter than the known serum half-life of immunoglobulin G1, which might be partly due to high clearance in association with target-mediated drug disposition (TMDD).
Summary of PK parameters of patients on the Q3W dosing regimen (≥90 μg/kg)
| Cohort, µg/kg | Arithmetic mean (CV%); n | |||||
|---|---|---|---|---|---|---|
| Cycle 1 | Cycle 2 | |||||
| CL, L/d | Vdβ, L | Thalf, d | CLSS, L/d | Vdβ, L | Thalf, d | |
| Conjugated Ab | ||||||
| 90 | 80.8 (131); 3 | 10.5 (121); 3 | 0.355 (141); 3 | 9.63 (130); 2 | 5.61 (-); 1 | 4.96 (-); 1 |
| 120 | 84.3 (121); 2 | 6.89 (45.5); 2 | 0.156 (105); 2 | 31.1 (119); 4 | 8.33 (65.1); 3 | 7.55 (136); 3 |
| 150 | 27.4 (45.2); 5 | 34.2 (104); 5 | 0.713 (83.6); 5 | 1.57 (46.2); 4 | 9.45 (55.7); 4 | 3.99 (18.1); 4 |
| Total Ab | ||||||
| 90 | 3.98 (—); 1 | 4.50 (—); 1 | 0.784 (—); 1 | 0.831 (—); 1 | 6.71 (—); 1 | 5.59 (—); 1 |
| 120 | — | — | — | 10.3 (162); 4 | 11.4 (0.891); 2 | 14.5 (113); 2 |
| 150 | 18.9 (115); 2 | 2.68 (11.5); 2 | 0.275 (111); 2 | 1.37 (47.9); 4 | 9.14 (65.2); 3 | 4.67 (35.9); 3 |
| SG3199 | ||||||
| 90 | — | — | — | — | — | — |
| 120 | — | — | — | 838 (—); 1 | 614 (—); 1 | 0.508 (—); 1 |
| 150 | — | — | — | — | — | — |
| Cohort, µg/kg | Arithmetic mean (CV%); n | |||||
|---|---|---|---|---|---|---|
| Cycle 1 | Cycle 2 | |||||
| CL, L/d | Vdβ, L | Thalf, d | CLSS, L/d | Vdβ, L | Thalf, d | |
| Conjugated Ab | ||||||
| 90 | 80.8 (131); 3 | 10.5 (121); 3 | 0.355 (141); 3 | 9.63 (130); 2 | 5.61 (-); 1 | 4.96 (-); 1 |
| 120 | 84.3 (121); 2 | 6.89 (45.5); 2 | 0.156 (105); 2 | 31.1 (119); 4 | 8.33 (65.1); 3 | 7.55 (136); 3 |
| 150 | 27.4 (45.2); 5 | 34.2 (104); 5 | 0.713 (83.6); 5 | 1.57 (46.2); 4 | 9.45 (55.7); 4 | 3.99 (18.1); 4 |
| Total Ab | ||||||
| 90 | 3.98 (—); 1 | 4.50 (—); 1 | 0.784 (—); 1 | 0.831 (—); 1 | 6.71 (—); 1 | 5.59 (—); 1 |
| 120 | — | — | — | 10.3 (162); 4 | 11.4 (0.891); 2 | 14.5 (113); 2 |
| 150 | 18.9 (115); 2 | 2.68 (11.5); 2 | 0.275 (111); 2 | 1.37 (47.9); 4 | 9.14 (65.2); 3 | 4.67 (35.9); 3 |
| SG3199 | ||||||
| 90 | — | — | — | — | — | — |
| 120 | — | — | — | 838 (—); 1 | 614 (—); 1 | 0.508 (—); 1 |
| 150 | — | — | — | — | — | — |
CL, clearance; CLss, clearance at steady state; Thalf, apparent terminal half-life; Vdβ, volume of distribution at terminal phase.
Exploratory analysis of the correlation between AUC of loncastuximab tesirine conjugated antibody during cycle 1 and the baseline peripheral CD19+ cell count revealed a potential inverse relationship, suggesting lower baseline CD19+ cell count might contribute to higher exposure of loncastuximab tesirine (supplemental Figure 1). As expected, it seemed that the baseline level of peripheral CD19+ B cells was lower in patients who had received prior B cell–directed therapy than in those who had not, with the exception of the 90 µg/kg cohort, where the baseline level of B cells was comparable regardless of prior B cell–directed therapy (supplemental Figure 2). Based on this, the effect of prior therapy on the PKs of loncastuximab tesirine was explored. Although the sample size was small, both Cmax and AUC during cycle 1 (supplemental Figures 3 and 4, respectively) seemed to be higher in patients who had received prior B cell–directed therapies than in those who had not received prior B cell–directed therapy in the lower-dose cohorts of 15 and 30 µg/kg.
By cycle 2, there was a decrease in apparent clearance and an increase in apparent half-life of conjugated antibody to ∼4 to 8 days (Table 4). The highest mean accumulations of PBD-conjugated and total antibody in cycle 2 relative to cycle 1 were ∼30% and ∼67%, respectively. Overall, the PKs of PBD-conjugated and total antibody seemed to be time and dose dependent in patients with B-ALL.
Samples from all 35 patients were tested for ADAs to loncastuximab tesirine, with 111 sample measures available overall. Only 1 patient had a confirmed positive ADA titer, which occurred at predose only. No other patients exhibited confirmed positive ADA responses. There was no evidence of immunogenicity (ie, no clinically relevant ADA induction effect).
Clinical activity
Formal assessment of potential antileukemic effect of loncastuximab tesirine was not performed because of the early termination of the study. Three patients had responses to loncastuximab tesirine, all achieving CRs, 1 patient each in the 30, 120, and 150 μg/kg Q3W dose groups. Two of the 3 patients had received previous CD19-directed therapy.
One patient treated at on 30 μg/kg Q3W schedule received a total of 10 cycles of treatment, with no response in cycle 2 but a CR in cycle 4, and remained in CR at all subsequent assessments. This patient had received 3 lines of prior therapy, including CD19-directed therapy (blinatumomab), with PR as the best outcome. Blinatumomab was discontinued because of toxicity. Before receiving loncastuximab tesirine, this patient had blast count of ≤5% in the bone marrow.
One patient in the 120 μg/kg Q3W schedule had a CR in cycle 2 and during the follow-up period after end of treatment. However, this patient later discontinued the treatment to undergo stem cell transplantation but later died as a result of anoxic brain injury 80 days after receiving the last dose. This patient had previously received 2 lines of chemotherapy and an additional line of CD19-directed therapy (blinatumomab), with no response to any prior therapy. Blinatumomab was discontinued because of toxicity. Before receiving loncastuximab tesirine, this patient had blast counts between 5% and 25% in the marrow.
One patient in the 150 μg/kg Q3W schedule had a PR in cycle 2, improving to CR in cycle 5. This patient received 5 cycles in total before discontinuing the treatment after having a dose delay of >30 days because of grade 3 fluid overload and hypoxia, both of which resolved and were considered unrelated to study drug. This patient had an additional event of grade 3 fluid overload considered related to study drug, but no action was taken with regard to study drug. The patient was still in CR but died suddenly 70 days after receiving the last dose of loncastuximab tesirine; no autopsy was performed, and cause of death was unknown. This patient had previously received 2 lines of chemotherapy in combination with the tyrosine kinase inhibitor dasatinib, achieving a CR with incomplete blood count recovery with secondline therapy, followed by thirdline chemotherapy and progressive disease. This patient had blast count of >50% before loncastuximab tesirine treatment.
Five patients received 50 μg/kg of loncastuximab tesirine QW, but no responses were seen on this schedule.
Discussion
Loncastuximab tesirine is a novel CD19-targeted humanized ADC delivering SG3199, a potent cytotoxic DNA minor groove interstrand cross-linking PBD dimer warhead.11,15 In this open-label, dose-escalation, phase 1 trial, 35 patients with R/R B-ALL received loncastuximab tesirine in the dose-escalation portion of the study. The study was terminated early because of slow accrual in this population. Of note, the recent approval of other novel therapies is already changing the management of R/R B-ALL.3-5,10
Loncastuximab tesirine had a manageable safety profile in R/R B-ALL. There was a trend toward increased severity of TEAEs and more study treatment–related TEAEs with increasing doses of loncastuximab tesirine. However, there was only 1 DLT, and the MTD was not determined. Because of the early termination of the study, dose expansion was not performed. Frequently observed TEAEs such as febrile neutropenia, nausea, and fatigue are typical for patients being treated for R/R B-ALL, and elevated liver test results and rash have previously been observed in patients treated with either PBD dimer cytotoxin alone or with other ADCs containing the same PBD dimer.16-19 Hematologic abnormalities by laboratory reporting were common because of the nature of the disease, with many patients having hematologic abnormalities at baseline. It is of note that the incidence of pleural and pericardial effusions and ascites was lower than that observed with loncastuximab tesirine in other indications, such as DLBCL.20 Loop diuretics were not effective in mitigating the edema in this or other trials of loncastuximab tesirine; therefore, future trials will incorporate dexamethasone as a premedication and recommend other diuretics for management.
PK data analysis showed nonlinear disposition of loncastuximab tesirine in the B-ALL population. Loncastuximab tesirine exposure and PK parameters displayed marked variability in B-ALL patients. The high variability may have been partly due to different levels of CD19 target in the B-ALL patient group, resulting in different clearance in association with TMDD. By cycle 2, the increase in apparent half-life of conjugated antibody may have been partly due to lower target presence and thus lower clearance. As would be expected, baseline peripheral CD19+ B-cell levels by dose cohort were generally lower in patients who had received prior B cell–directed therapies than in patients who had not received prior B cell–directed therapies. For the lower-dose cohorts of 15 and 30 µg/kg, both Cmax and AUC of loncastuximab tesirine during cycle 1 were higher in patients who had received prior B cell–directed therapies compared with those who had not, but the limited data warrant careful interpretation. Overall, it seemed that TMDD contributed to the dose- and time-dependent disposition of loncastuximab tesirine. This is in contrast to the PKs of loncastuximab tesirine in B-cell NHL, where exposure to PBD-conjugated antibody was sustained throughout the duration of the 3-week dosage interval, and loncastuximab tesirine had a relatively long half-life. Although B-cell NHL and B-ALL are distinct diseases, it is possible that these PK differences may have contributed to the lower activity signal for loncastuximab tesirine in B-ALL compared with DLBCL (overall response rate, 8.6% vs 40.2%).21
On the basis of the manageable safety and tolerability and short half-life of loncastuximab tesirine seen with the Q3W doing schedule in patients with R/R B-cell ALL, the DESC decided to explore a QW dosing schedule. Five patients received 50 μg/kg of loncastuximab tesirine on the QW schedule. Weekly dosing did not result in DLTs, but there was no change in the efficacy signal in this small group.
In the setting of this phase 1 study in heavily pretreated patients, there were signals of efficacy in B-ALL. Three of the 35 patients treated achieved a CR, 1 each in the 30, 120, and 150 μg/kg Q3W dose schedules. Two of these 3 patients had received prior blinatumomab and had discontinued blinatumomab because of toxicity. There was no pattern with regard to blast counts before receiving loncastuximab tesirine (≤5%, 5%-25%, and >50%, respectively). Although responses observed with loncastuximab tesirine gave an indication of preliminary efficacy in the B-ALL population, the efficacy overall was less promising than that of loncastuximab tesirine in other diseases under investigation, such as DLBCL (CR rate, 40.2%).21 Moreover, the response rates observed in this trial were lower than those seen with other novel agents recently approved for patients with R/R B-ALL, such as blinatumomab (CR rate, 44%), inotuzumab ozogamycin (CR rate, 80.7%), and tisagenleucel CAR-T cells (CR rate, 90%).22 However, the clinical activity of loncastuximab tesirine seen in the present study occurred in a heavily pretreated population, and its efficacy could potentially be improved if it could be safely combined with chemotherapy. Although data are limited and require further investigation, preliminary evidence exploring the correlation between PKs and clinical activity of loncastuximab tesirine suggests that high levels of B lymphocytes might limit exposure of B-ALL cells to this agent, and it is therefore possible that B-cell depletion before administration of loncastuximab tesirine in B-ALL might increase exposure and consequently efficacy. Loncastuximab tesirine could also be explored as a consolidation therapy.
The recommended dose of loncastuximab tesirine was not determined because of the early termination of the study. However, single-agent loncastuximab tesirine was well tolerated, with only 1 DLT and 3 CRs observed in a heavily pretreated R/R B-ALL population. Future studies could explore the efficacy of loncastuximab tesirine in combination with other treatments or determine a subpopulation of ALL patients who might be sensitive to this therapy.
Acknowledgments
This work was supported by ADC Therapeutics SA. Editorial assistance was provided by Becky Salisbury and Eshvendar Reddy Kasala at Fishawack Communications, funded by ADC Therapeutics SA. A.Z. is a Leukemia and Lymphoma Society Scholar in Clinical Research and is also supported by a National Cancer Institute (NCI) Cancer Clinical Investigator Team Leadership Award. Research reported in this publication was supported in part by the NCI of the National Institutes of Health (NIH) under Award P30 CA016359.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Authorship
Contribution: N.J., W.S., A.Z., E.A., J.M., L.H., B.T., B.B., J.F., D.U., G.C., Y.Q., H.K., and M.J.W. were involved in data acquisition, analysis, and interpretation; K.H. was involved in the basic design of study and acquisition and analysis of data; X.Z. was involved in data analysis and interpretation; all authors meet the International Committee of Medical Journal Editors criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole, contributed to the writing and reviewing of the manuscript, and have given final approval of the version to be published; and all authors had full access to all the data in this study and take complete responsibility for the integrity of the data and accuracy of the data analysis.
Conflict-of-interest disclosure: N.J. has received research funding and honoraria from ADC Therapeutics SA, research funding and honoraria from Servier, research funding and honoraria from Pfizer, research funding and honoraria from Precision Biosciences, research funding from Cellectis, research funding from Incyte, and research funding and honoraria from Adaptive Biotechnologies. A.Z. has received research funding (institutional) from Celgene, Acceleron, AbbVie, Otsuka, Pfizer, Medimmune/AstraZeneca, Boehringer Ingelheim, Trovagene, Incyte, Takeda, and ADC Therapeutics and has also had a consultancy agreement with and received honoraria from AbbVie, Otsuka, Pfizer, Celgene, Jazz, Ariad, Incyte, Agios, Boehringer Ingelheim, Novartis, Acceleron, Astellas, Daiichi Sankyo, Cardinal Health, Seattle Genetics, BeyondSpring, Trovagene, and Takeda (none of these relationships were related to the development of this manuscript). L.H. has received institutional research funding from ADC Therapeutics SA. N.J., H.K., and W.S. are advisory board members for ADC Therapeutics SA. J.F., D.U., G.C., X.Z., Y.Q., and K.H. are employees of ADC Therapeutics with stock options. B.B. is an advisory board member for Novartis and Astellas. The remaining authors declare no competing financial interests.
Correspondence: Nitin Jain, Department of Leukemia, The University of Texas MD Anderson Cancer Center, 1515 Holcombe Blvd, Unit 428, Houston, TX 77401; e-mail: njain@mdanderson.org.

